Strategies for Developing Functional Secretory Epithelia from Porcine Salivary Gland Explant Outgrowth Culture Models



Abstract
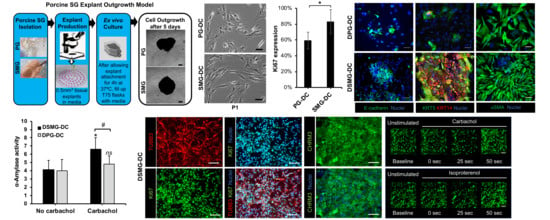
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Explant Outgrowth Culture of Porcine SG-Derived Primary Cells
2.2. Expansion and Differentiation of SMG-DC and PG-DC
2.3. Flow Cytometry Analysis
2.4. Transcriptome Analysis by Quantitative PCR
2.5. Histological Analysis
2.6. Immunofluorescence and Proteome Quantification
2.7. Western Blot Analysis
2.8. Salivary Gland Functional Assays
2.8.1. α-Amylase Enzymatic Activity
2.8.2. Epithelial Barrier Function
2.8.3. Intracellular Calcium Activity
2.9. Statistical Analysis
3. Results
3.1. Porcine PG and SMG Primary Cells Had Heterotypic Morphology
3.2. Primary Cell Subcultures Contained Large Putative SG Epithelial Stem/Progenitor Cell Subpopulations
3.3. Primary Cell Subcultures Had Up-Regulated Genes Associated with SG Epithelial Progenitors
3.4. Secretory Epithelial and Myoepithelial Cells Were Abundant in Differentiated SMG-DC and PG-DC
3.5. DSMG-DC Had Greater SG-Like Functional Properties
3.6. Differentiated SG Primary Cells Displayed Responses to Neurotransmitters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Affoo, R.H.; Foley, N.; Garrick, R.; Siqueira, W.L.; Martin, R.E. Meta-Analysis of Salivary Flow Rates in Young and Older Adults. J. Am. Geriatr. Soc. 2015, 63, 2142–2151. [Google Scholar] [CrossRef] [PubMed]
- Mandel, I.D. The role of saliva in maintaining oral homeostasis. J. Am. Dent. Assoc. 1989, 119, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Von Bultzingslowen, I.; Sollecito, T.P.; Fox, P.C.; Daniels, T.; Jonsson, R.; Lockhart, P.B.; Wray, D.; Brennan, M.T.; Carrozzo, M.; Gandera, B.; et al. Salivary dysfunction associated with systemic diseases: Systematic review and clinical management recommendations. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2007, 103, S57e1–S57e15. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.B.; Pedersen, A.M.; Vissink, A.; Andersen, E.; Brown, C.G.; Davies, A.N.; Dutilh, J.; Fulton, J.S.; Jankovic, L.; Lopes, N.N.; et al. A systematic review of salivary gland hypofunction and xerostomia induced by cancer therapies: Management strategies and economic impact. Support. Care Cancer 2010, 18, 1061–1079. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, O.; Mitchell, G.C.; Limesand, K.H. Sensitivity of salivary glands to radiation: From animal models to therapies. J. Dent. Res. 2009, 88, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Vissink, A.; Mitchell, J.B.; Baum, B.J.; Limesand, K.H.; Jensen, S.B.; Fox, P.C.; Elting, L.S.; Langendijk, J.A.; Coppes, R.P.; Reyland, M.E. Clinical management of salivary gland hypofunction and xerostomia in head-and-neck cancer patients: Successes and barriers. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, C.A.; Haddad, R.I.; Tishler, R.B.; Mahadevan, A.; Krane, J.F. Chemoradiation-induced cell loss in human submandibular glands. Laryngoscope 2005, 115, 958–964. [Google Scholar] [CrossRef]
- Franzen, L.; Funegard, U.; Ericson, T.; Henriksson, R. Parotid gland function during and following radiotherapy of malignancies in the head and neck. A consecutive study of salivary flow and patient discomfort. Eur. J. Cancer 1992, 28, 457–462. [Google Scholar] [CrossRef]
- Diaz-Arnold, A.M.; Marek, C.A. The impact of saliva on patient care: A literature review. J. Prosthet. Dent. 2002, 88, 337–343. [Google Scholar] [CrossRef]
- Atkinson, J.C.; Baum, B.J. Salivary enhancement: Current status and future therapies. J. Dent. Educ. 2001, 65, 1096–1101. [Google Scholar]
- Srinivasan, P.P.; Patel, V.N.; Liu, S.; Harrington, D.A.; Hoffman, M.P.; Jia, X.; Witt, R.L.; Farach-Carson, M.C.; Pradhan-Bhatt, S. Primary Salivary Human Stem/Progenitor Cells Undergo Microenvironment-Driven Acinar-Like Differentiation in Hyaluronate Hydrogel Culture. Stem Cells Transl. Med. 2017, 6, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Foraida, Z.I.; Kamaldinov, T.; Nelson, D.A.; Larsen, M.; Castracane, J. Elastin-PLGA hybrid electrospun nanofiber scaffolds for salivary epithelial cell self-organization and polarization. Acta Biomater. 2017, 62, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Pringle, S.; Maimets, M.; van der Zwaag, M.; Stokman, M.A.; van Gosliga, D.; Zwart, E.; Witjes, M.J.; de Haan, G.; van Os, R.; Coppes, R.P. Human Salivary Gland Stem Cells Functionally Restore Radiation Damaged Salivary Glands. Stem Cells 2016, 34, 640–652. [Google Scholar] [CrossRef] [PubMed]
- Adine, C.; Ng, K.K.; Rungarunlert, S.; Souza, G.R.; Ferreira, J.N. Engineering innervated secretory epithelial organoids by magnetic three-dimensional bioprinting for stimulating epithelial growth in salivary glands. Biomaterials 2018, 180, 52–66. [Google Scholar] [CrossRef]
- Maruyama, C.L.; Monroe, M.M.; Hunt, J.P.; Buchmann, L.; Baker, O.J. Comparing human and mouse salivary glands: A practice guide for salivary researchers. Oral Dis. 2019, 25, 403–415. [Google Scholar] [CrossRef]
- Maimets, M.; Bron, R.; de Haan, G.; van Os, R.; Coppes, R.P. Similar ex vivo expansion and post-irradiation regenerative potential of juvenile and aged salivary gland stem cells. Radiother. Oncol. 2015, 116, 443–448. [Google Scholar] [CrossRef]
- Leigh, N.J.; Nelson, J.W.; Mellas, R.E.; McCall, A.D.; Baker, O.J. Three-dimensional cultures of mouse submandibular and parotid glands: A comparative study. J. Tissue Eng. Regen. Med. 2017, 11, 618–626. [Google Scholar] [CrossRef]
- Wang, S.; Liu, Y.; Fang, D.; Shi, S. The miniature pig: A useful large animal model for dental and orofacial research. Oral Dis. 2007, 13, 530–537. [Google Scholar] [CrossRef]
- Wang, S.L.; Li, J.; Zhu, X.Z.; Sun, K.; Liu, X.Y.; Zhang, Y.G. Sialographic characterization of the normal parotid gland of the miniature pig. Dentomaxillofac. Radiol. 1998, 27, 178–181. [Google Scholar] [CrossRef]
- Radfar, L.; Sirois, D.A. Structural and functional injury in minipig salivary glands following fractionated exposure to 70 Gy of ionizing radiation: An animal model for human radiation-induced salivary gland injury. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2003, 96, 267–274. [Google Scholar] [CrossRef]
- Yan, X.; Voutetakis, A.; Zheng, C.; Hai, B.; Zhang, C.; Baum, B.J.; Wang, S. Sorting of transgenic secretory proteins in miniature pig parotid glands following adenoviral-mediated gene transfer. J. Gene Med. 2007, 9, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.M.; Huang, Y.; Zhang, K.; Qu, L.H.; Cong, X.; Su, J.Z.; Wu, L.L.; Yu, G.Y.; Zhang, Y. Expression patterns of tight junction proteins in porcine major salivary glands: A comparison study with human and murine glands. J. Anat. 2018, 233, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Lombaert, I.; Movahednia, M.M.; Adine, C.; Ferreira, J.N. Concise Review: Salivary Gland Regeneration: Therapeutic Approaches from Stem Cells to Tissue Organoids. Stem Cells 2017, 35, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.N.; Hasan, R.; Urkasemsin, G.; Ng, K.K.; Adine, C.; Muthumariappan, S.; Souza, G.R. A magnetic three-dimensional levitated primary cell culture system for the development of secretory salivary gland-like organoids. J. Tissue Eng. Regen. Med. 2019, 13, 495–508. [Google Scholar] [CrossRef]
- Shan, Z.; Li, J.; Zheng, C.; Liu, X.; Fan, Z.; Zhang, C.; Goldsmith, C.M.; Wellner, R.B.; Baum, B.J.; Wang, S. Increased fluid secretion after adenoviral-mediated transfer of the human aquaporin-1 cDNA to irradiated miniature pig parotid glands. Mol. Ther. 2005, 11, 444–451. [Google Scholar] [CrossRef]
- Ferreira, J.N.A.; Zheng, C.; Lombaert, I.M.A.; Goldsmith, C.M.; Cotrim, A.P.; Symonds, J.M.; Patel, V.N.; Hoffman, M.P. Neurturin Gene Therapy Protects Parasympathetic Function to Prevent Irradiation-Induced Murine Salivary Gland Hypofunction. Mol. Ther. Methods Clin. Dev. 2018, 9, 172–180. [Google Scholar] [CrossRef]
- Lv, F.J.; Tuan, R.S.; Cheung, K.M.; Leung, V.Y. Concise review: The surface markers and identity of human mesenchymal stem cells. Stem Cells 2014, 32, 1408–1419. [Google Scholar] [CrossRef]
- Togarrati, P.P.; Dinglasan, N.; Desai, S.; Ryan, W.R.; Muench, M.O. CD29 is highly expressed on epithelial, myoepithelial, and mesenchymal stromal cells of human salivary glands. Oral Dis. 2018, 24, 561–572. [Google Scholar] [CrossRef]
- Nanduri, L.S.; Baanstra, M.; Faber, H.; Rocchi, C.; Zwart, E.; de Haan, G.; van Os, R.; Coppes, R.P. Purification and ex vivo expansion of fully functional salivary gland stem cells. Stem Cell Rep. 2014, 3, 957–964. [Google Scholar] [CrossRef]
- Maria, O.M.; Maria, A.M.; Cai, Y.; Tran, S.D. Cell surface markers CD44 and CD166 localized specific populations of salivary acinar cells. Oral Dis. 2012, 18, 162–168. [Google Scholar] [CrossRef]
- Yi, T.; Lee, S.; Choi, N.; Shin, H.S.; Kim, J.; Lim, J.Y. Single Cell Clones Purified from Human Parotid Glands Display Features of Multipotent Epitheliomesenchymal Stem Cells. Sci. Rep. 2016, 6, 36303. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Li, Y.; Du, M.J.; Zhang, C.; Zhang, X.Y.; Tong, H.Z.; Liu, L.; Han, T.L.; Li, W.D.; Yan, L.; et al. Characterization of a Self-renewing and Multi-potent Cell Population Isolated from Human Minor Salivary Glands. Sci. Rep. 2015, 5, 10106. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.; Alston, N.; Ghazizadeh, S. Identification of Stem Cells in the Secretory Complex of Salivary Glands. J. Dent. Res. 2016, 95, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Aure, M.H.; Arany, S.; Ovitt, C.E. Salivary Glands: Stem Cells, Self-duplication, or Both? J. Dent. Res. 2015, 94, 1502–1507. [Google Scholar] [CrossRef]
- Kogata, N.; Howard, B.A. A protocol for studying embryonic mammary progenitor cells during mouse mammary primordial development in explant culture. Methods Mol. Biol. 2015, 1293, 51–62. [Google Scholar] [CrossRef]
- Nikolakis, G.; Seltmann, H.; Hossini, A.M.; Makrantonaki, E.; Knolle, J.; Zouboulis, C.C. Ex vivo human skin and SZ95 sebocytes exhibit a homoeostatic interaction in a novel coculture contact model. Exp. Dermatol. 2015, 24, 497–502. [Google Scholar] [CrossRef]
- Jang, S.I.; Ong, H.L.; Gallo, A.; Liu, X.; Illei, G.; Alevizos, I. Establishment of functional acinar-like cultures from human salivary glands. J. Dent. Res. 2015, 94, 304–311. [Google Scholar] [CrossRef]
- Li, J.; Shan, Z.; Ou, G.; Liu, X.; Zhang, C.; Baum, B.J.; Wang, S. Structural and functional characteristics of irradiation damage to parotid glands in the miniature pig. Int. J. Radiat. Oncol. Biol. Phys. 2005, 62, 1510–1516. [Google Scholar] [CrossRef]
- Hai, B.; Yan, X.; Voutetakis, A.; Zheng, C.; Cotrim, A.P.; Shan, Z.; Ding, G.; Zhang, C.; Xu, J.; Goldsmith, C.M.; et al. Long-term transduction of miniature pig parotid glands using serotype 2 adeno-associated viral vectors. J. Gene Med. 2009, 11, 506–514. [Google Scholar] [CrossRef]
- Hu, L.; Zhu, Z.; Hai, B.; Chang, S.; Ma, L.; Xu, Y.; Li, X.; Feng, X.; Wu, X.; Zhao, Q.; et al. Intragland Shh gene delivery mitigated irradiation-induced hyposalivation in a miniature pig model. Theranostics 2018, 8, 4321–4331. [Google Scholar] [CrossRef]
- Matsumoto, S.; Okumura, K.; Ogata, A.; Hisatomi, Y.; Sato, A.; Hattori, K.; Matsumoto, M.; Kaji, Y.; Takahashi, M.; Yamamoto, T.; et al. Isolation of tissue progenitor cells from duct-ligated salivary glands of swine. Cloning Stem Cells 2007, 9, 176–190. [Google Scholar] [CrossRef] [PubMed]
- Edgar, W.M. Saliva and dental health. Clinical implications of saliva: Report of a consensus meeting. Br. Dent. J. 1990, 169, 96–98. [Google Scholar] [CrossRef] [PubMed]
- Sommakia, S.; Baker, O.J. Neurons Self-Organize Around Salivary Epithelial Cells in Novel Co-Culture Model. J. Stem Cell Regen. Biol. 2016, 2, 1–11. [Google Scholar] [CrossRef][Green Version]
- Maimets, M.; Rocchi, C.; Bron, R.; Pringle, S.; Kuipers, J.; Giepmans, B.N.; Vries, R.G.; Clevers, H.; de Haan, G.; van Os, R.; et al. Long-Term In Vitro Expansion of Salivary Gland Stem Cells Driven by Wnt Signals. Stem Cell Rep. 2016, 6, 150–162. [Google Scholar] [CrossRef]
- Baker, O.J.; Schulz, D.J.; Camden, J.M.; Liao, Z.; Peterson, T.S.; Seye, C.I.; Petris, M.J.; Weisman, G.A. Rat parotid gland cell differentiation in three-dimensional culture. Tissue Eng. Part C Methods 2010, 16, 1135–1144. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urkasemsin, G.; Castillo, P.; Rungarunlert, S.; Klincumhom, N.; Ferreira, J.N. Strategies for Developing Functional Secretory Epithelia from Porcine Salivary Gland Explant Outgrowth Culture Models. Biomolecules 2019, 9, 657. https://doi.org/10.3390/biom9110657
Urkasemsin G, Castillo P, Rungarunlert S, Klincumhom N, Ferreira JN. Strategies for Developing Functional Secretory Epithelia from Porcine Salivary Gland Explant Outgrowth Culture Models. Biomolecules. 2019; 9(11):657. https://doi.org/10.3390/biom9110657
Chicago/Turabian StyleUrkasemsin, Ganokon, Phoebe Castillo, Sasitorn Rungarunlert, Nuttha Klincumhom, and Joao N. Ferreira. 2019. "Strategies for Developing Functional Secretory Epithelia from Porcine Salivary Gland Explant Outgrowth Culture Models" Biomolecules 9, no. 11: 657. https://doi.org/10.3390/biom9110657
APA StyleUrkasemsin, G., Castillo, P., Rungarunlert, S., Klincumhom, N., & Ferreira, J. N. (2019). Strategies for Developing Functional Secretory Epithelia from Porcine Salivary Gland Explant Outgrowth Culture Models. Biomolecules, 9(11), 657. https://doi.org/10.3390/biom9110657