Molecular Dynamics Simulations Suggest a Non-Doublet Decoding Model of −1 Frameshifting by tRNASer3



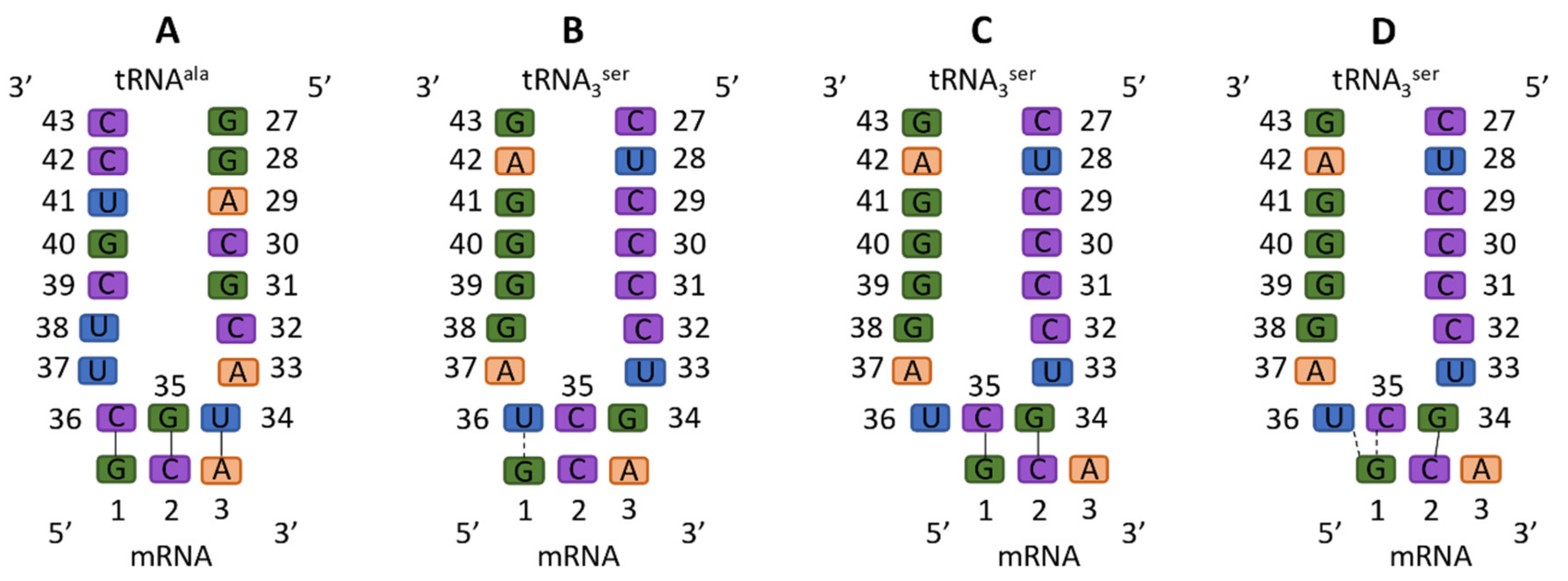{kind=link}
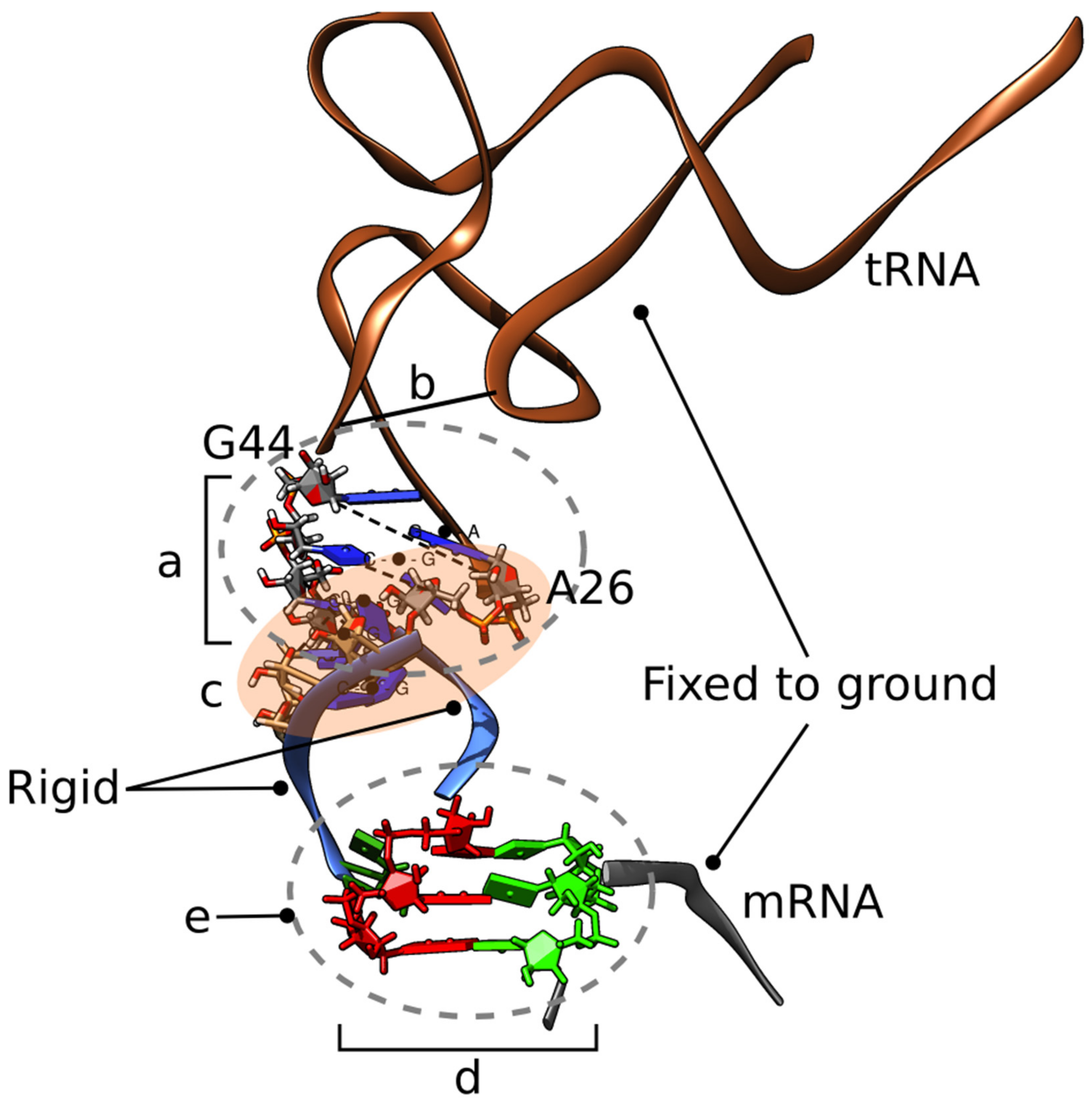{kind=link}
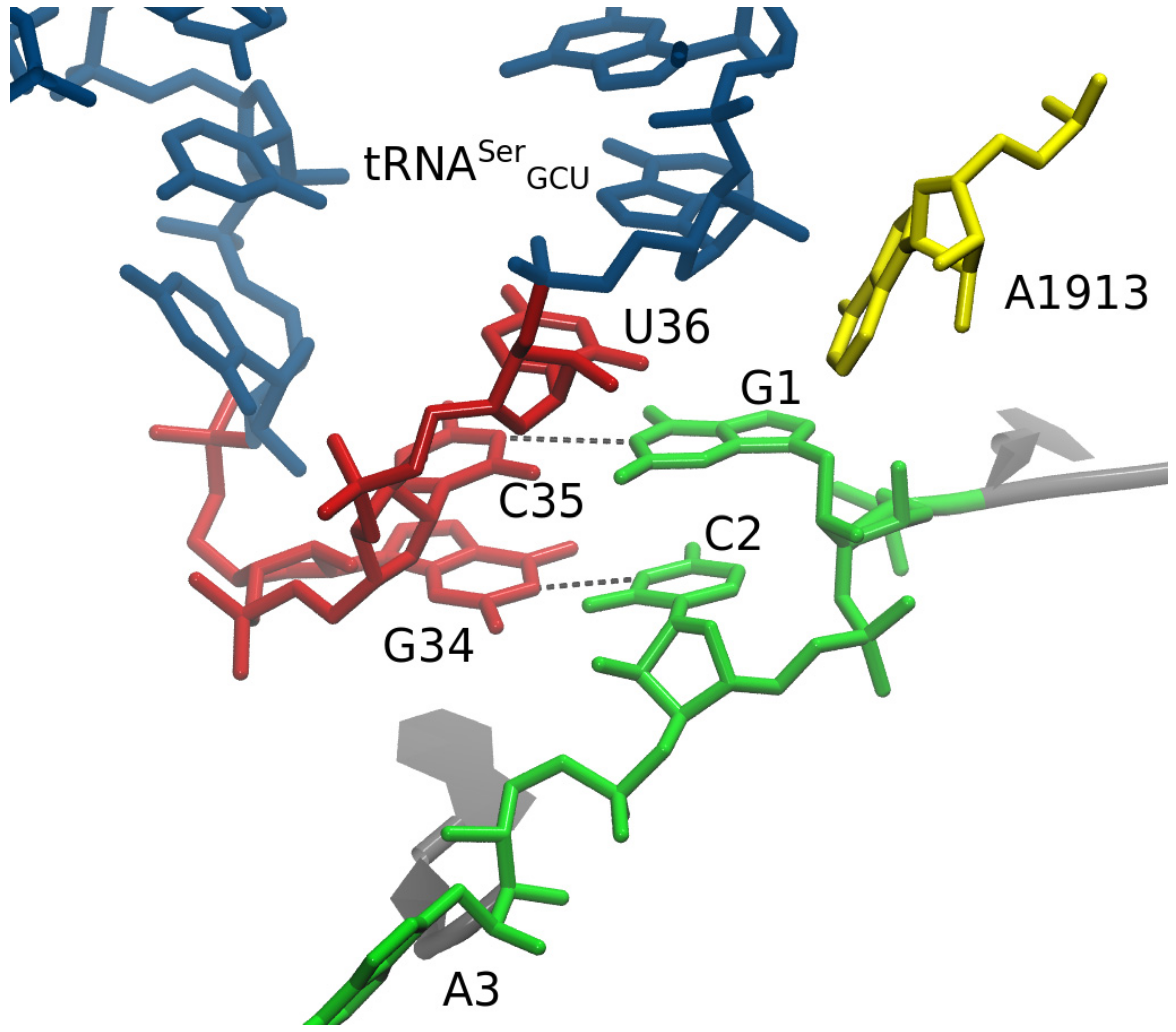{kind=link}
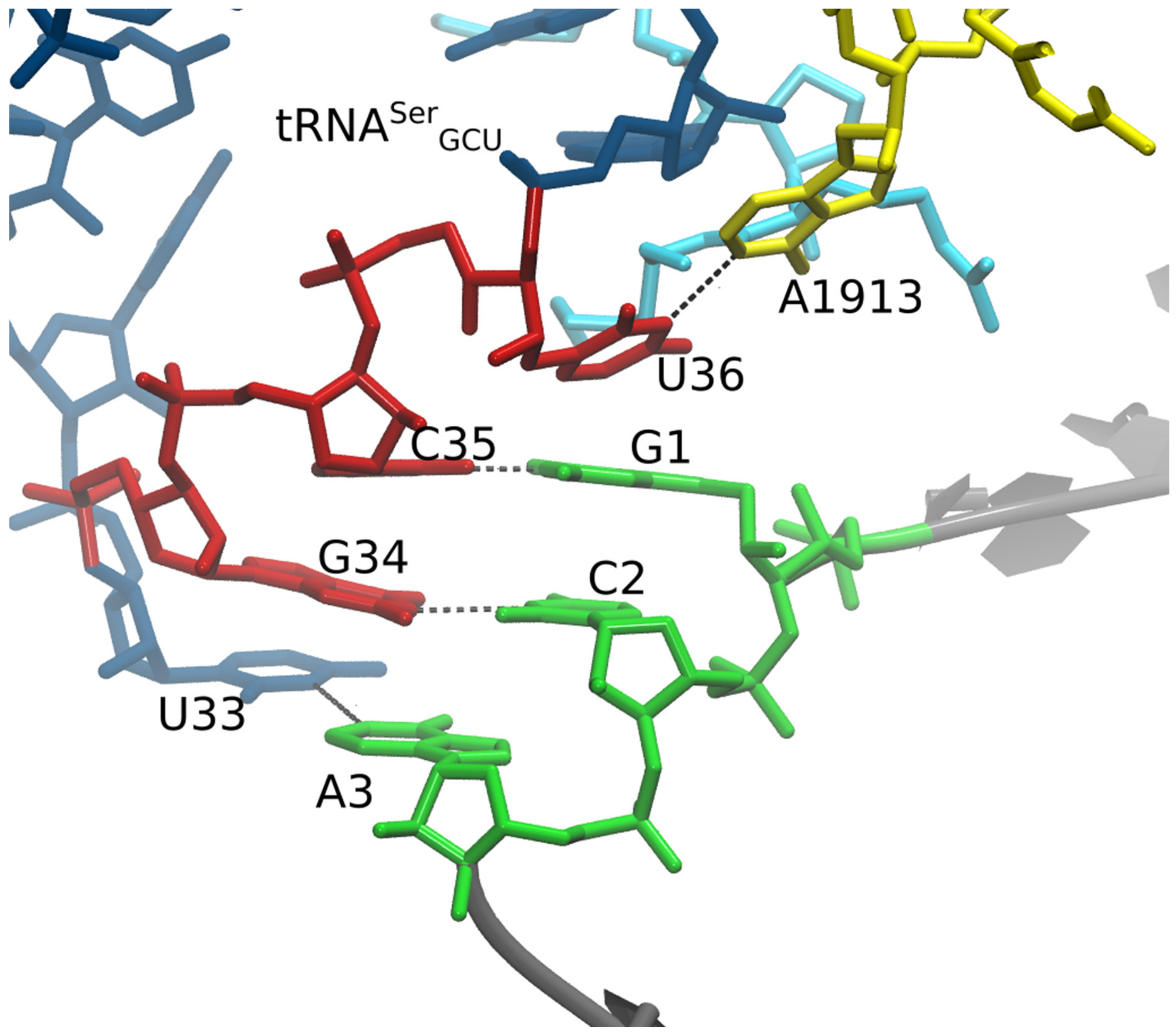{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. MMB Modeling
2.2. Molecular Dynamics (MD) Simulations
2.2.1. Mutant Ribosome Structure Preparation
2.2.2. Molecular Dynamics Simulation Protocol
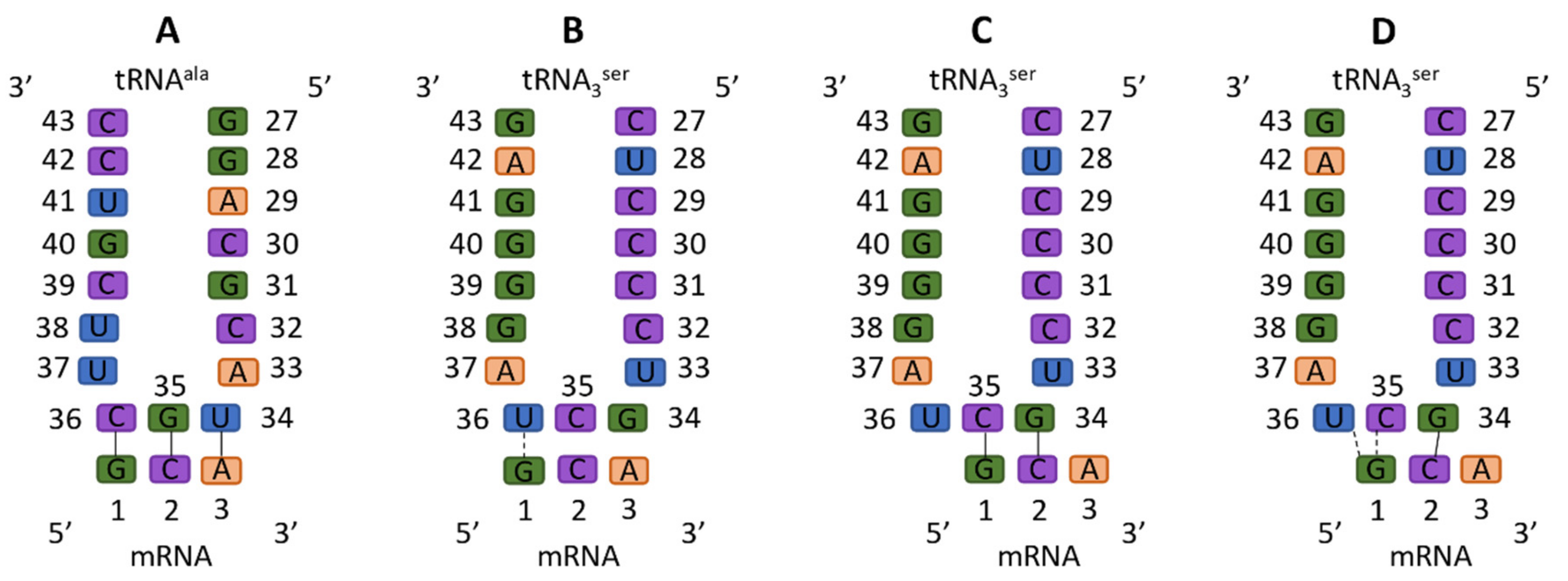
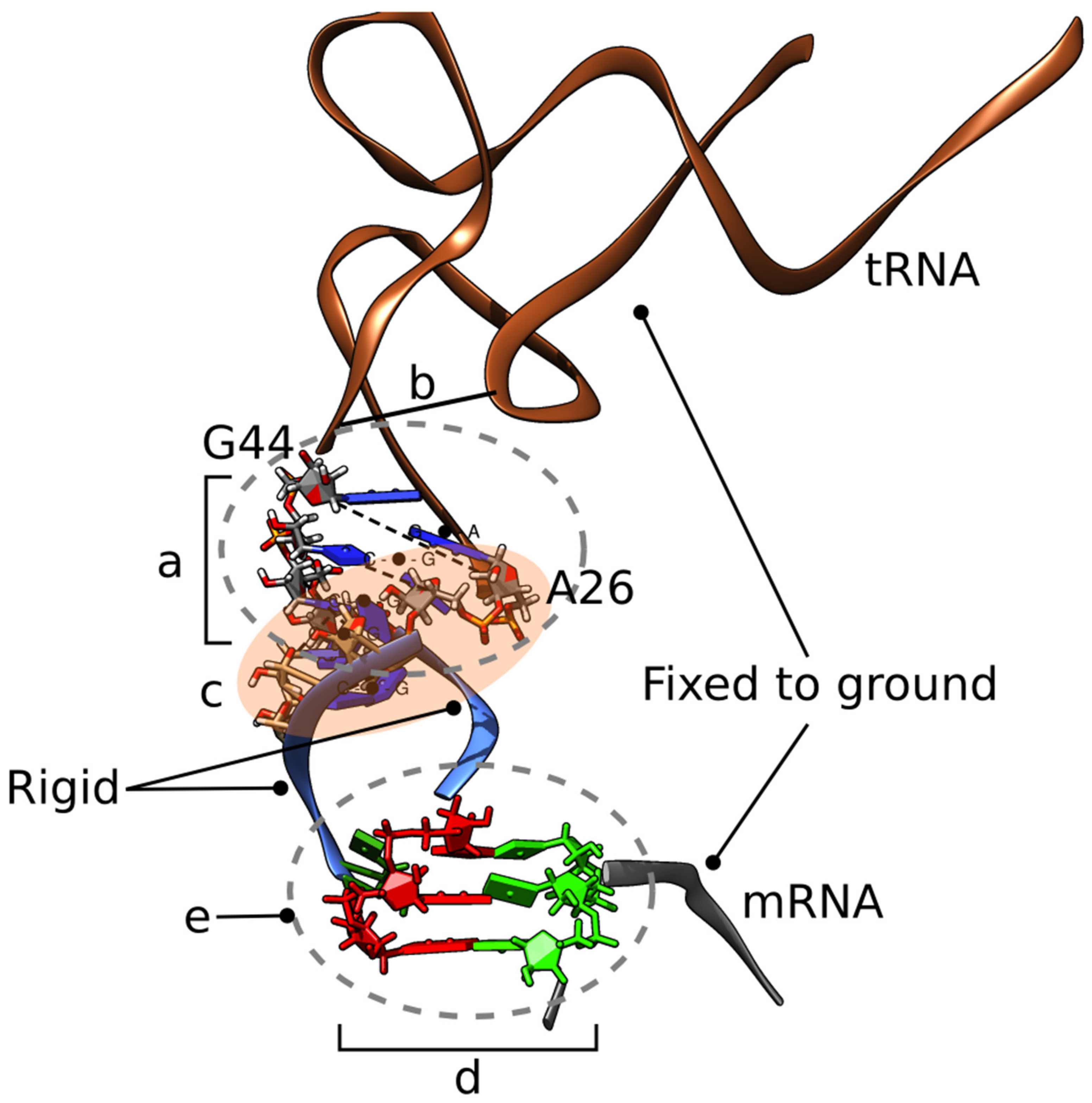
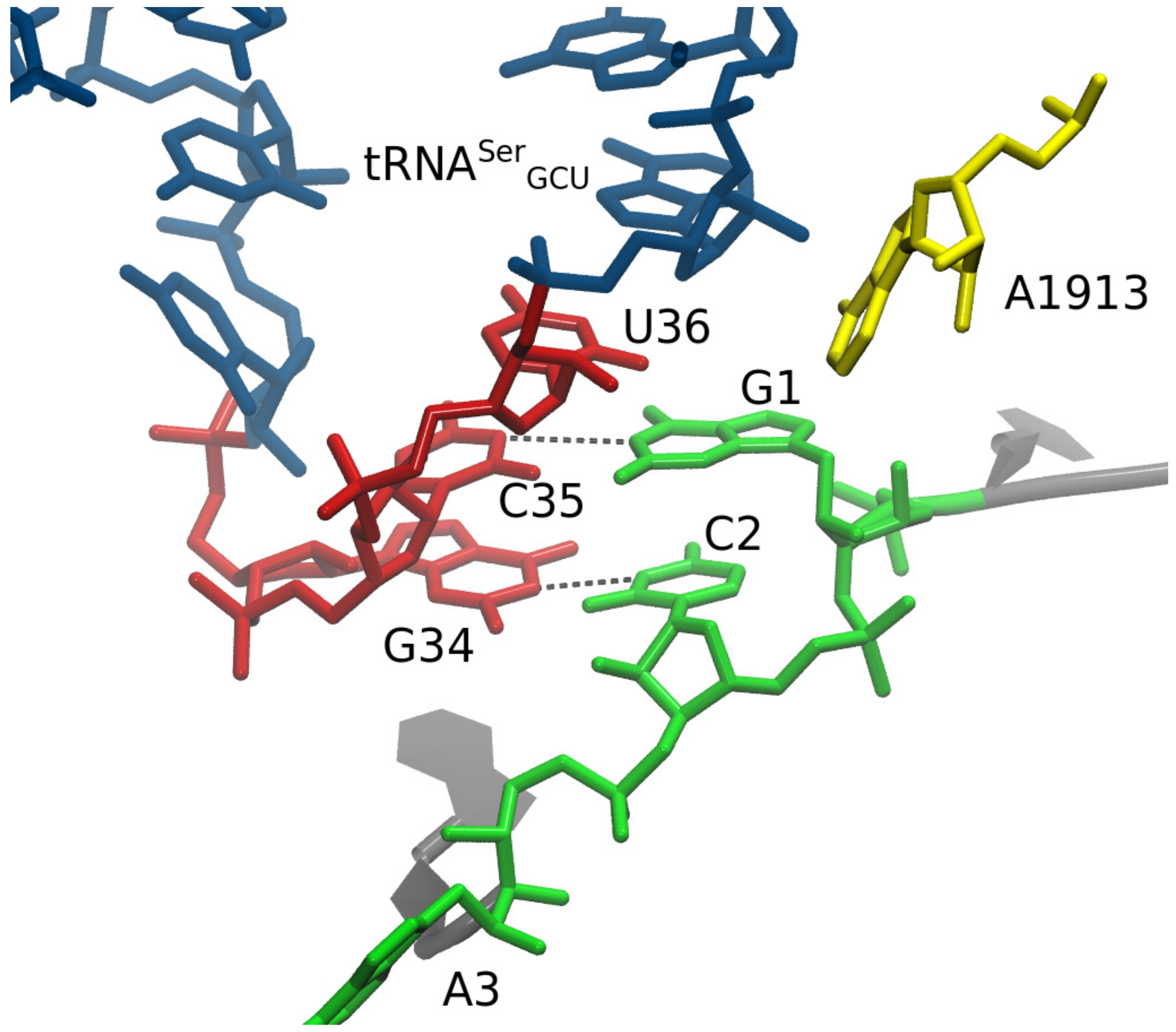
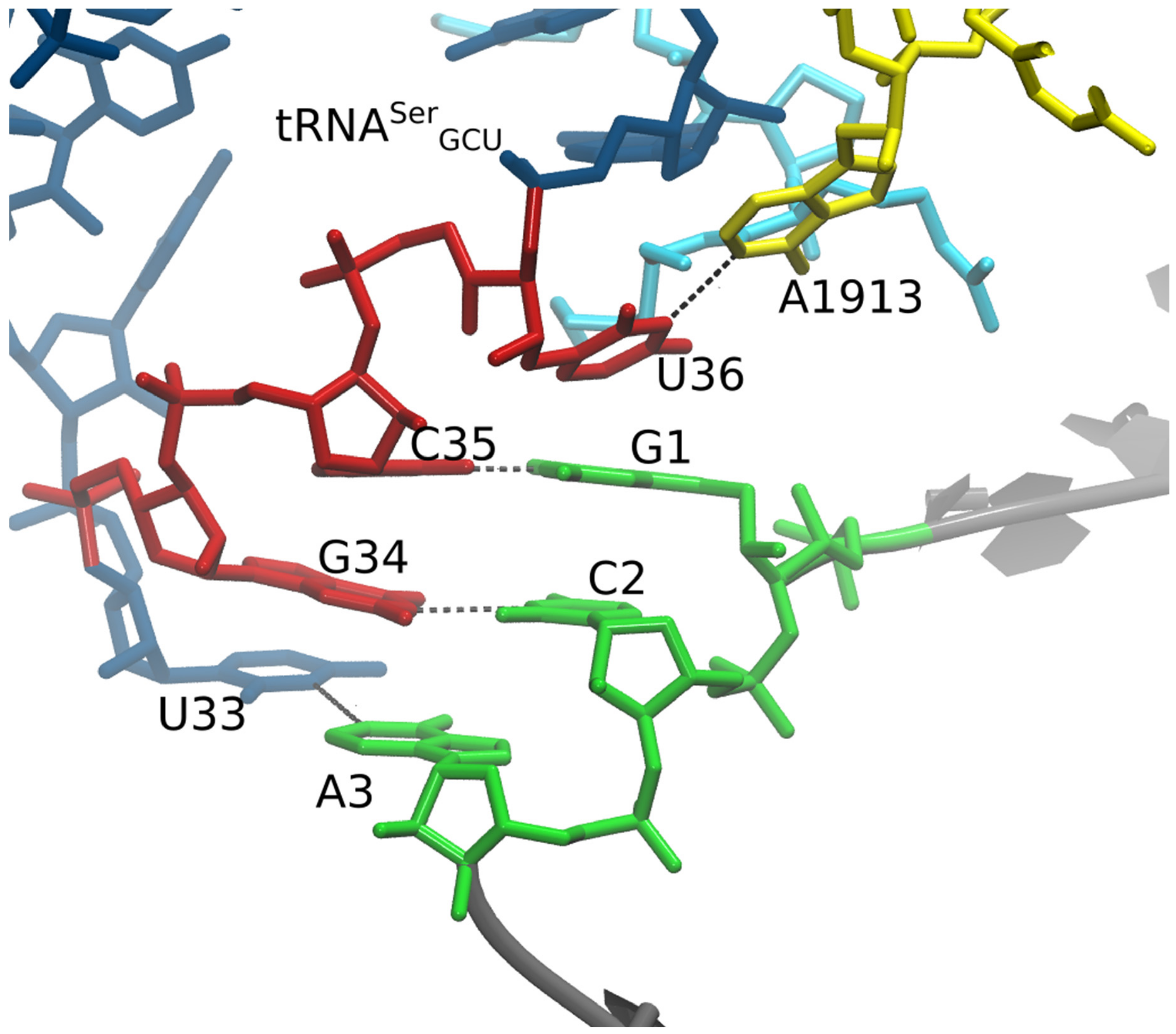
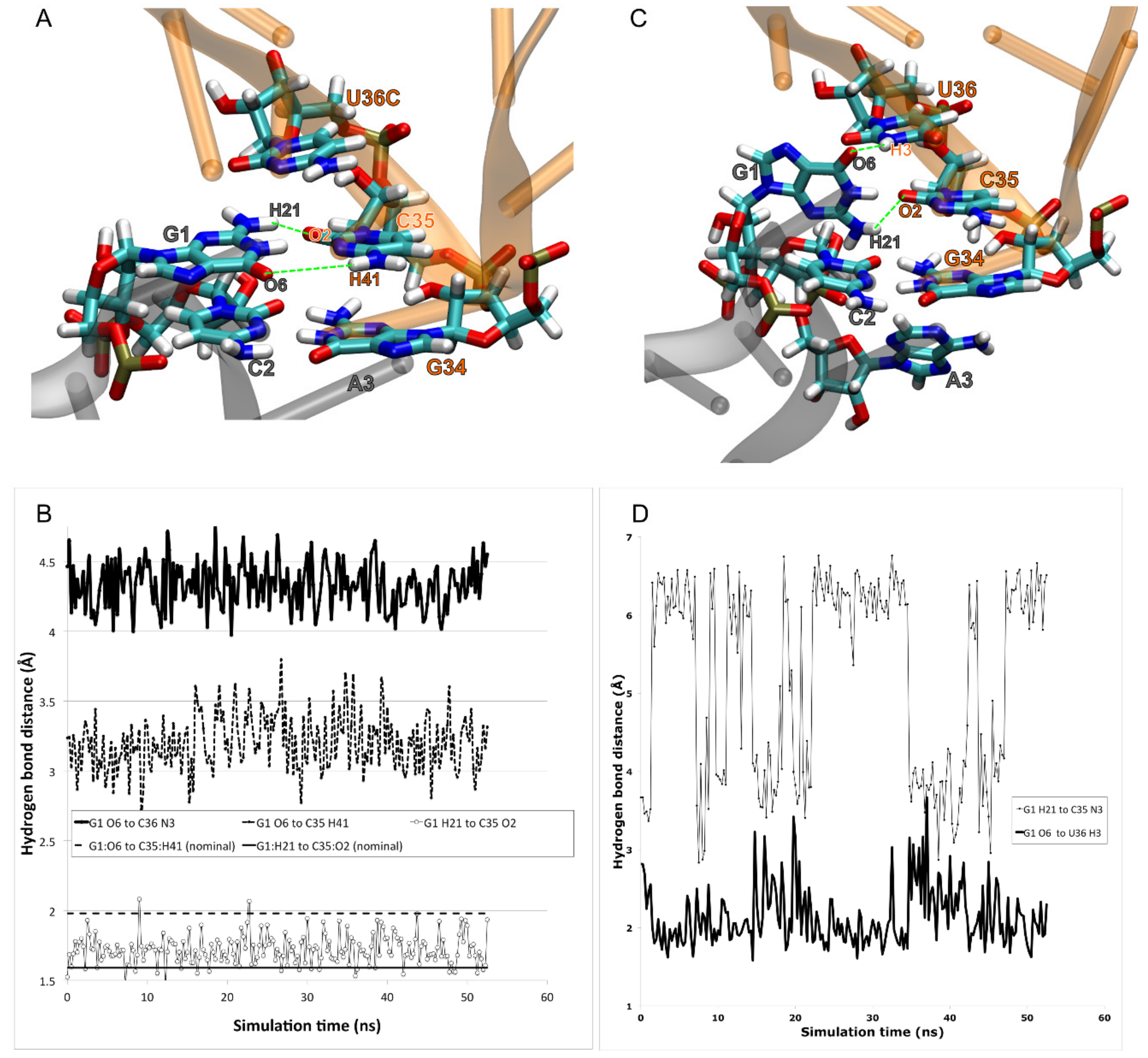
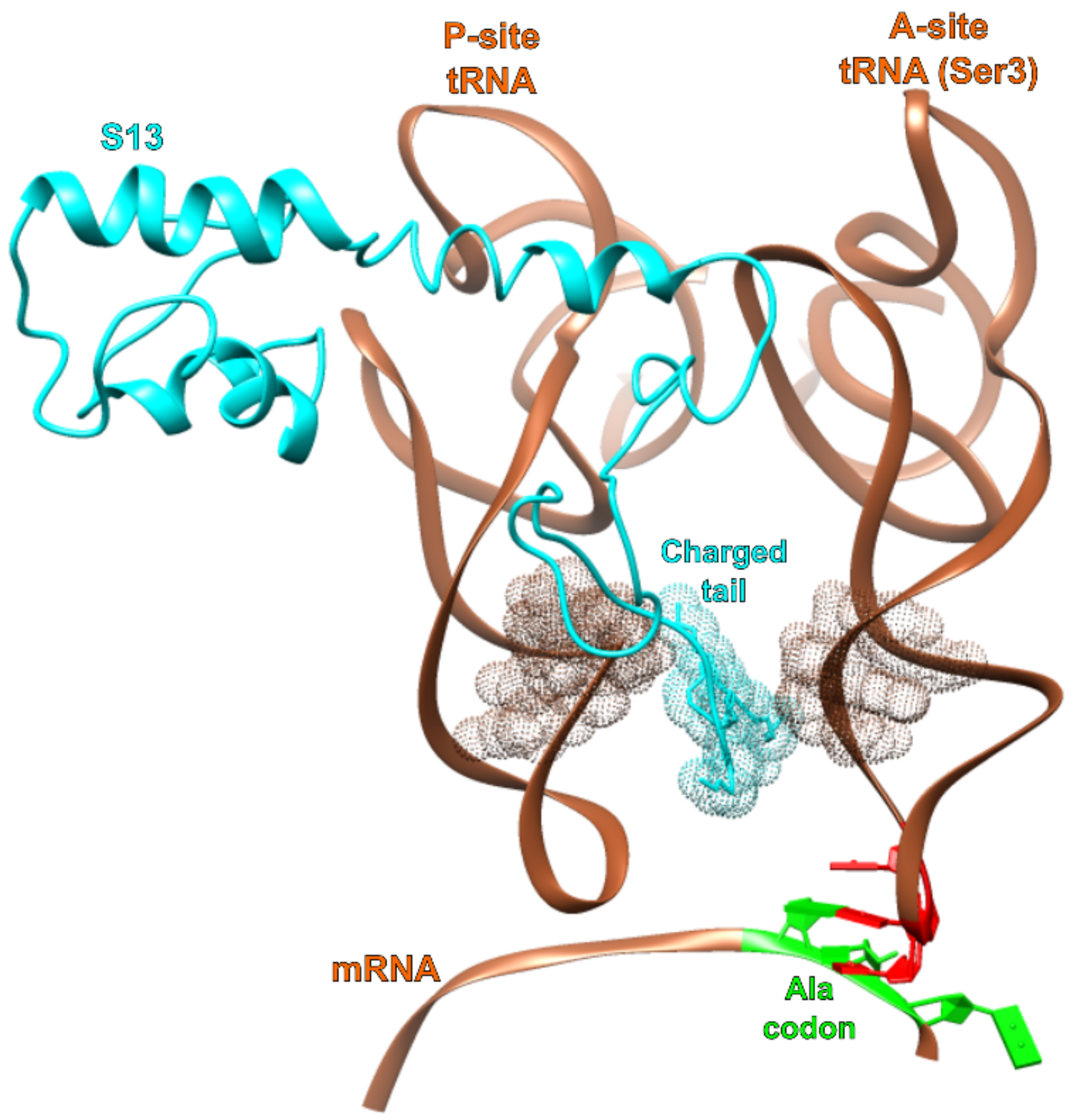
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kapur, M.; Ackerman, S.L. mRNA Translation Gone Awry: Translation Fidelity and Neurological Disease. Trends. Genet. 2018, 34, 218–231. [Google Scholar] [CrossRef]
- Atkins, J.F.; Loughran, G.; Bhatt, P.R.; Firth, A.E.; Baranov, P.V. Ribosomal frameshifting and transcriptional slippage: From genetic steganography and cryptography to adventitious use. Nucleic Acids Res. 2016, 44, 7007–7078. [Google Scholar] [CrossRef]
- Dabrowski, M.; Bukowy-Bieryllo, Z.; Zietkiewicz, E. Advances in therapeutic use of a drug-stimulated translational readthrough of premature termination codons. Mol. Med. 2018, 24, 25. [Google Scholar] [CrossRef] [PubMed]
- Demeshkina, N.; Jenner, L.; Westhof, E.; Yusupov, M.; Yusupova, G. A new understanding of the decoding principle on the ribosome. Nature 2012, 484, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Selmer, M.; Dunham, C.M.; Murphy, F.V., 4th; Weixlbaumer, A.; Petry, S.; Kelley, A.C.; Weir, J.R.; Ramakrishnan, V. Structure of the 70S ribosome complexed with mRNA and tRNA. Science 2006, 313, 1935–1942. [Google Scholar] [CrossRef] [PubMed]
- Manickam, N.; Nag, N.; Abbasi, A.; Patel, K.; Farabaugh, P.J. Studies of translational misreading in vivo show that the ribosome very efficiently discriminates against most potential errors. RNA 2014, 20, 9–15. [Google Scholar] [CrossRef]
- Ivanov, I.P.; Gesteland, R.F.; Matsufuji, S.; Atkins, J.F. Programmed frameshifting in the synthesis of mammalian antizyme is +1 in mammals, predominantly +1 in fission yeast, but -2 in budding yeast. RNA 1998, 4, 1230–1238. [Google Scholar] [CrossRef]
- Lin, Z.; Gilbert, R.J.; Brierley, I. Spacer-length dependence of programmed -1 or -2 ribosomal frameshifting on a U6A heptamer supports a role for messenger RNA (mRNA) tension in frameshifting. Nucleic Acids Res. 2012, 40, 8674–8689. [Google Scholar] [CrossRef]
- Weiss, R.B.; Dunn, D.M.; Atkins, J.F.; Gesteland, R.F. Slippery runs, shifty stops, backward steps, and forward hops: −2, −1, +1, +2, +5, and +6 ribosomal frameshifting. Cold Spring Harb. Symp. Quant. Biol. 1987, 52, 687–693. [Google Scholar] [CrossRef]
- Davies, J.E.; Rubinsztein, D.C. Polyalanine and polyserine frameshift products in Huntington’s disease. J. Med. Genet. 2006, 43, 893–896. [Google Scholar] [CrossRef]
- Girstmair, H.; Saffert, P.; Rode, S.; Czech, A.; Holland, G.; Bannert, N.; Ignatova, Z. Depletion of cognate charged transfer RNA causes translational frameshifting within the expanded CAG stretch in huntingtin. Cell Rep. 2013, 3, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Saffert, P.; Adamla, F.; Schieweck, R.; Atkins, J.F.; Ignatova, Z. An Expanded CAG Repeat in Huntingtin Causes +1 Frameshifting. J. Biol. Chem. 2016, 291, 18505–18513. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, C.; Jannatipour, M.; Dion, P.; Laganière, J.; Sequeiros, J.; Brais, B.; Rouleau, G.A. CAG tract of MJD-1 may be prone to frameshifts causing polyalanine accumulation. Hum. Mol. Genet. 2000, 9, 1957–1966. [Google Scholar] [CrossRef] [PubMed]
- Toulouse, A.; Au-Yeung, F.; Gaspar, C.; Roussel, J.; Dion, P.; Rouleau, G.A. Ribosomal frameshifting on MJD-1 transcripts with long CAG tracts. Hum. Mol. Genet. 2005, 14, 2649–2660. [Google Scholar] [CrossRef] [PubMed]
- Parker, J. Errors and alternatives in reading the universal genetic code. Microbiol. Rev. 1989, 53, 273–298. [Google Scholar]
- Farabaugh, P.J. Programmed translational frameshifting. Annu. Rev. Genet. 1996, 30, 507–528. [Google Scholar] [CrossRef]
- Chen, Y.; Chang, K.; Hu, H.; Chen, Y.; Lin, Y.; Hsu, C.; Chang, C.; Chang, K.; Wen, J. Coordination among tertiary base pairs results in an efficient frameshift-stimulating RNA pseudoknot. Nucleic Acids Res. 2017, 45, 6011–6022. [Google Scholar] [CrossRef]
- Zhong, Z.; Yang, L.; Zhang, H.; Shi, J.; Vandana, J.J.; Lam, D.T.U.H.; Rene, C.L.; Lu, O.L.; Chen, G. Mechanical unfolding kinetics of the SRV-1 gag-pro mRNA pseudoknot: possible implications for −1 ribosomal frameshifting stimulation. Sci. Rep. 2016, 6, 39549. [Google Scholar] [CrossRef]
- Meydan, S.; Klepacki, D.; Karthikeyan, S.; Margus, T.; Thomas, P.; Jones, J.E.; Khan, Y.; Briggs, J.; Dinman, J.D.; Vázquez-Laslop, N.; et al. Programmed Ribosomal Frameshifting Generates a Copper Transporter and a Copper Chaperone from the Same Gene. Mol. Cell. 2017, 65, 207–219. [Google Scholar] [CrossRef]
- Sundararajan, A.; Michaud, W.A.; Qian, Q.; Stahl, G.; Farabaugh, P.J. Near-cognate peptidyl-tRNAs promote +1 programmed translational frameshifting in yeast. Mol. Cell. 1999, 4, 1005–1015. [Google Scholar] [CrossRef]
- Clark, M.B.; Jänicke, M.; Gottesbühren, U.; Kleffmann, T.; Legge, M.; Poole, E.S.; Tate, W.P. Mammalian gene PEG10 expresses two reading frames by high efficiency -1 frameshifting in embryonic-associated tissues. J. Biol. Chem. 2007, 282, 37359–37369. [Google Scholar] [CrossRef] [PubMed]
- Saulquin, X.; Scotet, E.; Trautmann, L.; Peyrat, M.A.; Halary, F.; Bonneville, M.; Houssaint, E. +1 Frameshifting as a novel mechanism to generate a cryptic cytotoxic T lymphocyte epitope derived from human interleukin 10. J. Exp. Med. 2002, 195, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Mears, J.A.; Sharma, M.R.; Gutell, R.R.; McCook, A.S.; Richardson, P.E.; Caulfield, T.R.; Agrawal, R.K.; Harvey, S.C. A structural model for the large subunit of the mammalian mitochondrial ribosome. J. Mol. Biol. 2006, 358, 193–212. [Google Scholar] [CrossRef] [PubMed]
- Temperley, R.; Richter, R.; Dennerlein, S.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M. Hungry codons promote frameshifting in human mitochondrial ribosomes. Science 2010, 327, 301. [Google Scholar] [CrossRef]
- Dunham, C.M.; Selmer, M.; Phelps, S.S.; Kelley, A.C.; Suzuki, T.; Joseph, S.; Ramakrishnan, V. Structures of tRNAs with an expanded anticodon loop in the decoding center of the 30S ribosomal subunit. RNA 2007, 13, 817–823. [Google Scholar] [CrossRef]
- Fagan, C.E.; Maehigashi, T.; Dunkle, J.A.; Miles, S.J.; Dunham, C.M. Structural insights into translational recoding by frameshift suppressor tRNASufJ. RNA 2014, 20, 1944–1954. [Google Scholar] [CrossRef]
- Hong, S.; Sunita, S.; Maehigashi, T.; Hoffer, E.D.; Dunkle, J.A.; Dunham, C.M. Mechanism of tRNA-mediated +1 ribosomal frameshifting. Proc. Natl. Acad. Sci. USA 2018, 115, 11226–11231. [Google Scholar] [CrossRef]
- Maehigashi, T.; Dunkle, J.A.; Miles, S.J.; Dunham, C.M. Structural insights into +1 frameshifting promoted by expanded or modification-deficient anticodon stem loops. Proc. Natl. Acad. Sci. USA 2014, 111, 12740–12745. [Google Scholar] [CrossRef]
- Nguyen, H.A.; Hoffer, E.D.; Dunham, C.M. Importance of a tRNA anticodon loop modification and a conserved, noncanonical anticodon stem pairing in tRNACGGProfor decoding. J. Biol. Chem. 2019, 294, 5281–5291. [Google Scholar] [CrossRef]
- Atkins, J.F.; Gesteland, R.F.; Reid, B.R.; Anderson, C.W. Normal tRNAs promote ribosomal frameshifting. Cell 1979, 18, 1119–1131. [Google Scholar] [CrossRef]
- Bruce, A.G.; Atkins, J.F.; Gesteland, R.F. tRNA anticodon replacement experiments show that ribosomal frameshifting can be caused by doublet decoding. Proc. Natl. Acad. Sci. USA 1986, 83, 5062–5066. [Google Scholar] [CrossRef] [PubMed]
- Tek, A.; Korostelev, A.A.; Flores, S.C. MMB-GUI: A fast morphing method demonstrates a possible ribosomal tRNA translocation trajectory. Nucleic Acids Res. 2016, 44, 95–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, S.C.; Sherman, M.; Bruns, C.M.; Eastman, P.; Altman, R.B. Fast flexible modeling of RNA structure using internal coordinates. IEEE ACM Trans. Comput. Biol. Bioinform. 2011, 8, 1247–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leontis, N.B.; Stombaugh, J.; Westhof, E. The non-Watson-Crick base pairs and their associated isostericity matrices. Nucleic Acids Res. 2002, 30, 3497–3531. [Google Scholar] [CrossRef]
- Flores, S.C.; Altman, R.B. Turning limited experimental information into 3D models of RNA. RNA 2010, 16, 1769–1778. [Google Scholar] [CrossRef] [Green Version]
- Voorhees, R.M.; Weixlbaumer, A.; Loakes, D.; Kelley, A.C.; Ramakrishnan, V. Insights into substrate stabilization from snapshots of the peptidyl transferase center of the intact 70S ribosome. Nat. Struct. Mol. Biol. 2009, 16, 528–533. [Google Scholar] [CrossRef] [Green Version]
- Cavuzic, M.; Liu, Y. Biosynthesis of Sulfur-Containing tRNA Modifications: A Comparison of Bacterial, Archaeal, and Eukaryotic Pathways. Biomolecules 2017, 7, 27. [Google Scholar] [CrossRef] [Green Version]
- Cui, Z.; Wu, Y.; Mureev, S.; Alexandrov, K. Oligonucleotide-mediated tRNA sequestration enables one-pot sense codon reassignment in vitro. Nucleic Acids Res. 2018, 46, 6387–6400. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Laimer, J.; Hofer, H.; Fritz, M.; Wegenkittl, S.; Lackner, P. MAESTRO—Multi agent stability prediction upon point mutations. BMC Bioinform. 2015, 16, 116. [Google Scholar] [CrossRef] [Green Version]
- Watts, K.S.; Dalal, P.; Tebben, A.J.; Cheney, D.L.; Shelley, J.C. Macrocycle conformational sampling with MacroModel. J. Chem. Inf. Model 2014, 54, 2680–2696. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA—A self-parameterizing force field. Proteins 2002, 47, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Caulfield, T.; Devkota, B. Motion of transfer RNA from the A/T state into the A-site using docking and simulations. Proteins 2012, 80, 2489–2500. [Google Scholar] [CrossRef] [PubMed]
- Caulfield, T.; Medina-Franco, J.L. Molecular dynamics simulations of human DNA methyltransferase 3B with selective inhibitor nanaomycin A. J. Struct. Biol. 2011, 176, 185–191. [Google Scholar] [CrossRef]
- Caulfield, T.R. Inter-ring rotation of apolipoprotein A-I protein monomers for the double-belt model using biased molecular dynamics. J. Mol. Graph. Model 2011, 29, 1006–1014. [Google Scholar] [CrossRef]
- Caulfield, T.R.; Devkota, B.; Rollins, G.C. Examinations of tRNA Range of Motion Using Simulations of Cryo-EM Microscopy and X-Ray Data. J. Biophys. 2011, 2011, 219515. [Google Scholar] [CrossRef]
- Caulfield, T.R.; Fiesel, F.C.; Moussaud-Lamodière, E.L.; Dourado, D.F.; Flores, S.C.; Springer, W. Phosphorylation by PINK1 releases the UBL domain and initializes the conformational opening of the E3 ubiquitin ligase Parkin. PLoS Comput. Biol. 2014, 10, e1003935. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Chem. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Reblova, K.; Fadrná, E.; Sarzynska, J.; Kulinski, T.; Kulhánek, P.; Ennifar, E.; Koča, J.; Šponer, J. Conformations of flanking bases in HIV-1 RNA DIS kissing complexes studied by molecular dynamics. Biophys. J. 2007, 93, 3932–3949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reblova, K.; Lankaš, F.; Rázga, F.; Krasovska, M.V.; Koča, J.; Šponer, J. Structure, dynamics, and elasticity of free 16s rRNA helix 44 studied by molecular dynamics simulations. Biopolymers 2006, 82, 504–520. [Google Scholar] [CrossRef]
- Cheatham, T.E., III; Young, M.A. Molecular dynamics simulation of nucleic acids: Successes, limitations and promise. Biopolymers 2001, 56, 232–256. [Google Scholar] [CrossRef]
- Sponer, J.; Banáš, P.; Jurečka, P.; Zgarbová, M.; Kührová, P.; Havrila, M.; Krepl, M.; Stadlbauer, P.; Otyepka, M. Molecular Dynamics Simulations of Nucleic Acids from Tetranucleotides to the Ribosome. J. Phys. Chem. Lett. 2014, 5, 1771–1782. [Google Scholar]
- Kayode, O.; Wang, R.; Pendlebury, D.F.; Cohen, I.; Henin, R.D.; Hockla, A.; Soares, A.S.; Papo, N.; Caulfield, T.R.; Radisky, E.S. An Acrobatic Substrate Metamorphosis Reveals a Requirement for Substrate Conformational Dynamics in Trypsin Proteolysis. J. Biol. Chem. 2016, 291, 26304–26319. [Google Scholar] [CrossRef] [Green Version]
- Flores, S.C.; Bernauer, J.; Shin, S.; Zhou, R.; Huang, X. Multiscale modeling of macromolecular biosystems. Brief. Bioinform. 2012, 13, 395–405. [Google Scholar] [CrossRef] [Green Version]
- Flores, S.C.; Wan, Y.; Russell, R.; Altman, R.B. Predicting RNA structure by multiple template homology modeling. Pac. Symp. Biocomput. 2010, 216–227. [Google Scholar] [CrossRef] [Green Version]
- Flores, S.C. Fast fitting to low resolution density maps: Elucidating large-scale motions of the ribosome. Nucleic Acids Res. 2014, 42, e9. [Google Scholar] [CrossRef] [Green Version]
- Koripella, R.K.; Holm, M.; Dourado, D.; Mandava, C.S.; Flores, S.; Sanyal, S. A conserved histidine in switch-II of EF-G moderates release of inorganic phosphate. Sci. Rep. 2015, 5, 12970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cukras, A.R.; Green, R. Multiple effects of S13 in modulating the strength of intersubunit interactions in the ribosome during translation. J. Mol. Biol. 2005, 349, 47–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkins, J.F.; Bjork, G.R. A gripping tale of ribosomal frameshifting: extragenic suppressors of frameshift mutations spotlight P-site realignment. Microbiol. Mol. Biol. Rev. 2009, 73, 178–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caulfield, T.; Coban, M.; Tek, A.; Flores, S.C. Molecular Dynamics Simulations Suggest a Non-Doublet Decoding Model of −1 Frameshifting by tRNASer3. Biomolecules 2019, 9, 745. https://doi.org/10.3390/biom9110745
Caulfield T, Coban M, Tek A, Flores SC. Molecular Dynamics Simulations Suggest a Non-Doublet Decoding Model of −1 Frameshifting by tRNASer3. Biomolecules. 2019; 9(11):745. https://doi.org/10.3390/biom9110745
Chicago/Turabian StyleCaulfield, Thomas, Matt Coban, Alex Tek, and Samuel Coulbourn Flores. 2019. "Molecular Dynamics Simulations Suggest a Non-Doublet Decoding Model of −1 Frameshifting by tRNASer3" Biomolecules 9, no. 11: 745. https://doi.org/10.3390/biom9110745
APA StyleCaulfield, T., Coban, M., Tek, A., & Flores, S. C. (2019). Molecular Dynamics Simulations Suggest a Non-Doublet Decoding Model of −1 Frameshifting by tRNASer3. Biomolecules, 9(11), 745. https://doi.org/10.3390/biom9110745