A Computational Framework for Predicting Direct Contacts and Substructures within Protein Complexes

Abstract
1. Introduction
2. Materials and Methods
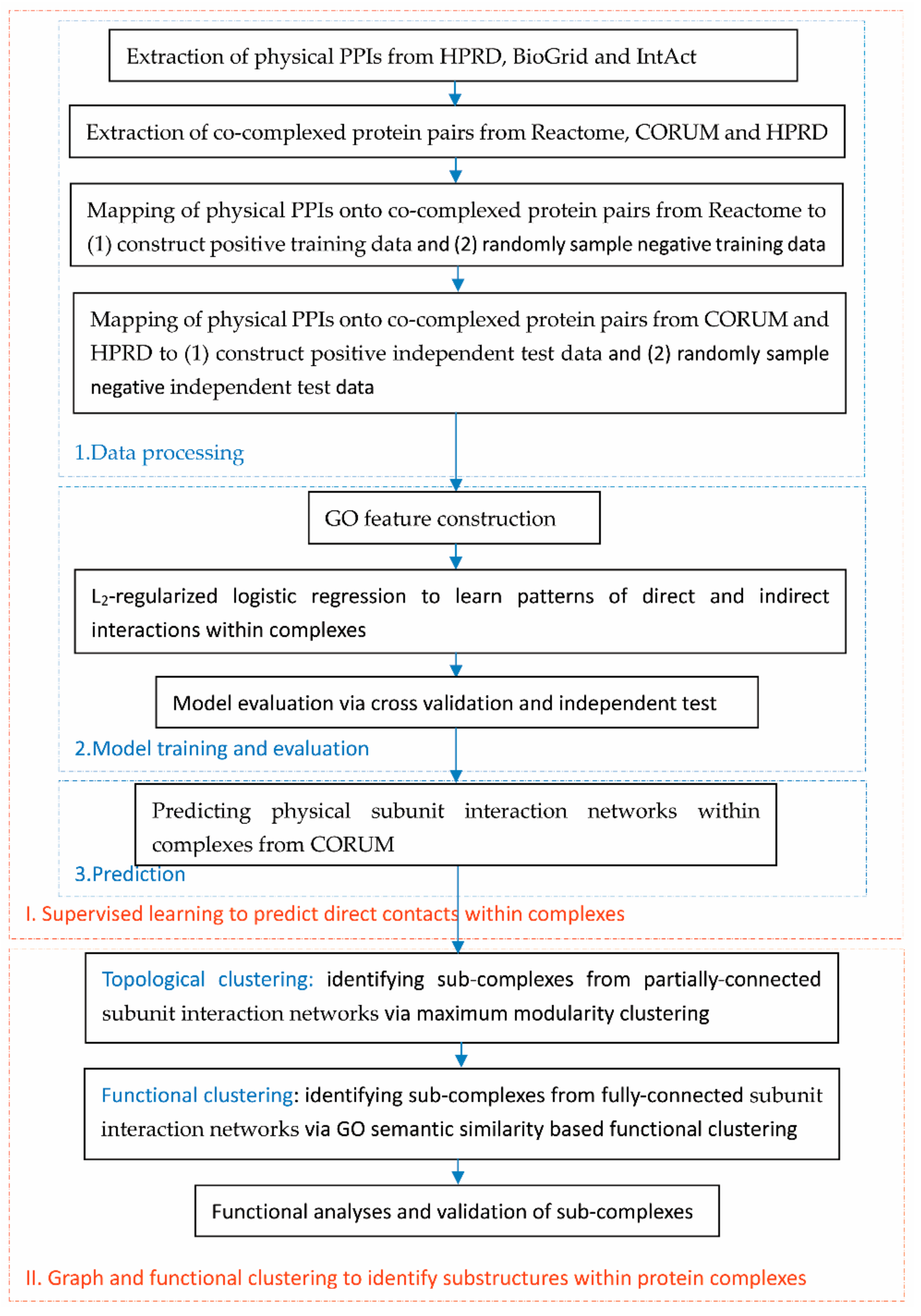
2.1. Flowchart of the Proposed Framework
2.2. Construction of Training and Independent Test Data
2.2.1. Positive Training and Independent Test Data
2.2.2. Negative Training and Independent Test Data
2.3. Supervised Learning for Predicting Direct Contacts within Protein Complexes
2.3.1. Feature Construction
2.3.2. Supervised Learning via L2-Regularized Logistic Regression
2.4. Graph Clustering for Sub-Complexes Discovery
2.4.1. Topological Clustering via Maximum Modularity Clustering
2.4.2. Functional Clustering via GO Semantic Similarities
2.5. Experimental Setting and Model Evaluation
2.5.1. Supervised Learning
2.5.2. Graph Clustering
3. Results
3.1. Performance of Predicting Physical Subunit Interactions within Complexes
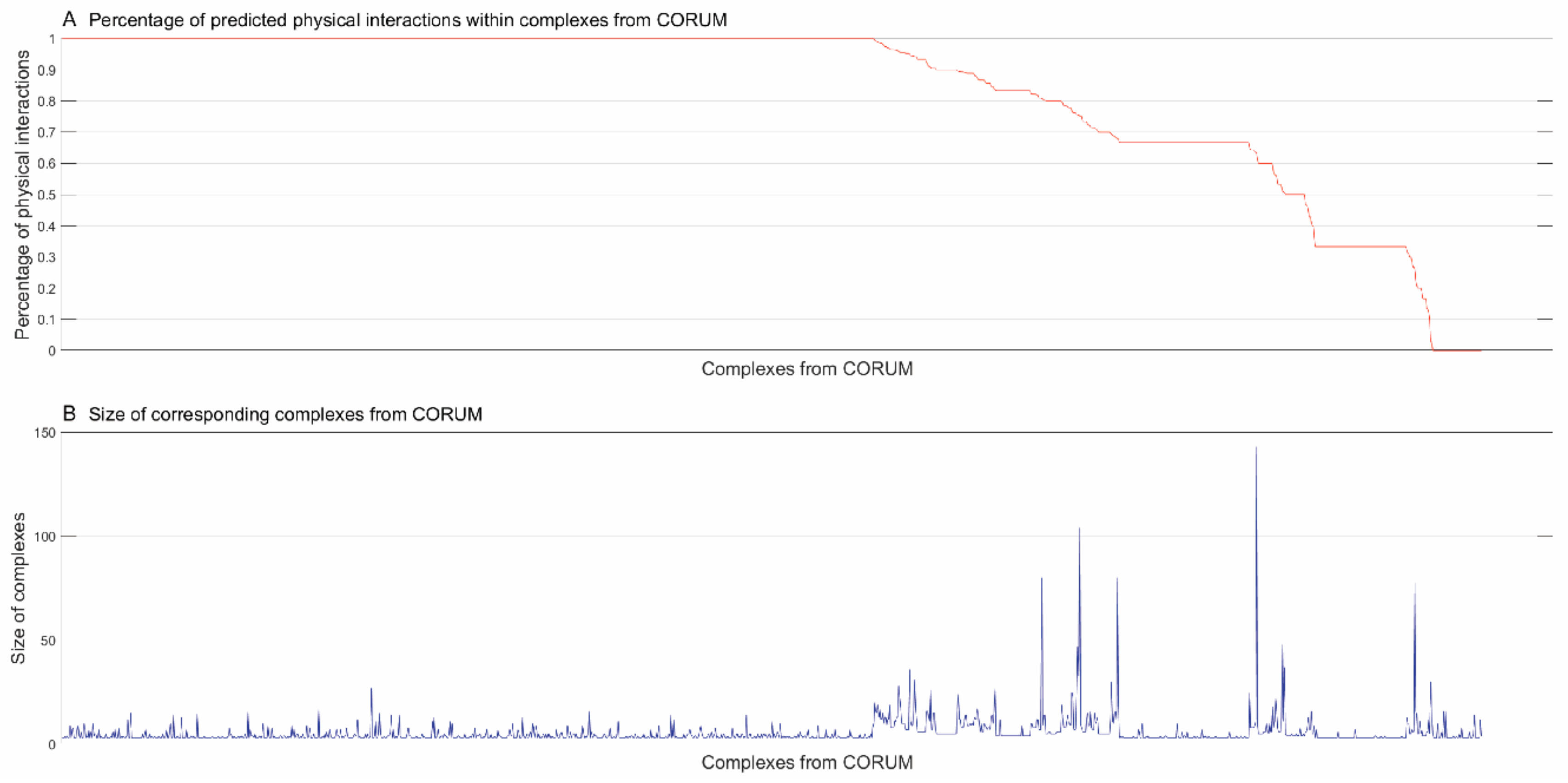
3.2. Inferring Substructures within Complexes from CORUM [4]
3.2.1. Identifying Physical Interactions within Complexes from CORUM [4]
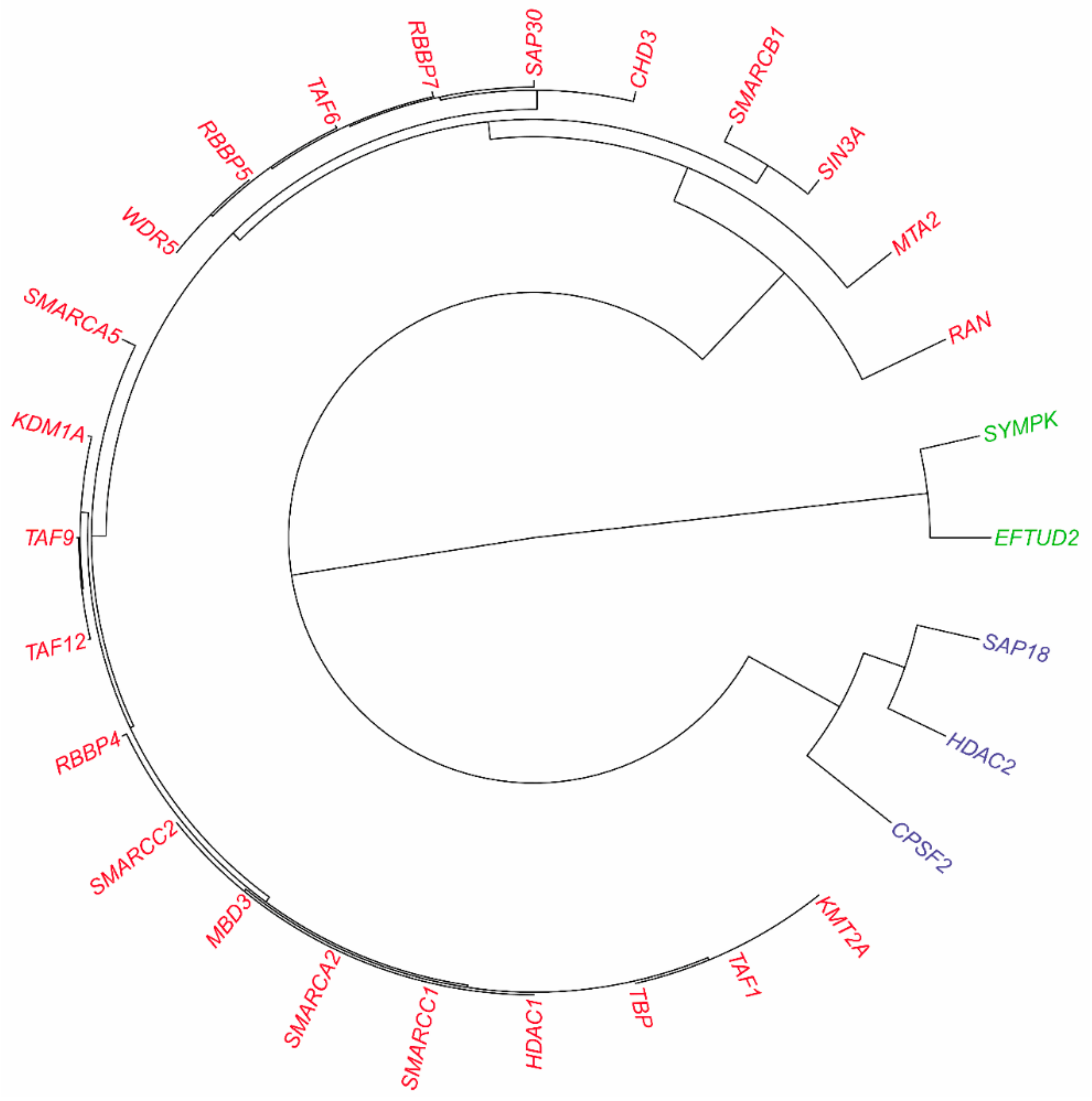
3.2.2. Inferring Substructures within Partially-Connected Complexes via Mmc Algorithm
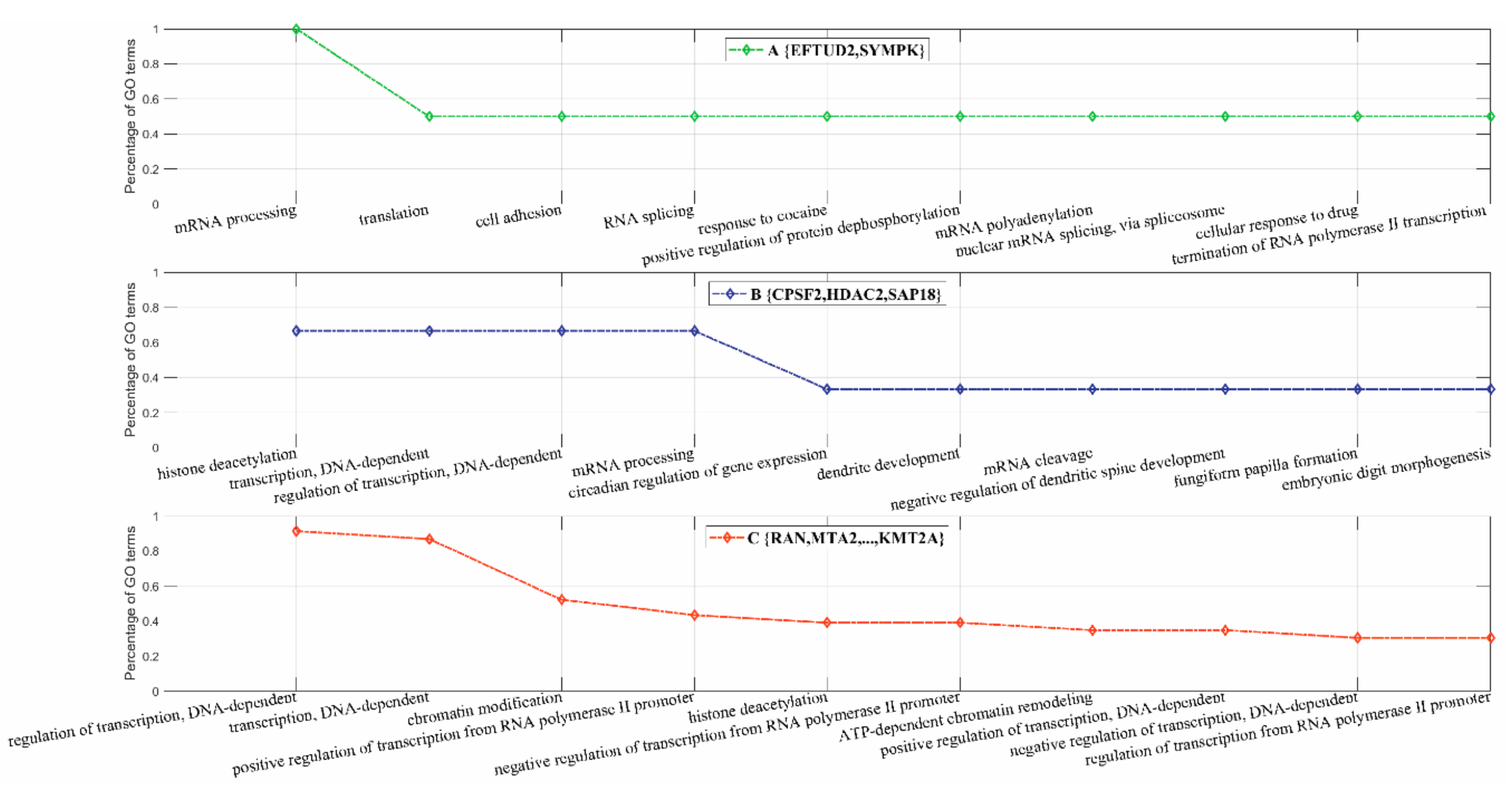
3.2.3. Inferring Substructures within Fully-Connected Complexes via Functional Clustering
3.3. Comparison with the Related Work
3.3.1. Predicting Physical Interactions within Complexes
3.3.2. Inferring Substructures within Complexes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gavin, A.-C.; Aloy, P.; Grandi, P.; Krause, R.; Boesche, M.; Marzioch, M.; Rau, C.; Jensen, L.J.; Bastuck, S.; Dümpelfeld, B.; et al. Proteome survey reveals modularity of the yeast cell machinery. Nature 2006, 440, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Lage, K.; Karlberg, E.O.; Størling, Z.M.; Ólason, P.Í.; Pedersen, A.G.; Rigina, O.; Hinsby, A.M.; Tümer, Z.; Pociot, F.; Tommerup, N.; et al. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat. Biotechnol. 2007, 25, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Mewes, H.W.; Amid, C.; Arnold, R.; Frishman, D.; Güldener, U.; Mannhaupt, G.; Münsterkötter, M.; Pagel, P.; Strack, N.; Stümpflen, V.; et al. MIPS: Analysis and annotation of proteins from whole genomes. Nucleic Acids Res. 2004, 32, D41–D44. [Google Scholar] [CrossRef] [PubMed]
- Ruepp, A.; Waegele, B.; Lechner, M.; Brauner, B.; Dunger-Kaltenbach, I.; Fobo, G.; Montrone, C.; Mewes, H.W. CORUM: The comprehensive resource of mammalian protein complexes--2009. Nucleic. Acids Res. 2010, 38, D497–D501. [Google Scholar] [CrossRef] [PubMed]
- Keshava Prasad, T.S.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human Protein Reference Database--2009 update. Nucleic. Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Feng, X.; Stein, L. A human functional protein interaction network and its application to cancer data analysis. Genome Biol. 2010, 11, R53. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Sidiropoulos, K.; Garapati, P.; Gillespie, M.; Hausmann, K.; Haw, R.; Jassal, B.; Jupe, S.; Korninger, F.; McKay, S.; et al. The Reactome pathway Knowledgebase. Nucleic. Acids Res. 2016, 44, D481–D487. [Google Scholar] [CrossRef]
- Li, X.; Wu, M.; Kwoh, C.-K.; Ng, S.-K. Computational approaches for detecting protein complexes from protein interaction networks: A survey. BMC Genom. 2010, 11, S3. [Google Scholar] [CrossRef]
- Srihari, S.; Yong, C.H.; Patil, A.; Wong, L. Methods for protein complex prediction and their contributions towards understanding the organisation, function and dynamics of complexes. FEBS Lett. 2015, 589, 2590–2602. [Google Scholar] [CrossRef]
- Aloy, P.; Böttcher, B.; Ceulemans, H.; Leutwein, C.; Mellwig, C.; Fischer, S.; Gavin, A.C.; Bork, P.; Superti-Furga, G.; Serrano, L.; et al. Structure-based assembly of protein complexes in yeast. Science 2004, 30, 2026–2029. [Google Scholar] [CrossRef]
- Drew, K.; Müller, C.L.; Bonneau, R.; Marcotte, E.M. Identifying direct contacts between protein complex subunits from their conditional dependence in proteomics datasets. PLoS Comput. Biol. 2017, 13, e1005625. [Google Scholar] [CrossRef] [PubMed]
- Friedel, C.C.; Zimmer, R. Identifying the topology of protein complexes from affinity purification assays. Bioinformatic 2009, 25, 2140–2146. [Google Scholar] [CrossRef] [PubMed]
- Soong, T.-T.; Wrzeszczynski, K.O.; Rost, B. Physical protein-protein interactions predicted from microarrays. Bioinformatic 2008, 24, 2608–2614. [Google Scholar] [CrossRef] [PubMed]
- Elefsinioti, A.; Saraç Ömer, S.; Hegele, A.; Plake, C.; Hubner, N.C.; Poser, I.; Sarov, M.; Hyman, A.; Mann, M.; Schroeder, M.; et al. Large-scale de novo prediction of physical protein-protein association. Mol. Cell. Proteom. 2011, 10, M111.010629. [Google Scholar]
- Mei, S.; Flemington, E.K.; Zhang, K. A computational framework for distinguishing direct versus indirect interactions in human functional protein-protein interaction networks. Integr. Biol. 2017, 9, 595–606. [Google Scholar] [CrossRef]
- Van Dongen, S. Graph Clustering by Flow Simulation; University of Utrecht: Utrecht, The Netherlands, 2000. [Google Scholar]
- Noack, A.; Rotta, R. Multi-level Algorithms for Modularity Clustering. In Experimental Algorithms; Vahrenhold, J., Ed.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 257–268. [Google Scholar]
- Chatr-Aryamontri, A.; Breitkreutz, B.J.; Oughtred, R.; Boucher, L.; Heinicke, S.; Chen, D.; Stark, C.; Breitkreutz, A.; Kolas, N.; O’Donnell, L.; et al. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2015, 43, D470–D4788. [Google Scholar] [CrossRef]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; del-Toro, N.; et al. The MIntAct project--IntAct as a common curation platform for 11 molecular interaction databases. Nucleic. Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic. Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Maetschke, S.; Simonsen, M.; Davis, M.; Ragan, M.A. Gene Ontology-driven inference of protein–protein interactions using inducers. Bioinformatics 2012, 28, 69–75. [Google Scholar] [CrossRef]
- Altschul, S. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Boeckmann, B. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Barrell, D.; Dimmer, E.; Huntley, R.P.; Binns, D.; O’Donovan, C.; Apweiler, R. The GOA database in 2009--an integrated Gene Ontology Annotation resource. Nucleic. Acids Res. 2009, 37, D396–D403. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-B.; Tang, Q.-R. Protein–protein interaction inference based on semantic similarity of Gene Ontology terms. J. Theor. Boil. 2016, 401, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Huang, F.; Lin, C. Dual coordinate descent methods for logistic regression and maximum entropy models. Mach. Learn 2011, 85, 41–75. [Google Scholar] [CrossRef]
- Fan, R.; Chang, K.; Hsieh, C.; Wang, X.; Lin, C. LIBLINEAR: A Library for Large Linear Classification. Mach. Learn Res. 2008, 9, 1871–1874. [Google Scholar]
- Newman, M.E.J. Finding community structure in networks using the eigenvectors of matrices. Phys. Rev. E 2006, 74, 036104. [Google Scholar] [CrossRef]
- Wang, J.Z.; Du, Z.; Payattakool, R.; Yu, P.S.; Chen, C.-F. A new method to measure the semantic similarity of GO terms. Bioinformatic 2007, 23, 1274–1281. [Google Scholar] [CrossRef]
- Schalch, T.; Steiner, F.A. Structure of centromere chromatin: From nucleosome to chromosomal architecture. Chromosoma 2017, 126, 443–455. [Google Scholar] [CrossRef]
- Tatsuya, T.; Mori, T.; Tada, S.; Krajewski, W.; Rozovskaia, T.; Wassell, R.; Dubois, G.; Mazo, A.; Croce, C.M.; Canaani, E. ALL-1 Is a Histone Methyltransferase that Assembles a Supercomplex of Proteins Involved in Transcriptional Regulation. Mol. Cell 2002, 5, 1119–1128. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cross Validation | Size | Combined-instance | Homolog-instance | Target-instance | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PR | SE | MCC | PR | SE | MCC | PR | SE | MCC | ||
| Direct contact | 9125 | 0.8553 | 0.8830 | 0.7627 | 0.8554 | 0.8830 | 0.7629 | 0.8613 | 0.8830 | 0.7673 |
| Indirect contact | 9125 | 0.8790 | 0.8506 | 0.7611 | 0.8792 | 0.8508 | 0.7613 | 0.8786 | 0.8562 | 0.7655 |
| (Acc; MCC) | (86.68%; 0.7616) | (86.69%; 0.7618) | (86.96%; 0.7663) | |||||||
| (ROC-AUC) | (0.9308) | (0.9309) | (0.9320) | |||||||
| F1 Score | 0.8740 | 0.8690 | 0.8720 | |||||||
| Independent test | HPRD | CORUM | KEGG | |||||||
| (+83.99%; −86.38%) | (+83.10%; −86.26%) | ( ; −75.00%) | ||||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mei, S.; Zhang, K. A Computational Framework for Predicting Direct Contacts and Substructures within Protein Complexes. Biomolecules 2019, 9, 656. https://doi.org/10.3390/biom9110656
Mei S, Zhang K. A Computational Framework for Predicting Direct Contacts and Substructures within Protein Complexes. Biomolecules. 2019; 9(11):656. https://doi.org/10.3390/biom9110656
Chicago/Turabian StyleMei, Suyu, and Kun Zhang. 2019. "A Computational Framework for Predicting Direct Contacts and Substructures within Protein Complexes" Biomolecules 9, no. 11: 656. https://doi.org/10.3390/biom9110656
APA StyleMei, S., & Zhang, K. (2019). A Computational Framework for Predicting Direct Contacts and Substructures within Protein Complexes. Biomolecules, 9(11), 656. https://doi.org/10.3390/biom9110656