Transcriptomic Analysis Reveals the Roles of Detoxification Systems in Response to Mercury in Chromera velia

 ,
, 

Abstract

1. Introduction
2. Materials and Methods
2.1. Chromera velia Culture and Treatment
2.2. Cell Count and Chlorophyll Quantification
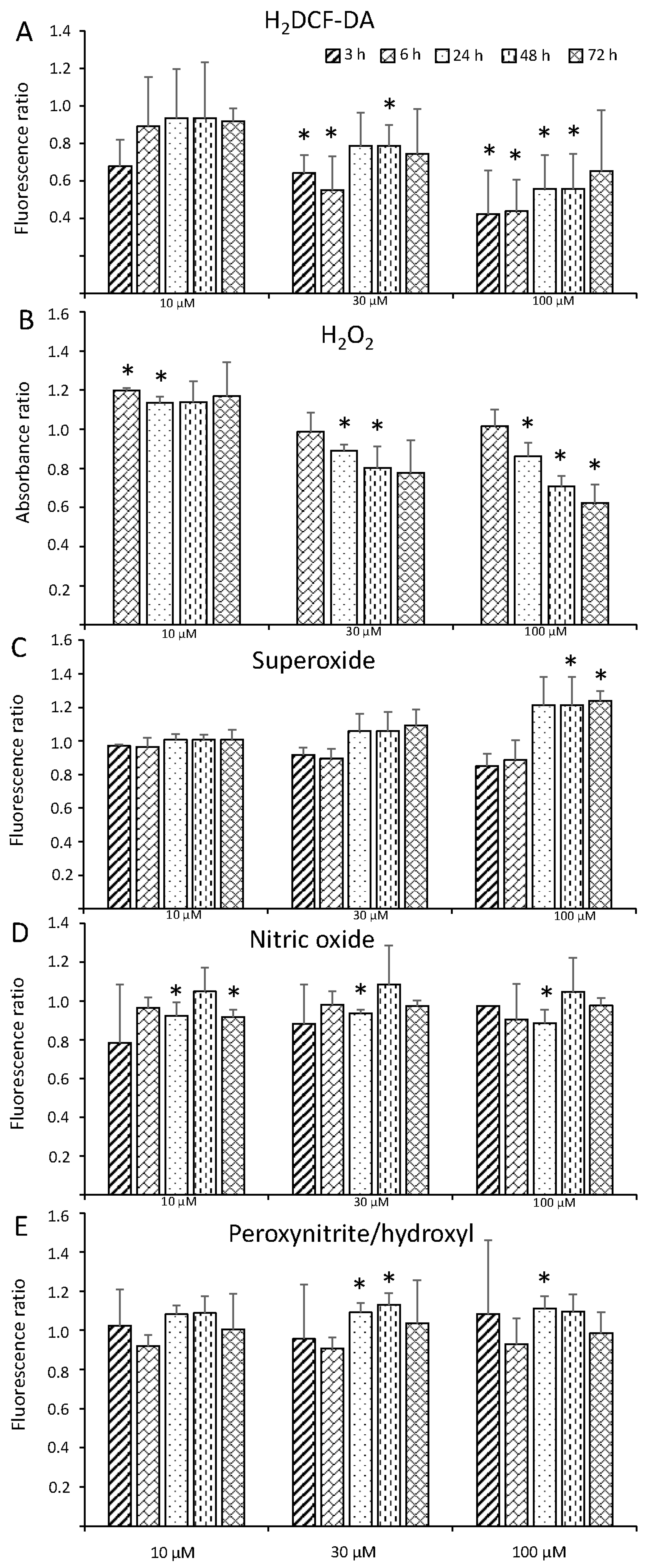
2.3. ROS and RNS Quantification
2.4. RNA Isolation, cDNA Library Construction, and Sequencing
2.5. Transcriptome Analysis
2.6. Quantification of Gene Expression Levels
2.7. Molecular Phylogeny
2.8. RT-qPCR Analysis
2.9. Data Deposition
3. Results and Discussions
3.1. Mercury Toxicity
3.2. ROS and RNS Response
3.3. Sequencing Output and Assembly
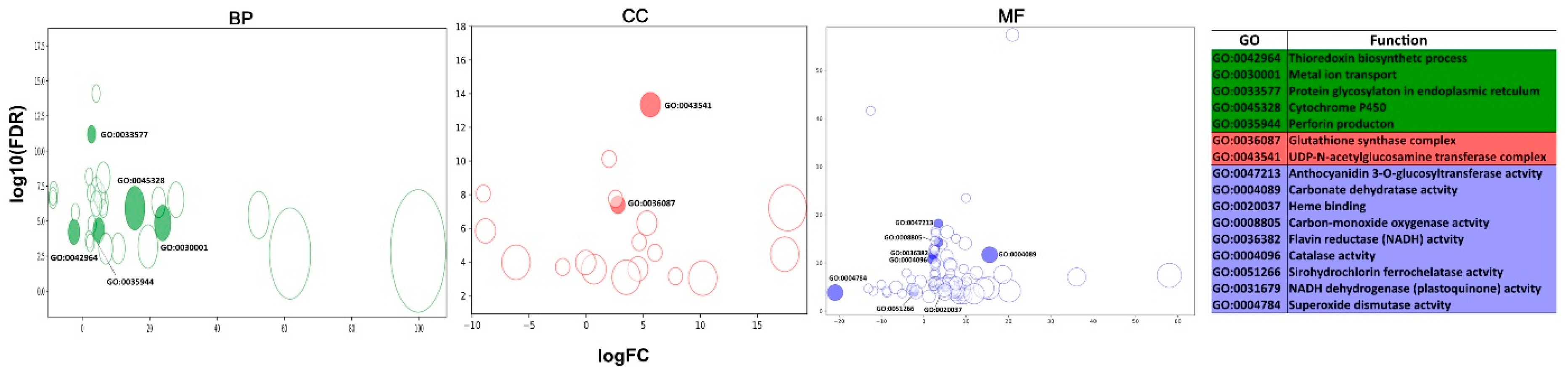
3.4. Functional Annotation
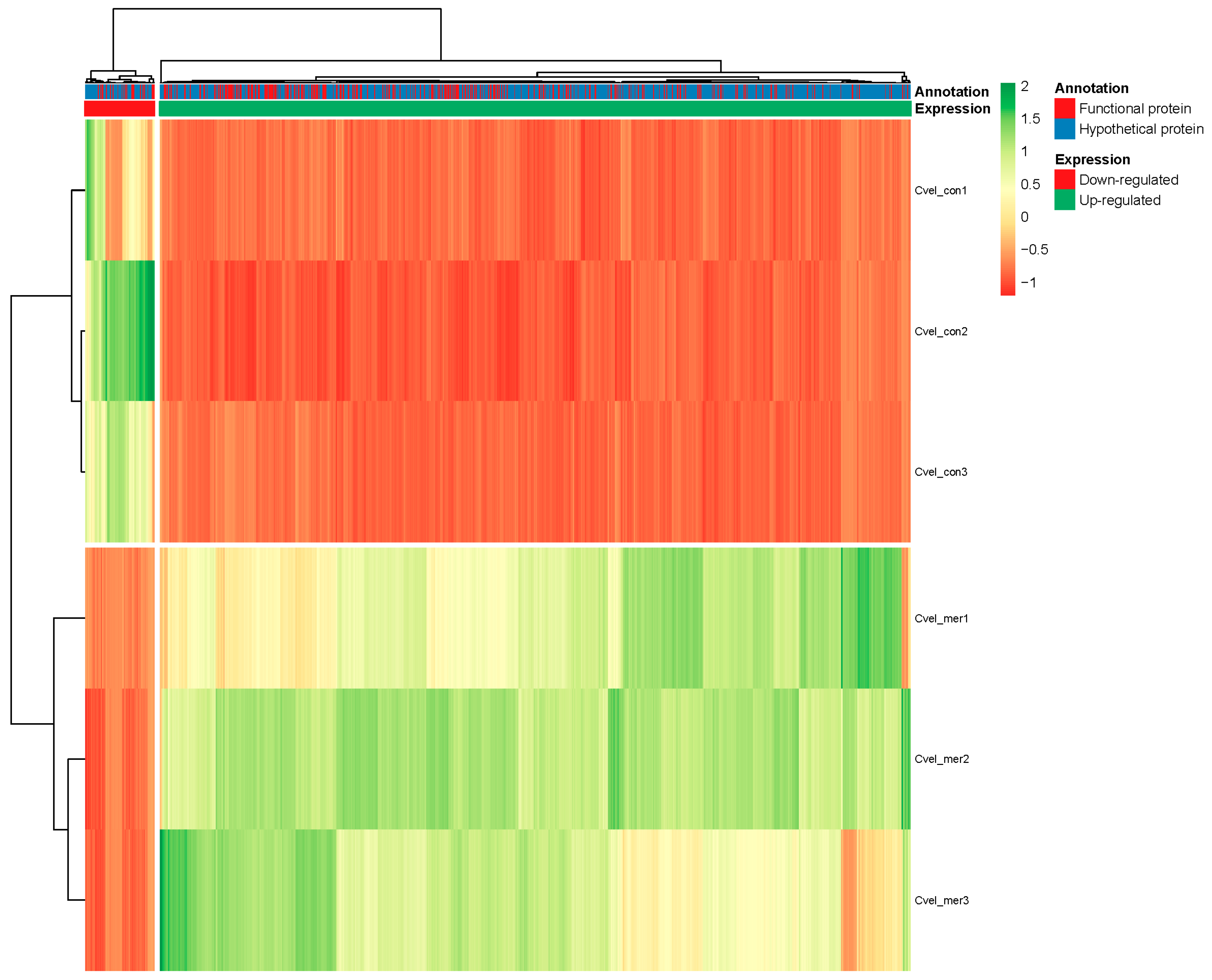
3.5. Differential Expression Analysis
3.6. Hg Exposure
3.6.1. ROS-Antioxidant Defense System Related Genes
3.6.2. Xenobiotics Detoxification-Related Genes
3.6.3. Heavy-Metal Stress Biomarkers
3.6.4. Phylogenetic Analyses
3.7. Transcription Factors in Relation to Hg Stress
3.8. Validation of the DEGs by Real-Time RT-PCR Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Harada, M. Minamata Disease: Methylmercury Poisoning in Japan Caused by Environmental Pollution. Crit. Rev. Toxicol. 1995, 25, 1–24. [Google Scholar] [CrossRef]
- Lamborg, C.H.; Hammerschmidt, C.R.; Bowman, K.L.; Swarr, G.J.; Munson, K.M.; Ohnemus, D.C.; Lam, P.J.; Heimbürger, L.-E.; Rijkenberg, M.J.A.; Saito, M.A. A global ocean inventory of anthropogenic mercury based on water column measurements. Nature 2014, 512, 65–68. [Google Scholar] [CrossRef]
- Cho, U.H.; Park, J.O. Mercury-induced oxidative stress in tomato seedlings. Plant Sci. 2000, 156, 1–9. [Google Scholar] [CrossRef]
- Israr, M.; Sahi, S.; Datta, R.; Sarkar, D. Bioaccumulation and physiological effects of mercury in Sesbania drummondii. Chemosphere 2006, 65, 591–598. [Google Scholar] [CrossRef]
- Lomonte, C.; Doronila, A.I.; Gregory, D.; Baker, A.J.M.; Kolev, S.D. Phytotoxicity of biosolids and screening of selected plant species with potential for mercury phytoextraction. J. Hazard. Mater. 2010, 173, 494–501. [Google Scholar] [CrossRef]
- Beauvais-Flück, R.; Slaveykova, V.; Cosio, C. Molecular Effects of Inorganic and Methyl Mercury in Aquatic Primary Producers: Comparing Impact to a Macrophyte and a Green Microalga in Controlled Conditions. Geosciences 2018, 8, 393. [Google Scholar] [CrossRef]
- Bodaly, R.A.; Jansen, W.A.; Majewski, A.R.; Fudge, R.J.P.; Strange, N.E.; Derksen, A.J.; Green, D.J. Postimpoundment time course of increased mercury concentrations in fish in hydroelectric reservoirs of northern Manitoba, Canada. Arch. Environ. Contam. Toxicol. 2007, 53, 379–389. [Google Scholar] [CrossRef]
- Elbaz, A.; Wei, Y.Y.; Meng, Q.; Zheng, Q.; Yang, Z.M. Mercury-induced oxidative stress and impact on antioxidant enzymes in Chlamydomonas reinhardtii. Ecotoxicology 2010, 19, 1285–1293. [Google Scholar] [CrossRef]
- Kamal, M.; Ghaly, A.E.; Mahmoud, N.; CoteCôté, R. Phytoaccumulation of heavy metals by aquatic plants. Environ. Int. 2004, 29, 1029–1039. [Google Scholar] [CrossRef]
- Narula, P.; Mahajan, A.; Gurnani, C.; Kumar, V.; Mukhija, S. Microalgae as an indispensable tool against heavy metals toxicity to plants: A review. Int. J. Pharm. Sci. Rev. Res. 2015, 31, 86–93. [Google Scholar]
- He, S.; Yang, X.; He, Z.; Baligar, V.C. Morphological and Physiological Responses of Plants to Cadmium Toxicity: A Review. Pedosphere 2017, 27, 421–438. [Google Scholar] [CrossRef]
- Azevedo, R.; Rodriguez, E. Phytotoxicity of Mercury in Plants: A Review. J. Bot. 2012, 2012, 1–6. [Google Scholar] [CrossRef]
- Chen, J.; Yang, Z.M. Mercury toxicity, molecular response and tolerance in higher plants. BioMetals 2012, 25, 847–857. [Google Scholar] [CrossRef]
- Apel, K.; Hirt, H. Reactive Oxygen Species: Metabolism, Oxidative Stress, and Signal Transduction. Annu. Rev. Plant Biol. 2004, 55, 373–399. [Google Scholar] [CrossRef]
- Gupta, D.K.; Pena, L.B.; Romero-Puertas, M.C.; Hernández, A.; Inouhe, M.; Sandalio, L.M. NADPH oxidases differentially regulate ROS metabolism and nutrient uptake under cadmium toxicity. Plant Cell Environ. 2017, 40, 509–526. [Google Scholar] [CrossRef]
- Del Río, L.A. ROS and RNS in plant physiology: An overview. J. Exp. Bot. 2015, 66, 2827–2837. [Google Scholar] [CrossRef]
- Martins, L.L.; Mourato, M.P.; Cardoso, A.I.; Pinto, A.P.; Mota, A.M.; Maria, M.L.; de Varennes, A.; de Lurdes, S.; Gonçalves, M.; de Varennes, A. Oxidative stress induced by cadmium in Nicotiana tabacum L.: Effects on growth parameters, oxidative damage and antioxidant responses in different plant parts. Acta Physiol. Plant. 2011, 33, 1375–1383. [Google Scholar] [CrossRef]
- Sytar, O.; Kumar, A.; Latowski, D.; Kuczynska, P.; Strzałka, K.; Prasad, M.N.V. Heavy metal-induced oxidative damage, defense reactions, and detoxification mechanisms in plants. Acta Physiol. Plant. 2013, 35, 985–999. [Google Scholar] [CrossRef]
- Gupta, D.K.; Palma, J.M.; Corpas, F.J. Reactive Oxygen Species and Oxidative Damage in Plants under Stress; Springer International Publishing: Cham, Switzerland, 2015; ISBN 978-3-319-20420-8. [Google Scholar]
- De Michele, R.; Vurro, E.; Rigo, C.; Costa, A.; Elviri, L.; Di Valentin, M.; Careri, M.; Zottini, M.; Sanita di Toppi, L.; Lo Schiavo, F. Nitric Oxide Is Involved in Cadmium-Induced Programmed Cell Death in Arabidopsis Suspension Cultures. Plant Physiol. 2009, 150, 217–228. [Google Scholar] [CrossRef]
- Chen, J.; Shiyab, S.; Han, F.X.; Monts, D.L.; Waggoner, C.A.; Yang, Z.; Su, Y. Bioaccumulation and physiological effects of mercury in Pteris vittata and Nephrolepis exaltata. Ecotoxicology 2009, 18, 110. [Google Scholar] [CrossRef]
- Ortega-Villasante, C.; Rellán-Álvarez, R.; Del Campo, F.F.; Carpena-Ruiz, R.O.; Hernández, L.E. Cellular damage induced by cadmium and mercury in Medicago sativa. J. Exp. Bot. 2005, 56, 2239–2251. [Google Scholar] [CrossRef]
- Ortega-Villasante, C.; Hernández, L.E.; Rellán-Álvarez, R.; Del Campo, F.F.; Carpena-Ruiz, R.O. Rapid alteration of cellular redox homeostasis upon exposure to cadmium and mercury in alfalfa seedlings. New Phytol. 2007, 176, 96–107. [Google Scholar] [CrossRef]
- Zhou, Z.S.; Wang, S.J.; Yang, Z.M. Biological detection and analysis of mercury toxicity to alfalfa (Medicago sativa) plants. Chemosphere 2008, 70, 1500–1509. [Google Scholar] [CrossRef]
- Zhou, Z.S.; Huang, S.Q.; Guo, K.; Mehta, S.K.; Zhang, P.C.; Yang, Z.M. Metabolic adaptations to mercury-induced oxidative stress in roots of Medicago sativa L. J. Inorg. Biochem. 2007, 101, 1–9. [Google Scholar] [CrossRef]
- Kováčik, J.; Rotková, G.; Bujdoš, M.; Babula, P.; Peterková, V.; Matúš, P. Ascorbic acid protects Coccomyxa subellipsoidea against metal toxicity through modulation of ROS/NO balance and metal uptake. J. Hazard. Mater. 2017, 339, 200–207. [Google Scholar] [CrossRef]
- Rellán-Álvarez, R.; Ortega-Villasante, C.; Álvarez-Fernández, A.; del Campo, F.F.; Hernández, L.E. Stress Responses of Zea mays to Cadmium and Mercury. Plant Soil 2006, 279, 41–50. [Google Scholar] [CrossRef]
- DellaGreca, M.; Pinto, G.; Pollio, A.; Previtera, L.; Temussi, F. Biotransformation of sinapic acid by the green algae Stichococcus bacillaris 155LTAP and Ankistrodesmus braunii C202.7a. Tetrahedron Lett. 2003, 44, 2779–2780. [Google Scholar] [CrossRef]
- Kirso, U.; Irha, N. Role of algae in fate of carcinogenic polycyclic aromatic hydrocarbons in the aquatic environment. Ecotoxicol. Environ. Saf. 1998, 41, 83–89. [Google Scholar] [CrossRef]
- Pflugmacher, S.; Wiencke, C.; Sandermann, H. Activity of phase I and phase II detoxication enzymes in Antarctic and Arctic macroalgae. Mar. Environ. Res. 1999, 48, 23–36. [Google Scholar] [CrossRef]
- Thies, F.; Grimme, L.H. In vivo O-dealkylation of resorufin and coumarin ethers by the green alga Chlorella fusca analysed by a rapid and sensitive high-performance liquid chromatographic assay. J. Chromatogr. A 1994, 684, 168–173. [Google Scholar] [CrossRef]
- Warshawsky, D.; Cody, T.; Radike, M.; Reilman, R.; Schumann, B.; LaDow, K.; Schneider, J. Biotransformation of benzo[a]pyrene and other polycyclic aromatic hydrocarbons and heterocyclic analogs by several green algae and other algal species under gold and white light. Chem. Biol. Interact. 1995, 97, 131–148. [Google Scholar] [CrossRef]
- Barque, J.P.; Abahamid, A.; Flinois, J.P.; Beaune, P.; Bonaly, J. Constitutive overexpression of immunoidentical forms of PCP-induced Euglena gracilis CYP-450. Biochem. Biophys. Res. Commun. 2002, 298, 277–281. [Google Scholar] [CrossRef]
- Pflugmacher, S.; Sandermann, H. Cytochrome P450 Monooxygenases for Fatty Acids and Xenobiotics in Marine Macroalgae. Plant Physiol. 1998, 117, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Thies, F.; Backhaus, T.; Bossmann, B.; Grimme, L.H. Xenobiotic Biotransformation in Unicellular Green Algae (Involvement of Cytochrome P450 in the Activation and Selectivity of the Pyridazinone Pro-Herbicide Metflurazon). Plant Physiol. 1996, 112, 361–370. [Google Scholar] [CrossRef]
- Zangar, R.C.; Davydov, D.R.; Verma, S. Mechanisms that regulate production of reactive oxygen species by cytochrome P450. Toxicol. Appl. Pharmacol. 2004, 199, 316–331. [Google Scholar] [CrossRef]
- Pflugmacher, S.; Schröder, P.; Sandermann, H. Taxonomic distribution of plant glutathione S-transferases acting on xenobiotics. Phytochemistry 2000, 54, 267–273. [Google Scholar] [CrossRef]
- Reilman, R.; Keenan, T.H.; Cody, T.E.; Radike, M.J.; Warshawsky, D. Conjugation of benzo[a]pyrene metabolites by freshwater green alga Selenastrum capricornutum. Chem. Biol. Interact. 1990, 74, 93–105. [Google Scholar]
- Sandermann, H. Molecular ecotoxicology of plants. Trends Plant Sci. 2004, 9, 406–413. [Google Scholar] [CrossRef]
- Avery, S.V.; Codd, G.A.; Gadd, G.M. Characterisation of caesium transport in the microalga Chlorella salina. Biochem. Soc. Trans. 1995, 23, 468S. [Google Scholar] [CrossRef]
- Avilés, C.; Torres-Márquez, M.E.; Mendoza-Cózatl, D.; Moreno-Sánchez, R. Time-course development of the Cd2+ hyper-accumulating phenotype in Euglena gracilis. Arch. Microbiol. 2005, 184, 83–92. [Google Scholar] [CrossRef]
- Jabusch, T.W.; Swackhamer, D.L. Subcellular accumulation of polychlorinated biphenyls in the green alga Chlamydomonas reinhardtii. Environ. Toxicol. Chem. 2004, 23, 2823–2830. [Google Scholar] [CrossRef] [PubMed]
- Smital, T.; Sauerborn, R.; Hackenberger, B.K. Inducibility of the P-glycoprotein transport activity in the marine mussel Mytilus galloprovincialis and the freshwater mussel Dreissena polymorpha. Aquat. Toxicol. 2003, 65, 443–465. [Google Scholar] [CrossRef]
- Torres, M.A.; Barros, M.P.; Campos, S.C.G.; Pinto, E.; Rajamani, S.; Sayre, R.T.; Colepicolo, P. Biochemical biomarkers in algae and marine pollution: A review. Ecotoxicol. Environ. Saf. 2008, 71, 1–15. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Chan, Z. ROS Regulation during Abiotic Stress Responses in Crop Plants. Front. Plant Sci. 2015, 6, 1092. [Google Scholar] [CrossRef] [PubMed]
- Jalmi, S.K.; Bhagat, P.K.; Verma, D.; Noryang, S.; Tayyeba, S.; Singh, K.; Sharma, D.; Sinha, A.K. Traversing the Links between Heavy Metal Stress and Plant Signaling. Front. Plant Sci. 2018, 9, 12. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, C.; Liang, Y.; Wang, C.; Yang, C.; Liu, G. A novel bZIP gene from Tamarix hispida mediates physiological responses to salt stress in tobacco plants. J. Plant Physiol. 2010, 167, 222–230. [Google Scholar] [CrossRef]
- Hong, C.; Cheng, D.; Zhang, G.; Zhu, D.; Chen, Y.; Tan, M. The role of ZmWRKY4 in regulating maize antioxidant defense under cadmium stress. Biochem. Biophys. Res. Commun. 2017, 482, 1504–1510. [Google Scholar] [CrossRef]
- Moore, R.B.; Oborník, M.; Janouškovec, J.; Chrudimský, T.; Vancová, M.; Green, D.H.; Wright, S.W.; Davies, N.W.; Bolch, C.J.S.; Heimann, K.; et al. A photosynthetic alveolate closely related to apicomplexan parasites. Nature 2008, 451, 959–963. [Google Scholar] [CrossRef]
- Oborník, M.; Vancová, M.; Lai, D.H.; Janouškovec, J.; Keeling, P.J.; Lukeš, J. Morphology and ultrastructure of multiple life cycle stages of the photosynthetic relative of apicomplexa, Chromera velia. Protist 2011, 162, 115–130. [Google Scholar] [CrossRef]
- Vazač, J.; Füssy, Z.; Hladová, I.; Killi, S.; Oborník, M. Ploidy and Number of Chromosomes in the Alveolate Alga Chromera velia. Protist 2018, 169, 53–63. [Google Scholar] [CrossRef]
- Kotabová, E.; Jarešová, J.; Kaňa, R.; Sobotka, R.; Bína, D.; Prášil, O. Novel type of red-shifted chlorophyll a antenna complex from Chromera velia. I. Physiological relevance and functional connection to photosystems. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Hoppenz, P.; Jakob, T.; Weisheit, W.; Mittag, M.; Wilhelm, C.; Goss, R. Unusual features of the high light acclimation of Chromera velia. Photosynth. Res. 2014, 122, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Quigg, A.; Kotabová, E.; Jarešová, J.; Kaňa, R.; Šetlík, J.; Šedivá, B.; Komárek, O.; Prášil, O. Photosynthesis in Chromera velia Represents a Simple System with High Efficiency. PLoS ONE 2012, 7, e47036. [Google Scholar] [CrossRef] [PubMed]
- Sobotka, R.; Esson, H.J.; Koník, P.; Trsková, E.; Moravcová, L.; Horák, A.; Dufková, P.; Oborník, M. Extensive gain and loss of photosystem I subunits in chromerid algae, photosynthetic relatives of apicomplexans. Sci. Rep. 2017, 7, 13214. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.M.; Xing, Y.M.; Zhang, D.W.; Guo, S.X. Transcriptome analysis of genes involved in defence response in Polyporus umbellatus with Armillaria mellea infection. Sci. Rep. 2015, 5, 16075. [Google Scholar] [CrossRef] [PubMed]
- Dittami, S.M.; Scornet, D.; Petit, J.-L.L.; Ségurens, B.; Da Silva, C.; Corre, E.; Dondrup, M.; Glatting, K.-H.H.; König, R.; Sterck, L.; et al. Global expression analysis of the brown alga Ectocarpus siliculosus (Phaeophyceae) reveals large-scale reprogramming of the transcriptome in response to abiotic stress. Genome Biol. 2009, 10, R66. [Google Scholar] [CrossRef]
- Holzinger, A.; Pichrtová, M. Abiotic Stress Tolerance of Charophyte Green Algae: New Challenges for Omics Techniques. Front. Plant Sci. 2016, 7, 678. [Google Scholar] [CrossRef]
- Khraiwesh, B.; Qudeimat, E.; Thimma, M.; Chaiboonchoe, A.; Jijakli, K.; Alzahmi, A.; Arnoux, M.; Salehi-Ashtiani, K. Genome-wide expression analysis offers new insights into the origin and evolution of Physcomitrella patens stress response. Sci. Rep. 2015, 5, 17434. [Google Scholar] [CrossRef]
- Matthijs, M.; Fabris, M.; Broos, S.; Vyverman, W.; Goossens, A. Profiling of the Early Nitrogen Stress Response in the Diatom Phaeodactylum tricornutum Reveals a Novel Family of RING-Domain Transcription Factors. Plant Physiol. 2016, 170, 489–498. [Google Scholar] [CrossRef]
- De Michele, R.; Formentin, E.; Todesco, M.; Toppo, S.; Carimi, F.; Zottini, M.; Barizza, E.; Ferrarini, A.; Delledonne, M.; Fontana, P.; et al. Transcriptome analysis of Medicago truncatula leaf senescence: Similarities and differences in metabolic and transcriptional regulations as compared with Arabidopsis, nodule senescence and nitric oxide signalling. New Phytol. 2009, 181, 563–575. [Google Scholar] [CrossRef]
- Wintermans, J.F.G.M.; De Mots, A. Spectrophotometric characteristics of chlorophylls a and b and their phenophytins in ethanol. Biochim. Biophys. Acta Biophys. Incl. Photosynth. 1965, 109, 448–453. [Google Scholar] [CrossRef]
- Chen, X.; Zhong, Z.; Xu, Z.; Chen, L.; Wang, Y. 2′,7′-Dichlorodihydrofluorescein as a fluorescent probe for reactive oxygen species measurement: Forty years of application and controversy. Free Radic. Res. 2010, 44, 587–604. [Google Scholar] [CrossRef] [PubMed]
- Nazarewicz, R.R.; Bikineyeva, A.; Dikalov, S.I. Rapid and Specific Measurements of Superoxide Using Fluorescence Spectroscopy. J. Biomol. Screen. 2013, 18, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Urano, Y.; Kikuchi, K.; Higuchi, T.; Hirata, Y.; Nagano, T. Fluorescent Indicators for Imaging Nitric Oxide Production. Angew. Chem. Int. Ed. 1999, 38, 3209–3212. [Google Scholar] [CrossRef]
- Setsukinai, K.; Urano, Y.; Kakinuma, K.; Majima, H.J.; Nagano, T. Development of Novel Fluorescence Probes That Can Reliably Detect Reactive Oxygen Species and Distinguish Specific Species. J. Biol. Chem. 2003, 278, 3170–3175. [Google Scholar] [CrossRef]
- Bellincampi, D.; Dipierro, N.; Salvi, G.; Cervone, F.; De Lorenzo, G. Extracellular H(2)O(2) induced by oligogalacturonides is not involved in the inhibition of the auxin-regulated rolB gene expression in tobacco leaf explants. Plant Physiol. 2000, 122, 1379–1386. [Google Scholar] [CrossRef]
- Hu, L.; Li, H.; Chen, L.; Lou, Y.; Amombo, E.; Fu, J. RNA-seq for gene identification and transcript profiling in relation to root growth of bermudagrass (Cynodon dactylon) under salinity stress. BMC Genom. 2015, 16, 575. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 299, 644. [Google Scholar] [CrossRef]
- Bryant, D.M.; Johnson, K.; DiTommaso, T.; Tickle, T.; Couger, M.B.; Payzin-Dogru, D.; Lee, T.J.; Leigh, N.D.; Kuo, T.-H.; Davis, F.G.; et al. A Tissue-Mapped Axolotl De Novo Transcriptome Enables Identification of Limb Regeneration Factors. Cell Rep. 2017, 18, 762–776. [Google Scholar] [CrossRef]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinfor. 2014, 12, 323. [Google Scholar]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [PubMed]
- Boratyn, G.M.; Schäffer, A.A.; Agarwala, R.; Altschul, S.F.; Lipman, D.J.; Madden, T.L. Domain enhanced lookup time accelerated BLAST. Biol. Direct 2012, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.H.; Ansari, H.; Otto, T.D.; Linger, C.M.K.; Olisko, M.K.; Michálek, J.; Saxena, A.; Shanmugam, D.; Tayyrov, A.; Veluchamy, A.; et al. Chromerid genomes reveal the evolutionary path from photosynthetic algae to obligate intracellular parasites. Elife 2015, 4, 1–41. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, H.; Cantor, M.; Dusheyko, S.; Hua, S.; Poliakov, A.; Shabalov, I.; Smirnova, T.; Grigoriev, I.V.; Dubchak, I. The genome portal of the Department of Energy Joint Genome Institute: 2014 updates. Nucleic Acids Res. 2014, 42, D26–D31. [Google Scholar] [CrossRef] [PubMed]
- Bendtsen, J.D.; Nielsen, H.; Von Heijne, G.; Brunak, S. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 2004, 340, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Gruber, A.; Rocap, G.; Kroth, P.G.; Armbrust, E.V.; Mock, T. Plastid proteome prediction for diatoms and other algae with secondary plastids of the red lineage. Plant J. 2015, 81, 519–528. [Google Scholar] [CrossRef]
- Emanuelsson, O.; Brunak, S.; von Heijne, G.; Nielsen, H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2007, 2, 953–971. [Google Scholar] [CrossRef]
- Bannai, H.; Tamada, Y.; Maruyama, O.; Nakai, K.; Miyano, S. Extensive feature detection of N-terminal protein sorting signals. Bioinformatics 2002, 18, 298–305. [Google Scholar] [CrossRef]
- Horton, P.; Park, K.J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef] [PubMed]
- Claros, M.G.; Vincens, P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 1996, 241, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Almagro Armenteros, J.J.; Sønderby, C.K.; Sønderby, S.K.; Nielsen, H.; Winther, O. DeepLoc: Prediction of protein subcellular localization using deep learning. Bioinformatics 2017, 33, 3387–3395. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- de Juan, D.; Pazos, F.; Valencia, A.; Evaluation, P.M.; Rehbein, P.; Schwalbe, H.; Jones, D.T.; Buchan, D.W.A.; Cozzetto, D.; Pontil, M.; et al. ProtTest: Selection of best-fit models of protein evolution. Bioinformatics 2015, 21, 2104–2105. [Google Scholar]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Nowicka, B.; Pluciński, B.; Kuczyńska, P.; Kruk, J. Physiological characterization of Chlamydomonas reinhardtii acclimated to chronic stress induced by Ag, Cd, Cr, Cu and Hg ions. Ecotoxicol. Environ. Saf. 2016, 130, 133–145. [Google Scholar] [CrossRef]
- Cargnelutti, D.; Tabaldi, L.A.; Spanevello, R.M.; de Oliveira Jucoski, G.; Battisti, V.; Redin, M.; Linares, C.E.B.; Dressler, V.L.; de Moraes Flores, É.M.; Nicoloso, F.T.; et al. Mercury toxicity induces oxidative stress in growing cucumber seedlings. Chemosphere 2006, 65, 999–1006. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes11Edited by F. Cohen. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Pinto, E.; Sigaud-kutner, T.C.S.; Leitao, M.A.S.; Okamoto, O.K.; Morse, D.; Colepicolo, P. HEAVY METAL-INDUCED OXIDATIVE STRESS IN ALGAE1. J. Phycol. 2003, 39, 1008–1018. [Google Scholar] [CrossRef]
- Sabatini, S.E.; Juárez, Á.B.; Eppis, M.R.; Bianchi, L.; Luquet, C.M.; de Molina, M.D.C.R. Oxidative stress and antioxidant defenses in two green microalgae exposed to copper. Ecotoxicol. Environ. Saf. 2009, 72, 1200–1206. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.T.; Weatherby, K.; Carter, D.; Šlapeta, J. Effect of nutrient concentration and salinity on immotile-motile transformation of Chromera velia. J. Eukaryot. Microbiol. 2010, 57, 444–446. [Google Scholar] [CrossRef] [PubMed]
- Kabeláčová, K. The Fitness of Three Strains of the Alga Chromera velia—Salinity and pH; University of South Bohemia: Budejovice, Ceske, 2018. [Google Scholar]
- Perez-Perez, M.E.; Lemaire, S.D.; Crespo, J.L. Reactive Oxygen Species and Autophagy in Plants and Algae. Plant Physiol. 2012, 160, 156–164. [Google Scholar] [CrossRef]
- Shanura Fernando, I.P.; Asanka Sanjeewa, K.K.; Samarakoon, K.W.; Lee, W.W.; Kim, H.S.; Kim, E.A.; Gunasekara, U.K.; Abeytunga, D.T.U.; Nanayakkara, C.; De Silva, E.D.; et al. FTIR characterization and antioxidant activity of water soluble crude polysaccharides of Sri Lankan marine algae. Algae 2017, 32, 75–86. [Google Scholar] [CrossRef]
- Fan, Y.; Bose, J.; Zhou, M.; Shabala, S. ROS Production, Scavenging, and Signaling under Salinity Stress. In Managing Salt Tolerance in Plants; Wani, S.H., Hossain, M.A., Eds.; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar]
- Gratão, P.L.; Monteiro, C.C.; Tezotto, T.; Carvalho, R.F.; Alves, L.R.; Peters, L.P.; Azevedo, R.A. Cadmium stress antioxidant responses and root-to-shoot communication in grafted tomato plants. BioMetals 2015, 28, 803–816. [Google Scholar] [CrossRef]
- Shahabivand, S.; Zare Maivan, H.; Mahmoudi, E.; Soltani, B.M.; Sharifi, M.; Aliloo, A.A. Antioxidant activity and gene expression associated with cadmium toxicity in wheat affected by mycorrhizal fungus. Zemdirb. Agric. 2016, 103, 53–60. [Google Scholar] [CrossRef]
- Cassier-Chauvat, C.; Chauvat, F. Responses to oxidative and heavy metal stresses in cyanobacteria: Recent advances. Int. J. Mol. Sci. 2015, 16, 871–886. [Google Scholar] [CrossRef]
- Narainsamy, K.; Marteyn, B.; Sakr, S.; Cassier-Chauvat, C.; Chauvat, F. Genomics of the Pleïotropic Glutathione System in Cyanobacteria. Adv. Bot. Res. 2013, 65, 157–188. [Google Scholar]
- Ken, C.F.; Hsiung, T.M.; Huang, Z.X.; Juang, R.H.; Lin, C.T. Characterization of Fe/Mn-superoxide dismutase from diatom Thallassiosira weissflogii: Cloning, expression, and property. J. Agric. Food Chem. 2005, 53, 1470–1474. [Google Scholar] [CrossRef] [PubMed]
- Fridovich, I. Superoxide anion radical (O 2), superoxide dismutases, and related matters. J. Biol. Chem. 1997, 272, 18515–18517. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, O.K.; Robertson, D.L.; Fagan, T.F.; Hastings, J.W.; Colepicolo, P. Different Regulatory Mechanisms Modulate the Expression of a Dinoflagellate Iron-Superoxide Dismutase. J. Biol. Chem. 2001, 276, 19989–19993. [Google Scholar] [CrossRef] [PubMed]
- Barros, M.P.; Pinto, E.; Sigaud-Kutner, T.C.S.; Cardozo, K.H.M.; Colepicolo, P. Rhythmicity and oxidative/nitrosative stress in algae. Biol. Rhythm Res. 2005, 36, 67–82. [Google Scholar] [CrossRef]
- Murthy, K.N.C.; Vanitha, A.; Rajesha, J.; Swamy, M.M.; Sowmya, P.R.; Ravishankar, G.A. In vivo antioxidant activity of carotenoids from Dunaliella salina—A green microalga. Life Sci. 2005, 76, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, B.N.; Mehta, S.K.; Amar, A.; Gaur, J.P. Oxidative stress in Scenedesmus sp. during short- and long-term exposure to Cu2+ and Zn2+. Chemosphere 2006, 62, 538–544. [Google Scholar] [CrossRef]
- Yamada, Y.; Fujiwara, T.; Sato, T.; Igarashi, N.; Tanaka, N. The 2.0 å crystal structure of catalase-peroxidase from haloarcula marismortui. Nat. Struct. Biol. 2002, 9, 691. [Google Scholar] [CrossRef]
- Welinder, K.G. Superfamily of plant, fungal and bacterial peroxidases. Curr. Opin. Struct. Biol. 1992, 2, 388–393. [Google Scholar] [CrossRef]
- Passardi, F.; Bakalovic, N.; Teixeira, F.K.; Margis-Pinheiro, M.; Penel, C.; Dunand, C. Prokaryotic origins of the non-animal peroxidase superfamily and organelle-mediated transmission to eukaryotes. Genomics 2007, 89, 567–579. [Google Scholar] [CrossRef]
- Klotz, M.G.; Loewen, P.C. The molecular evolution of catalatic hydroperoxidases: Evidence for multiple lateral transfer of genes between prokaryota and from bacteria into eukaryota. Mol. Biol. Evol. 2003, 20, 1098–1112. [Google Scholar] [CrossRef]
- Yu, R.; Tang, Y.; Liu, C.; Du, X.; Miao, C.; Shi, G. Comparative transcriptomic analysis reveals the roles of ROS scavenging genes in response to cadmium in two pak choi cultivars. Sci. Rep. 2017, 7, 9217. [Google Scholar] [CrossRef] [PubMed]
- Zang, X.; Geng, X.; Wang, F.; Liu, Z.; Zhang, L.; Zhao, Y.; Tian, X.; Ni, Z.; Yao, Y.; Xin, M.; et al. Overexpression of wheat ferritin gene TaFER-5B enhances tolerance to heat stress and other abiotic stresses associated with the ROS scavenging. BMC Plant Biol. 2017, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Zok, A.; Oláh, R.; Hideg, É.; Horváth, V.G.; Kós, P.B.; Majer, P.; Váradi, G.; Szegedi, E. Effect of Medicago sativa ferritin gene on stress tolerance in transgenic grapevine. Plant Cell. Tissue Organ Cult. 2010, 100, 339–344. [Google Scholar] [CrossRef]
- Hannemann, F.; Bichet, A.; Ewen, K.M.; Bernhardt, R. Cytochrome P450 systems-biological variations of electron transport chains. Biochim. Biophys. Acta Gen. Subj. 2007, 1770, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.L.; Kelly, D.E. Microbial cytochromes P450: Biodiversity and biotechnology. Where do cytochromes P450 come from, what do they do and what can they do for us? Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120476. [Google Scholar] [CrossRef]
- Schenkman, J.B.; Jansson, I. The many roles of cytochrome b5. Pharmacol. Ther. 2003, 97, 139–152. [Google Scholar] [CrossRef]
- Kandel, S.E.; Lampe, J.N. Role of Protein–Protein Interactions in Cytochrome P450-Mediated Drug Metabolism and Toxicity. Chem. Res. Toxicol. 2014, 27, 1474–1486. [Google Scholar] [CrossRef]
- Jamakhandi, A.P.; Kuzmic, P.; Sanders, D.E.; Miller, G.P. Global Analysis of Protein−Protein Interactions Reveals Multiple CYP2E1−Reductase Complexes †. Biochemistry 2007, 46, 10192–10201. [Google Scholar] [CrossRef][Green Version]
- Lane, T.S.; Rempe, C.S.; Davitt, J.; Staton, M.E.; Peng, Y.; Soltis, D.E.; Melkonian, M.; Deyholos, M.; Leebens-Mack, J.H.; Chase, M.; et al. Diversity of ABC transporter genes across the plant kingdom and their potential utility in biotechnology. BMC Biotechnol. 2016, 16, 47. [Google Scholar] [CrossRef]
- Ben Chekroun, K.; Baghour, M. The role of algae in phytoremediation of heavy metals: A review. J. Mater. Environ. Sci. 2013, 4, 873–880. [Google Scholar]
- Hossain, M.A.; Piyatida, P.; da Silva, J.A.T.; Fujita, M. Molecular Mechanism of Heavy Metal Toxicity and Tolerance in Plants: Central Role of Glutathione in Detoxification of Reactive Oxygen Species and Methylglyoxal and in Heavy Metal Chelation. J. Bot. 2012, 2012, 37. [Google Scholar] [CrossRef]
- Yamuna, A.; Kabila, V.; Geraldine, P. Expression of heat shock protein 70 in freshwater prawn Macrobrachium malcolmsonii (H. Milne Edwards) following exposure to Hg and Cu. Indian J. Exp. Biol. 2000, 38, 921–925. [Google Scholar] [PubMed]
- Wei, Y.Y.; Zheng, Q.; Liu, Z.P.; Yang, Z.M. Regulation of tolerance of chlamydomonas reinhardtii to heavy metal toxicity by heme oxygenase-1 and carbon monoxide. Plant Cell Physiol. 2011, 52, 1665–1675. [Google Scholar] [CrossRef]
- Sanità Di Toppi, L.; Prasad, M.N.V.; Ottonello, S. Metal Chelating Peptides and Proteins in Plants. In Physiology and Biochemistry of Metal Toxicity and Tolerance in Plants; Prasad, M.N.V., Ed.; Springer: Dordrecht, The Netherlands, 2002; pp. 59–93. [Google Scholar]
- Zémocký, M. Phylogenetic relationships in class I of the superfamily of bacterial, fungal, and plant peroxidases. Eur. J. Biochem. 2004, 271, 3297–3309. [Google Scholar] [CrossRef] [PubMed]
- Theologis, A.; Ecker, J.R.; Palm, C.J.; Federspiel, N.A.; Kaul, S.; White, O.; Alonso, J.; Altafi, H.; Araujo, R.; Bowman, C.L.; et al. Sequence and analysis of chromosome 1 of the plant Arabidopsis thaliana. Nature 2000, 408, 816–820. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Chen, C.; Du, J.; Liu, H.; Cui, Y.; Zhang, Y.; He, Y.; Wang, Y.; Chu, C.; Feng, Z.; et al. Co-Overexpression FIT with AtbHLH38 or AtbHLH39 in Arabidopsis-Enhanced Cadmium Tolerance via Increased Cadmium Sequestration in Roots and Improved Iron Homeostasis of Shoots 1. Plant Physiol. 2012, 158, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wang, C.; Wang, Y.; Guo, Y.; Zhao, Y.; Yang, C.; Gao, C. Overexpression of ThVHAc1 and its potential upstream regulator, ThWRKY7, improved plant tolerance of Cadmium stress. Sci. Rep. 2016, 6, 18752. [Google Scholar] [CrossRef]
- Zhang, H.; Li, A.; Zhang, Z.; Huang, Z.; Lu, P.; Zhang, D.; Liu, X.; Zhang, Z.F.; Huang, R. Ethylene Response Factor TERF1, Regulated by ETHYLENE-INSENSITIVE3-like Factors, Functions in Reactive Oxygen Species (ROS) Scavenging in Tobacco (Nicotiana tabacum L.). Sci. Rep. 2016, 6, 29948. [Google Scholar] [CrossRef]
- Yadav, N.R.; Taunk, J.; Rani, A.; Aneja, B.; Yadav, R.C. Role of Transcription Factors in Abiotic Stress Tolerance in Crop Plants. In Climate Change and Plant Abiotic Stress Tolerance; Tuteja, N., Gill, S.S., Eds.; Wiley-VCH GmbH & Co. KGaA: Weinheim, Germany, 2013; pp. 605–640. [Google Scholar]
- Moustafa, A.; Beszteri, B.; Maier, U.G.; Bowler, C.; Valentin, K.; Bhattacharya, D. Genomic footprints of a cryptic plastid endosymbiosis in diatoms. Science 2009, 324, 1724–1726. [Google Scholar] [CrossRef]
- Thiriet-Rupert, S.; Carrier, G.; Chénais, B.; Trottier, C.; Bougaran, G.; Cadoret, J.P.; Schoefs, B.; Saint-Jean, B. Transcription factors in microalgae: Genome-wide prediction and comparative analysis. BMC Genom. 2016, 17, 282. [Google Scholar] [CrossRef]
- Ritter, A.; Dittami, S.M.; Goulitquer, S.; Correa, J.A.; Boyen, C.; Potin, P.; Tonon, T. Transcriptomic and metabolomic analysis of copper stress acclimation in Ectocarpus siliculosus highlights signaling and tolerance mechanisms in brown algae. BMC Plant Biol. 2014, 14, 116. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Wang, T.; Persson, S.; Mueller-Roeber, B.; Schippers, J.H.M. Transcriptional control of ROS homeostasis by KUODA1 regulates cell expansion during leaf development. Nat. Commun. 2014, 5, 3767. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Dong, L.; Han, D.; Zhang, F.; Wu, J.; Jiang, L.; Cheng, Q.; Li, R.; Lu, W.; Meng, F.; et al. GmWRKY31 and GmHDL56 Enhances Resistance to Phytophthora sojae by Regulating Defense-Related Gene Expression in Soybean. Front. Plant Sci. 2017, 8, 781. [Google Scholar] [CrossRef] [PubMed]
- Sewelam, N.; Kazan, K.; Thomas-Hall, S.R.; Kidd, B.N.; Manners, J.M.; Schenk, P.M. Ethylene Response Factor 6 Is a Regulator of Reactive Oxygen Species Signaling in Arabidopsis. PLoS ONE 2013, 8, e70289. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yang, L.; Yan, X.; Liu, Y.; Wang, R.; Fan, T.; Ren, Y.; Tang, X.; Xiao, F.; Liu, Y.; et al. Zinc-Finger Transcription Factor ZAT6 Positively Regulates Cadmium Tolerance through Glutathione-dependent Pathway in Arabidopsis. Plant Physiol. 2016, 171, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Zhuang, K.Y.; Liu, Z.M.; Yang, D.Y.; Ma, N.N.; Meng, Q.W. Overexpression of a novel NAC-type tomato transcription factor, SlNAM1, enhances the chilling stress tolerance of transgenic tobacco. J. Plant Physiol. 2016, 204, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.P.; Way, H.M.; Richardson, T.; Drenth, J.; Joyce, P.A.; McIntyre, C.L. Overexpression of TaNAC69 leads to enhanced transcript levels of stress up-regulated genes and dehydration tolerance in bread wheat. Mol. Plant 2011, 4, 697–712. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Condition | Raw Reads | Clean Reads | Total Mapped |
|---|---|---|---|---|
| Cvel_cont1 | Control | 509,407.39 | 499,612.65 | 435,130,47 (87.09%) |
| Cvel_cont2 | Control | 602,350.18 | 593,301.55 | 440,098,77 (74.18%) |
| Cvel_cont3 | Control | 539,187.31 | 530,589.42 | 464,339,73 (87.51%) |
| Subtotal | 165,094,488 | 162,350,362 | 133,956,897 (82.51%) | |
| Cvel_mer1 | Mercury | 475,722.8 | 467,895.2 | 408,397,57 (87.28%) |
| Cvel_mer2 | Mercury | 532,104.78 | 523,476.04 | 456,650,32 (87.23%) |
| Cvel_mer3 | Mercury | 47,120.479 | 462,513.04 | 400,187,51 (86.52%) |
| Subtotal | 147,903,237 | 145,388,428 | 126,523,540 (87.02%) | |
| Total | 312,997,725 | 307,738,790 |
| Database | Number of Unigenes | Percentage (%) |
|---|---|---|
| CryptoDB | 52,404 | 58.32 |
| InterPro | 52,388 | 58.3 |
| NCBI non-redundant (NR) | 11,173 | 12.43 |
| UniProt | 16,625 | 18.5 |
| Pfam | 3456 | 3.85 |
| GeneOntology (GO) | 1005 | 1.12 |
| TmHMM | 1461 | 1.62 |
| SignalP | 1055 | 1.17 |
| Annotated in at least one Database | 68,713 | 76.47 |
| Total Unigenes | 89,853 |
| Transcription Factors Family | Numbers |
|---|---|
| Zinc finger C2H2-type | 12 |
| Zinc finger C3H-type | 9 |
| Basic helix-loop-helix (bHLH) | 6 |
| Basic Leucine Zipper (bZIP) | 5 |
| Trihelix | 5 |
| ERF | 3 |
| WRKY | 3 |
| B3 | 3 |
| GATA | 3 |
| NAC | 3 |
| ADP-ribosylation factor (ARF) | 2 |
| Homeodomain-leucine zipper (HD-ZIP) | 1 |
| MYB | 1 |
| Total | 56 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharaf, A.; De Michele, R.; Sharma, A.; Fakhari, S.; Oborník, M. Transcriptomic Analysis Reveals the Roles of Detoxification Systems in Response to Mercury in Chromera velia. Biomolecules 2019, 9, 647. https://doi.org/10.3390/biom9110647
Sharaf A, De Michele R, Sharma A, Fakhari S, Oborník M. Transcriptomic Analysis Reveals the Roles of Detoxification Systems in Response to Mercury in Chromera velia. Biomolecules. 2019; 9(11):647. https://doi.org/10.3390/biom9110647
Chicago/Turabian StyleSharaf, Abdoallah, Roberto De Michele, Ayush Sharma, Safieh Fakhari, and Miroslav Oborník. 2019. "Transcriptomic Analysis Reveals the Roles of Detoxification Systems in Response to Mercury in Chromera velia" Biomolecules 9, no. 11: 647. https://doi.org/10.3390/biom9110647
APA StyleSharaf, A., De Michele, R., Sharma, A., Fakhari, S., & Oborník, M. (2019). Transcriptomic Analysis Reveals the Roles of Detoxification Systems in Response to Mercury in Chromera velia. Biomolecules, 9(11), 647. https://doi.org/10.3390/biom9110647