Novel Series of Methyl 3-(Substituted Benzoyl)-7-Substituted-2-Phenylindolizine-1-Carboxylates as Promising Anti-Inflammatory Agents: Molecular Modeling Studies

 ,
,  , , ,
, , ,  , , , ,
, , , ,  ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. General
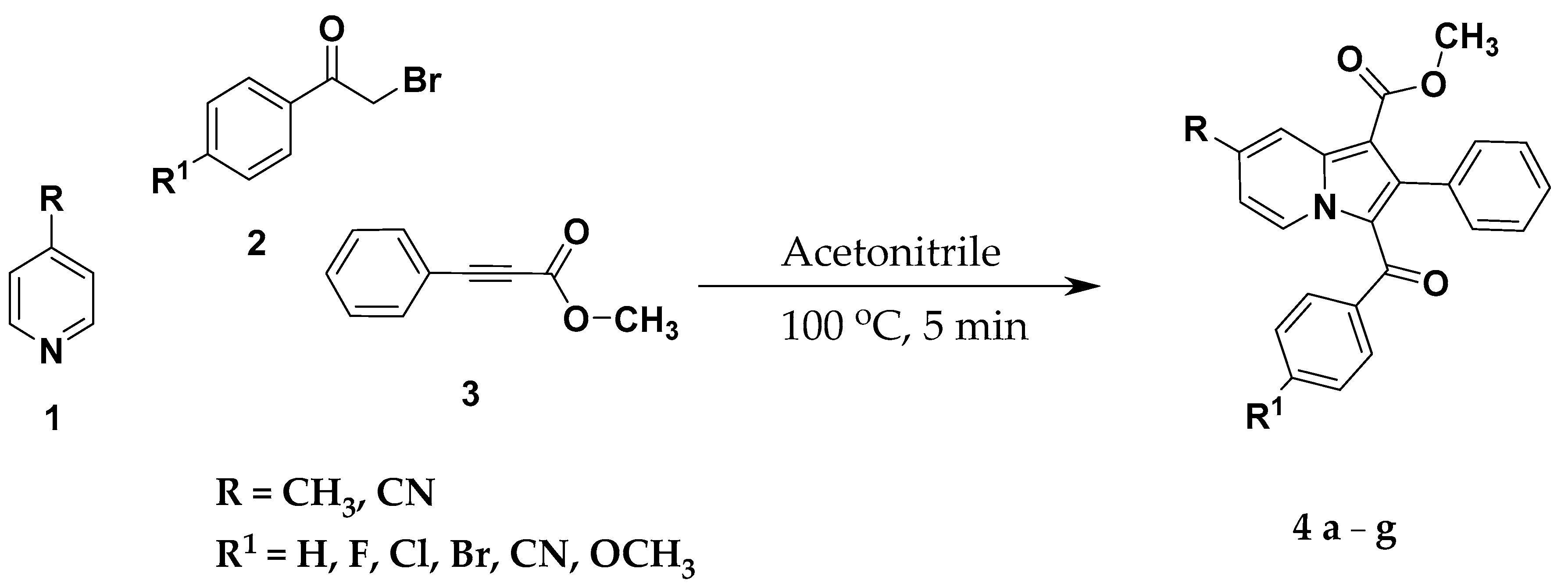
2.2. General Procedure for the Synthesis of Methyl 3-(Substituted Benzoyl)-7-Substituted-2-Phenylindolizine-1-Carboxylate Analogues (4a–g)
2.3. Crystallography
2.4. Pharmacology
2.4.1. COX-2 Inhibition Assay
2.4.2. Statistical Analysis
2.5. Computational Studies
2.5.1. Molecular Docking
2.5.2. ADME Prediction
2.5.3. Toxicity Prediction
3. Results and Discussion
3.1. Chemistry
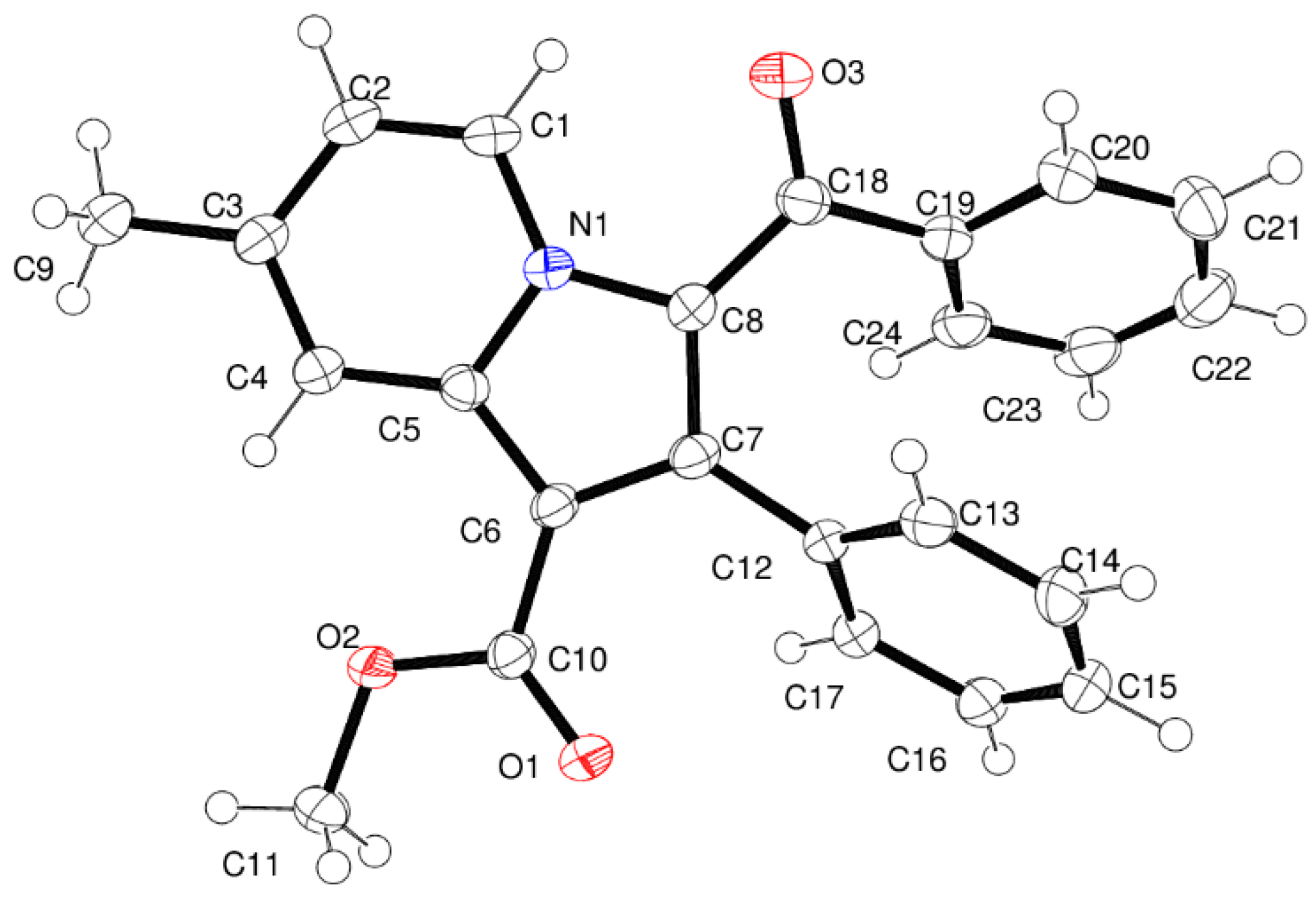
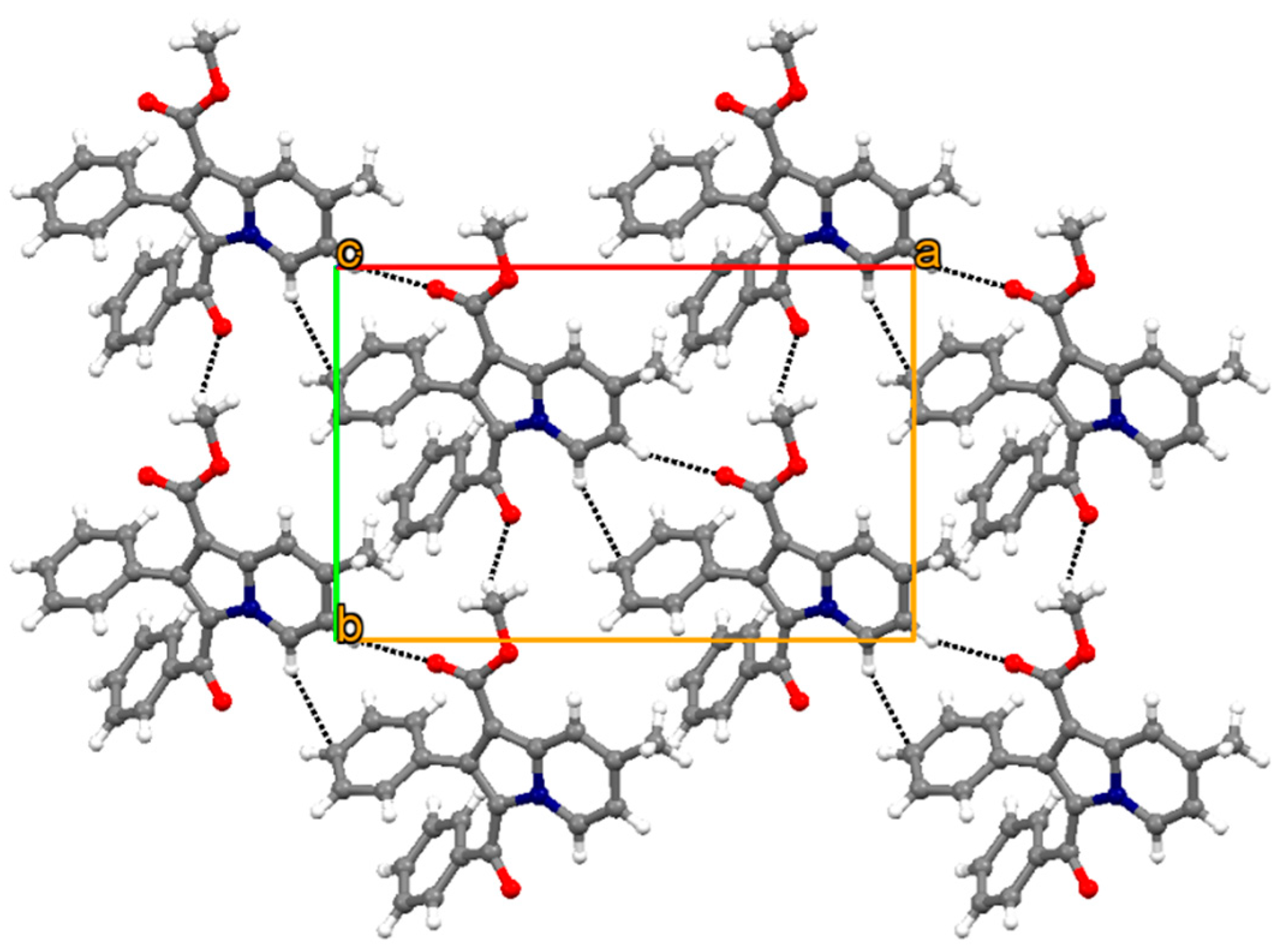
3.2. Crystallography
3.3. Pharmacology
COX-2 Inhibition
3.4. Computational Studies
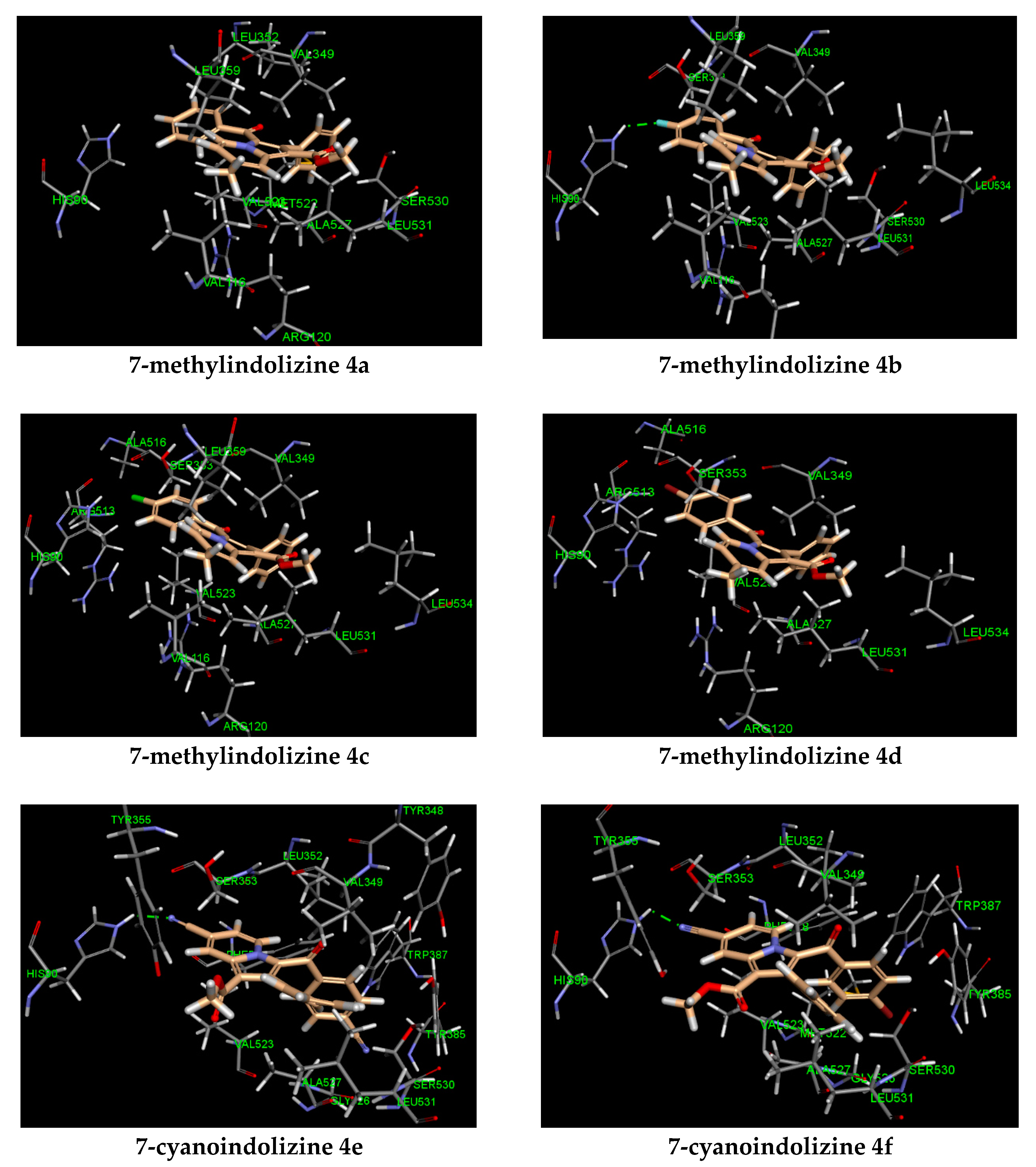
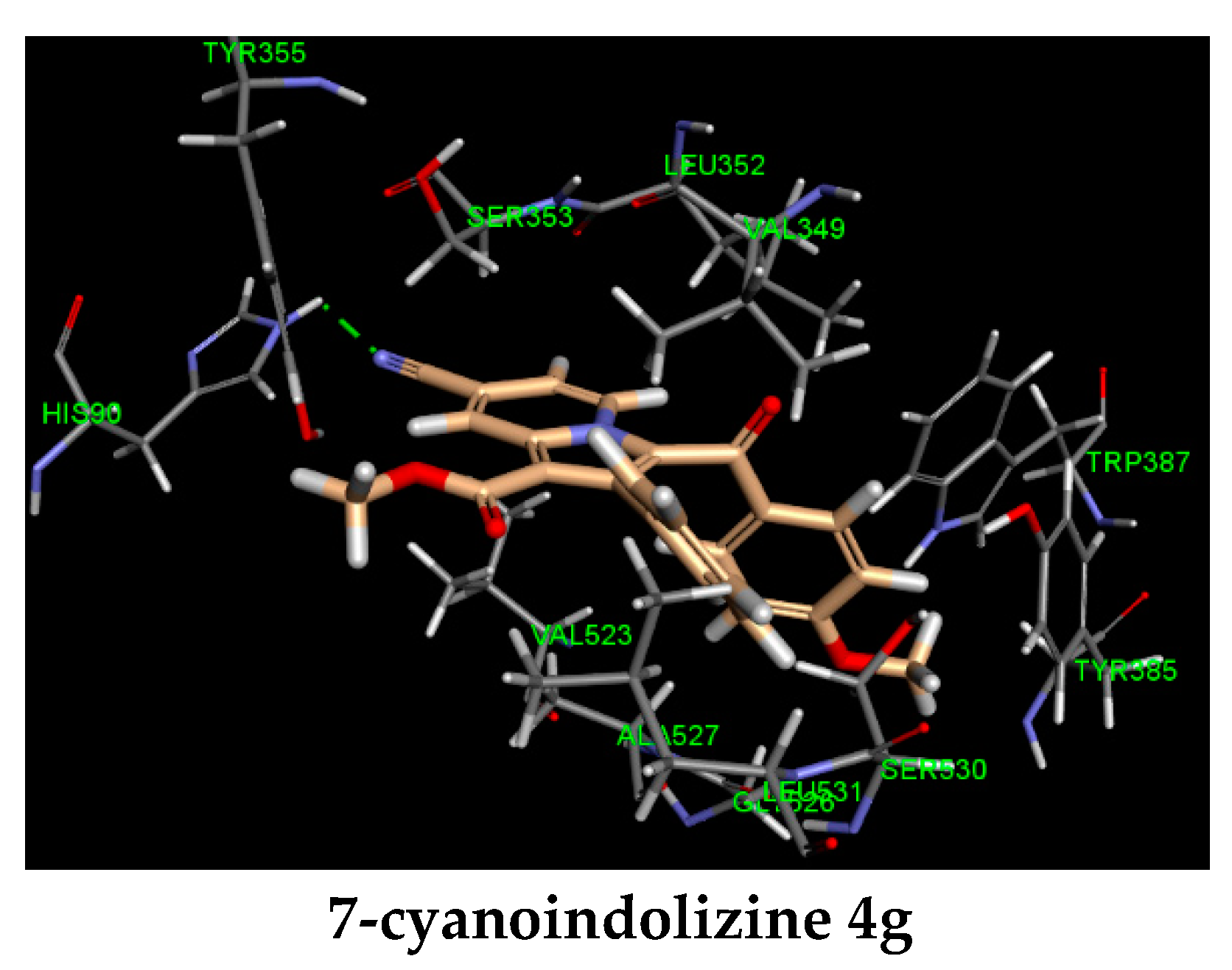
3.4.1. Molecular Modeling
3.4.2. ADME Prediction
3.4.3. Toxicity Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lipscomb, J. Management of Nonsteroidal Anti-inflammatory Drug-Induced Hypersensitivity Reactions. US Pharm. 2019, 44, 22–26. [Google Scholar]
- Ricciotti, E.; Tang, S.-Y.; Barekat, K.; Veglia, F.; Maseda, D.; Bittinger, K.; Bushman, F.; FitzGerald, G.A. The impact of cyclooxygenase-2 selective and non-isoform selective NSAIDs on the gut microbiota. FASEB J. 2019, 33, 516.511. [Google Scholar]
- Moro, M.G.; Oliveira, M.D.d.S.; Oliveira, L.R.d.; Teixeira, S.A.; Muscará, M.N.; Spolidorio, L.C.; Holzhausen, M. Effects of Selective Versus Non-Selective COX-2 Inhibition on Experimental Periodontitis. Braz. Dent. J. 2019, 30, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Amagase, K. Roles of cyclooxygenase, prostaglandin E2 and EP receptors in mucosal protection and ulcer healing in the gastrointestinal tract. Curr. Pharm. Des. 2018, 24, 2002–2011. [Google Scholar] [CrossRef] [PubMed]
- Pannunzio, A.; Coluccia, M. Cyclooxygenase-1 (COX-1) and COX-1 inhibitors in cancer: A review of oncology and medicinal chemistry literature. Pharmaceuticals 2018, 11, 101. [Google Scholar] [CrossRef] [PubMed]
- Mirshafiey, A.; Mortazavi-Jahromi, S.S.; Taeb, M.; Cuzzocrea, S.; Esposito, E. Evaluation of the effect of α-L-guluronic acid (G2013) on COX-1, COX-2 activity and gene expression for introducing this drug as a novel NSAID with immunomodulatory property. Recent Pat. Inflamm. Allergy Drug Discov. 2018, 12, 162–168. [Google Scholar] [CrossRef]
- Zarghi, A.; Zebardast, T.; Daraie, B.; Hedayati, M. Design and synthesis of new 1, 3-benzthiazinan-4-one derivatives as selective cyclooxygenase (COX-2) inhibitors. Bioorg. Med. Chem. 2009, 17, 5369–5373. [Google Scholar] [CrossRef]
- Omar, Y.M.; Abdu-Allah, H.H.; Abdel-Moty, S.G. Synthesis, biological evaluation and docking study of 1, 3, 4-thiadiazole-thiazolidinone hybrids as anti-inflammatory agents with dual inhibition of COX-2 and 15-LOX. Bioorg. Chem. 2018, 80, 461–471. [Google Scholar] [CrossRef]
- Gubin, J.; de Vogelaer, H.; Inion, H.; Houben, C.; Lucchetti, J.; Mahaux, J.; Rosseels, G.; Peiren, M.; Clinet, M. Novel heterocyclic analogs of the new potent class of calcium entry blockers: 1-[[4-(aminoalkoxy) phenyl] sulfonyl] indolizines. J. Med. Chem. 1993, 36, 1425–1433. [Google Scholar] [CrossRef]
- Hagishita, S.; Yamada, M.; Shirahase, K.; Okada, T.; Murakami, Y.; Ito, Y.; Matsuura, T.; Wada, M.; Kato, T.; Ueno, M. Potent inhibitors of secretory phospholipase A2: Synthesis and inhibitory activities of indolizine and indene derivatives. J. Med. Chem. 1996, 39, 3636–3658. [Google Scholar] [CrossRef]
- Chaniyara, R.; Tala, S.; Chen, C.-W.; Zang, X.; Kakadiya, R.; Lin, L.-F.; Chen, C.-H.; Chien, S.-I.; Chou, T.-C.; Tsai, T.-H. Novel antitumor indolizino [6, 7-b] indoles with multiple modes of action: DNA cross-linking and topoisomerase I and II inhibition. J. Med. Chem. 2013, 56, 1544–1563. [Google Scholar] [CrossRef] [PubMed]
- Artico, M.; Massa, S.; Stefancich, G.; Silvestri, R.; Di Santo, R.; Corelli, F. Potential antitumor agents. III. Synthesis of pyrazolo [3, 4-e] pyrrolo [3, 4-g] indolizine and 1H-pyrazolo [3, 4-e] indolizine derivatives. J. Heterocycl. Chem. 1989, 26, 503–507. [Google Scholar] [CrossRef]
- Shrivastava, S.K.; Srivastava, P.; Bandresh, R.; Tripathi, P.N.; Tripathi, A. Design, synthesis, and biological evaluation of some novel indolizine derivatives as dual cyclooxygenase and lipoxygenase inhibitor for anti-inflammatory activity. Bioorg. Med. Chem. 2017, 25, 4424–4432. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekharappa, S.; Venugopala, K.N.; Tratrat, C.; Mahomoodally, F.M.; Aldhubiab, B.E.; Haroun, M.; Venugopala, R.; Mohan, M.K.; Kulkarni, R.S.; Attimarad, M.V. Efficient synthesis and characterization of novel indolizines: Exploration of in vitro COX-2 inhibitory activity and molecular modelling studies. New J. Chem. 2018, 42, 4893–4901. [Google Scholar] [CrossRef]
- Sandeep, C.; Venugopala, K.; Khedr, M.; Padmashali, B.; Kulkarni, R.; Rashmi, V.; Odhav, B. Design and synthesis of novel indolizine analogues as COX-2 inhibitors: Computational perspective and in vitro screening. Indian J. Pharm. Educ. Res. 2017, 51, 452–460. [Google Scholar] [CrossRef]
- SAINT Version 7.60a; Bruker AXS Inc.: Madison, WI, USA, 2006.
- Sheldrick, G.M. SHELXS-97, SHELXL-2014 and SADABS Version 2.05; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Barbour, L.J. X-Seed—A Software Tool for Supramolecular Crystallography; Elsevier: Amsterdam, The Netherlands, 2001. [Google Scholar]
- Jerry, L.A.; Leonard, J.B. Molecular Graphics: From Science to Art. Cryst. Growth Des. 2003, 3, 3–8. [Google Scholar]
- Barbour, L. X-Seed, Graphical Interface to SHELX-97 and POV-Ray; University of Missouri–Columbia: Columbia, MO, USA, 1999. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Pollastri, M.P. Overview on the Rule of Five. Curr. Protoc. Pharmacol. 2010, 49, 9.12.11–19.12.18. [Google Scholar]
- Yu, D.K. The contribution of P-glycoprotein to pharmacokinetic drug-drug interactions. J. Clin. Pharmacol. 1999, 39, 1203–1211. [Google Scholar] [CrossRef]
- Fromm, M. Importance of P-glycoprotein for drug disposition in humans. Eur. J. Clin. Invest. 2003, 33, 6–9. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H. CYP induction-mediated drug interactions: In vitro assessment and clinical implications. Pharm. Res. 2006, 23, 1089–1116. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.Ccdc.Cam.Ac.Uk (accessed on 20 August 2019).
- Farrugia, L. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Kalgutkar, A.S.; Crews, B.C.; Rowlinson, S.W.; Marnett, A.B.; Kozak, K.R.; Remmel, R.P.; Marnett, L.J. Biochemically based design of cyclooxygenase-2 (COX-2) inhibitors: Facile conversion of nonsteroidal antiinflammatory drugs to potent and highly selective COX-2 inhibitors. Proc. Natl. Acad. Sci. USA 2000, 97, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, D.K.; Lecomte, M.; Rieke, C.J.; Garavito, R.M.; Smith, W.L. Involvement of arginine 120, glutamate 524, and tyrosine 355 in the binding of arachidonate and 2-phenylpropionic acid inhibitors to the cyclooxygenase active site of ovine prostaglandin endoperoxide H synthase-1. J. Biol. Chem. 1996, 271, 2179–2184. [Google Scholar] [CrossRef]
- Greig, G.M.; Francis, D.A.; Falgueyret, J.-P.; Ouellet, M.; Percival, M.D.; Roy, P.; Bayly, C.; Mancini, J.A.; O’Neill, G.P. The interaction of arginine 106 of human prostaglandin G/H synthase-2 with inhibitors is not a universal component of inhibition mediated by nonsteroidal anti-inflammatory drugs. Mol. Pharmacol. 1997, 52, 829–838. [Google Scholar] [CrossRef]
- Lombardo, F.; Gifford, E.; Shalaeva, M.Y. In silico ADME prediction: Data, models, facts and myths. Mini Rev. Med. Chem. 2003, 3, 861–875. [Google Scholar] [CrossRef]
- Smith, P.; Sorich, M.; Low, L.; McKinnon, R.; Miners, J. Towards integrated ADME prediction: Past, present and future directions for modelling metabolism by UDP-glucuronosyltransferases. J. Mol. Graph. Model. 2004, 22, 507–517. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Code | Mol Formula (Mol Weight) | R | R1 | Yield (%) a | M.P. (°C) | CLogP b |
|---|---|---|---|---|---|---|
| 4a | C24H19NO3 (369) | CH3 | H | 87 | 173–174 | 6.2094 |
| 4b | C24H18FNO3 (387) | CH3 | F | 85 | 167–168 | 6.3602 |
| 4c | C24H18ClNO3 (403) | CH3 | Cl | 89 | 154–155 | 6.9302 |
| 4d | C24H18BrNO3 (447) | CH3 | Br | 85 | 137–138 | 7.0802 |
| 4e | C25H15N3O3 (405) | CN | CN | 76 | 235–236 | 4.6463 |
| 4f | C24H15BrN2O3 (458) | CN | Br | 84 | 208–209 | 6.0300 |
| 4g | C25H18N2O4 (410) | CN | OCH3 | 81 | 166–167 | 5.3301 |
| DATA | Compound 4a |
|---|---|
| Formula | C24H19 N O3 |
| Formula weight | 369.40 |
| Temperature/K | 153 (2) |
| Wavelength (Å) | 0.71073 |
| Crystal system | monoclinic |
| Space group | C 2/c |
| a (Å) | 16.550 (4) |
| b (Å) | 10.391 (2) |
| c (Å) | 22.179 (4) |
| α (°) | 90 |
| β (°) | 104.468 (4) |
| γ (°) | 90 |
| V (Å3) | 3693.1 (13) |
| Z’, Z | 1, 8 |
| Density (g cm−1) | 1.329 |
| μ (mm−1) | 0.088 |
| F (000) | 1552 |
| θ (min, max) | 2.336, 25.413 |
| hmin, max; kmin, max; lmin, max. | −19, 19; −12, 12; −26, 26 |
| No. of refl. | 3375 |
| No of unique ref./Obs. ref. | 3375, 2340 |
| No. parameters | 255 |
| Rall, Robs | 0.0462, 0.0815 |
| wRall, wRobs | 0.113, 0.099 |
| Δρmin, max (eÅ−3) | −0.272, 0.195 |
| G.O.F. | 1.020 |
| D–X···A | D–X (Å) | X···A (Å) | D···A (Å) | <D–X···A (Å) | Symmetry Code |
|---|---|---|---|---|---|
| C2–H2···O1 | 0.95 | 2.56 | 3.450 (3) | 156 | 1/2 + x, 1/2 + y, z |
| C11–H11A···O3 | 0.98 | 2.53 | 3.383 (3) | 146 | x, −1 + y, z |
| C17–H17···Cg | 0.98 | 2.83 | 3.678 | 149 | 1/2 − x, −1/2 + y, 1/2 − z |
| Entry | R1 | R2 | IC50 (μM)a | CDocker E. (kcal/mol) | Hydrogen Bonding (Interacting Atom, Å) | Pi Interaction |
|---|---|---|---|---|---|---|
| 4a | CH3 | H | 41.59 ± 0.03 a | −31 | ARG 120 (pi–cation) | |
| 4b | CH3 | F | 27.08 ± 0.03 c,d | −34 | HIS 90 (F, 2.09) | ARG 120 (pi–cation) |
| 4c | CH3 | Cl | 38.11 ± 0.03 b,d | −33 | ARG 120 (pi–cation) | |
| 4d | CH3 | Br | 37.66 ± 0.03 d | −32 | ARG 120 (pi–cation) | |
| 4e | CN | CN | 6.71 ± 0.03 b | −35 | HIS 90 (CN, 2.02) | |
| 4f | CN | Br | 13.55 ± 0.03 b,d | −34 | HIS 90 (CN, 2.01) | |
| 4g | CN | OCH3 | 9.62 ± 0.03 c | −35 | HIS 90 (CN, 2.07) | |
| IND | 6.84 ± 0.03 b,c | −49 | ARG 120 (anion–cation) | |||
| CLB | 0.05 ± 0.03 b | −43 | HIS 90 (SO2, 3.05) PHE 158 (SO2, 2.92) GLN 192 (NH2, 2.69) LEU 352 (NH2, 1.95) ARG120 (CF3, 2.38) | ARG 120 (pi–cation) PHE 158 (pi–sulfur) ALA 527 (pi–amide) |
| Compound ID | 4a | 4b | 4c | 4d | 4e | 4f | 4g |
|---|---|---|---|---|---|---|---|
| Rotatable bonds | 5 | 5 | 5 | 5 | 5 | 5 | 6 |
| Molecular weight | 369.41 | 387.4 | 403.86 | 448.31 | 405.4 | 459.29 | 410.42 |
| H-bond acceptors | 3 | 4 | 3 | 3 | 5 | 4 | 5 |
| H-bond donors | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| MLOGP | 3.43 | 3.8 | 3.91 | 4.01 | 1.86 | 3.11 | 2.2 |
| GI absorption | High | High | High | High | High | High | High |
| BBB permeant | Yes | Yes | Yes | Yes | No | No | No |
| P-gp substrate | No | No | No | No | No | No | No |
| Lipinski violations | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Compound ID | 4a | 4b | 4c | 4d | 4e | 4f | 4g |
|---|---|---|---|---|---|---|---|
| Mutagenic | none | none | none | none | none | none | none |
| Tumorigenic | none | none | none | none | none | none | none |
| Reproductive Effective | none | none | none | none | none | none | none |
| Irritant | none | none | none | none | none | none | none |
| CYP1A2 inhibitor | Yes | Yes | Yes | Yes | Yes | No | Yes |
| CYP2D6 inhibitor | No | No | No | No | No | No | No |
| CYP3A4 inhibitor | No | No | No | No | Yes | Yes | No |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venugopala, K.N.; Al-Attraqchi, O.H.A.; Tratrat, C.; Nayak, S.K.; Morsy, M.A.; Aldhubiab, B.E.; Attimarad, M.; Nair, A.B.; Sreeharsha, N.; Venugopala, R.; et al. Novel Series of Methyl 3-(Substituted Benzoyl)-7-Substituted-2-Phenylindolizine-1-Carboxylates as Promising Anti-Inflammatory Agents: Molecular Modeling Studies. Biomolecules 2019, 9, 661. https://doi.org/10.3390/biom9110661
Venugopala KN, Al-Attraqchi OHA, Tratrat C, Nayak SK, Morsy MA, Aldhubiab BE, Attimarad M, Nair AB, Sreeharsha N, Venugopala R, et al. Novel Series of Methyl 3-(Substituted Benzoyl)-7-Substituted-2-Phenylindolizine-1-Carboxylates as Promising Anti-Inflammatory Agents: Molecular Modeling Studies. Biomolecules. 2019; 9(11):661. https://doi.org/10.3390/biom9110661
Chicago/Turabian StyleVenugopala, Katharigatta N., Omar H.A. Al-Attraqchi, Christophe Tratrat, Susanta K. Nayak, Mohamed A. Morsy, Bandar E. Aldhubiab, Mahesh Attimarad, Anroop B. Nair, Nagaraja Sreeharsha, Rashmi Venugopala, and et al. 2019. "Novel Series of Methyl 3-(Substituted Benzoyl)-7-Substituted-2-Phenylindolizine-1-Carboxylates as Promising Anti-Inflammatory Agents: Molecular Modeling Studies" Biomolecules 9, no. 11: 661. https://doi.org/10.3390/biom9110661
APA StyleVenugopala, K. N., Al-Attraqchi, O. H. A., Tratrat, C., Nayak, S. K., Morsy, M. A., Aldhubiab, B. E., Attimarad, M., Nair, A. B., Sreeharsha, N., Venugopala, R., Haroun, M., Girish, M. B., Chandrashekharappa, S., Alwassil, O. I., & Odhav, B. (2019). Novel Series of Methyl 3-(Substituted Benzoyl)-7-Substituted-2-Phenylindolizine-1-Carboxylates as Promising Anti-Inflammatory Agents: Molecular Modeling Studies. Biomolecules, 9(11), 661. https://doi.org/10.3390/biom9110661