Whole Genome Sequencing of Familial Non-Medullary Thyroid Cancer Identifies Germline Alterations in MAPK/ERK and PI3K/AKT Signaling Pathways

 ,
,  ,
, 

 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Approval
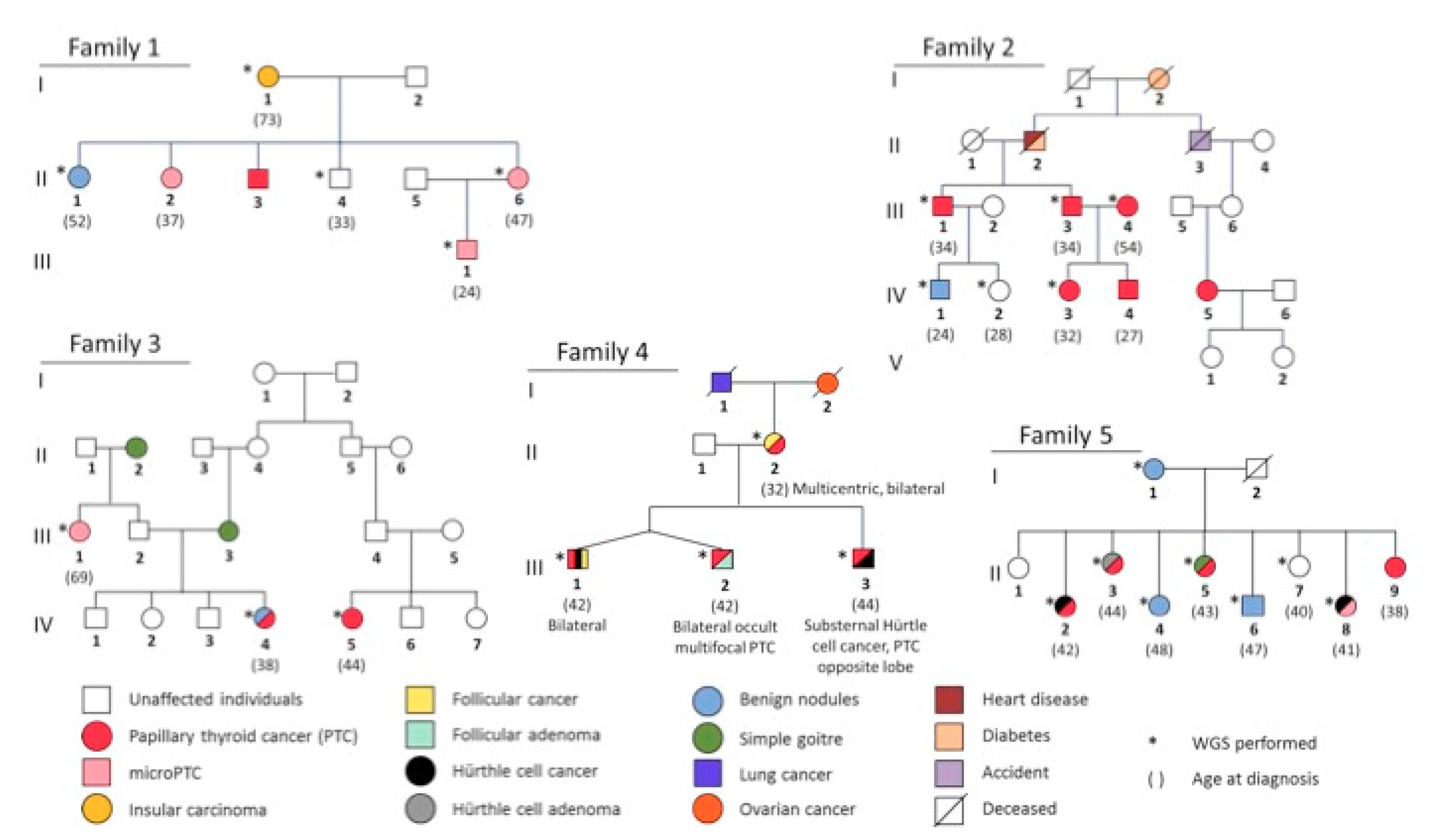
2.2. NMTC Families
2.3. Whole Genome Sequencing and Variant Evaluation
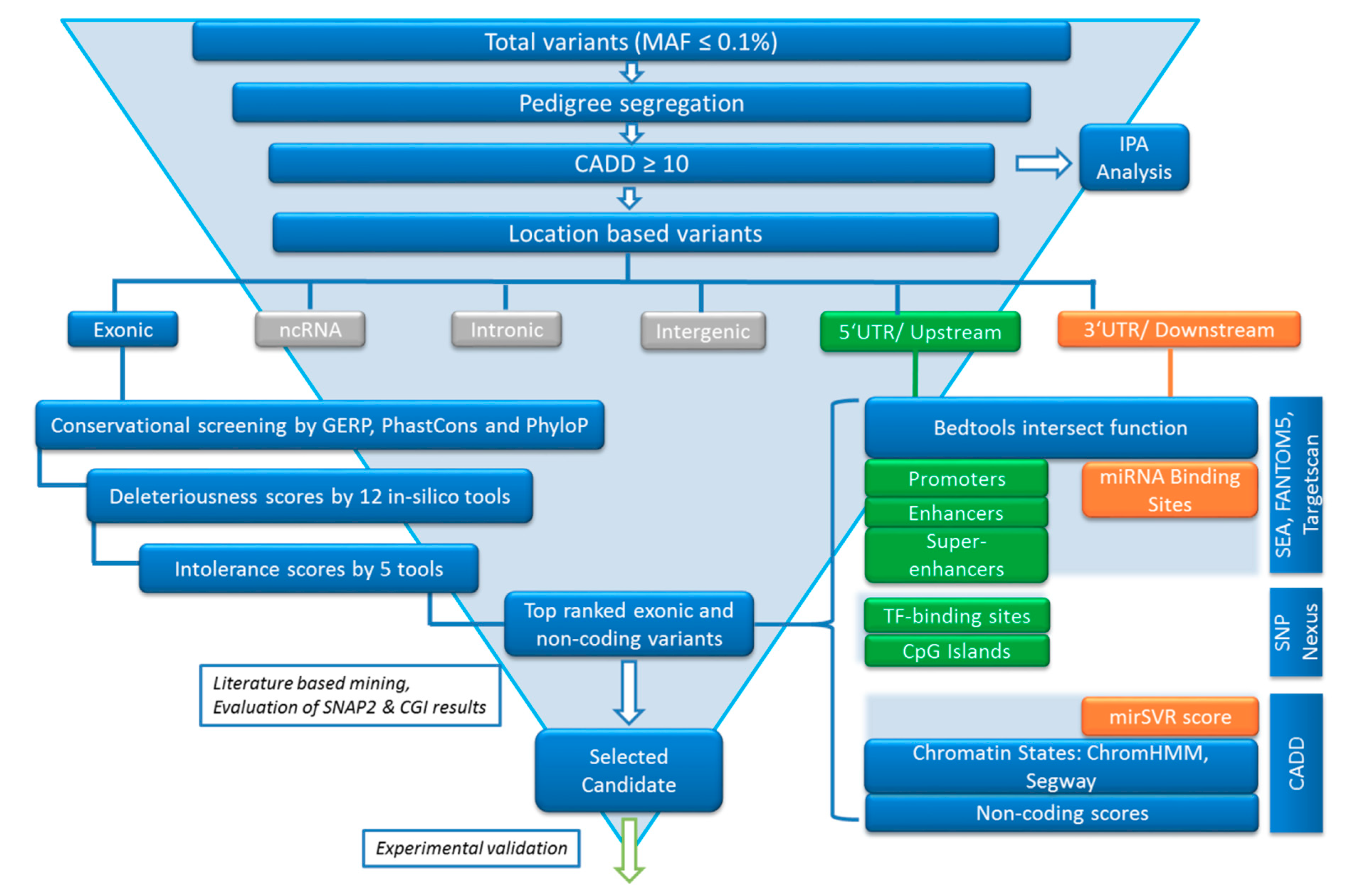
2.4. Variant Calling Annotation and Filtering
2.5. Variant Filtering following the FCVPPv2
2.5.1. Segregation in Pedigrees
2.5.2. Variant Ranking Using In Silico Tools
2.5.3. Analysis of Non-Coding Variants
2.6. Variant Validation
2.7. Ingenuity Pathway Analysis (IPA)
2.8. STRING Analysis
3. Results
3.1. Whole Genome Sequencing
3.2. Final Prioritization of Candidates according to the FCVPPv2
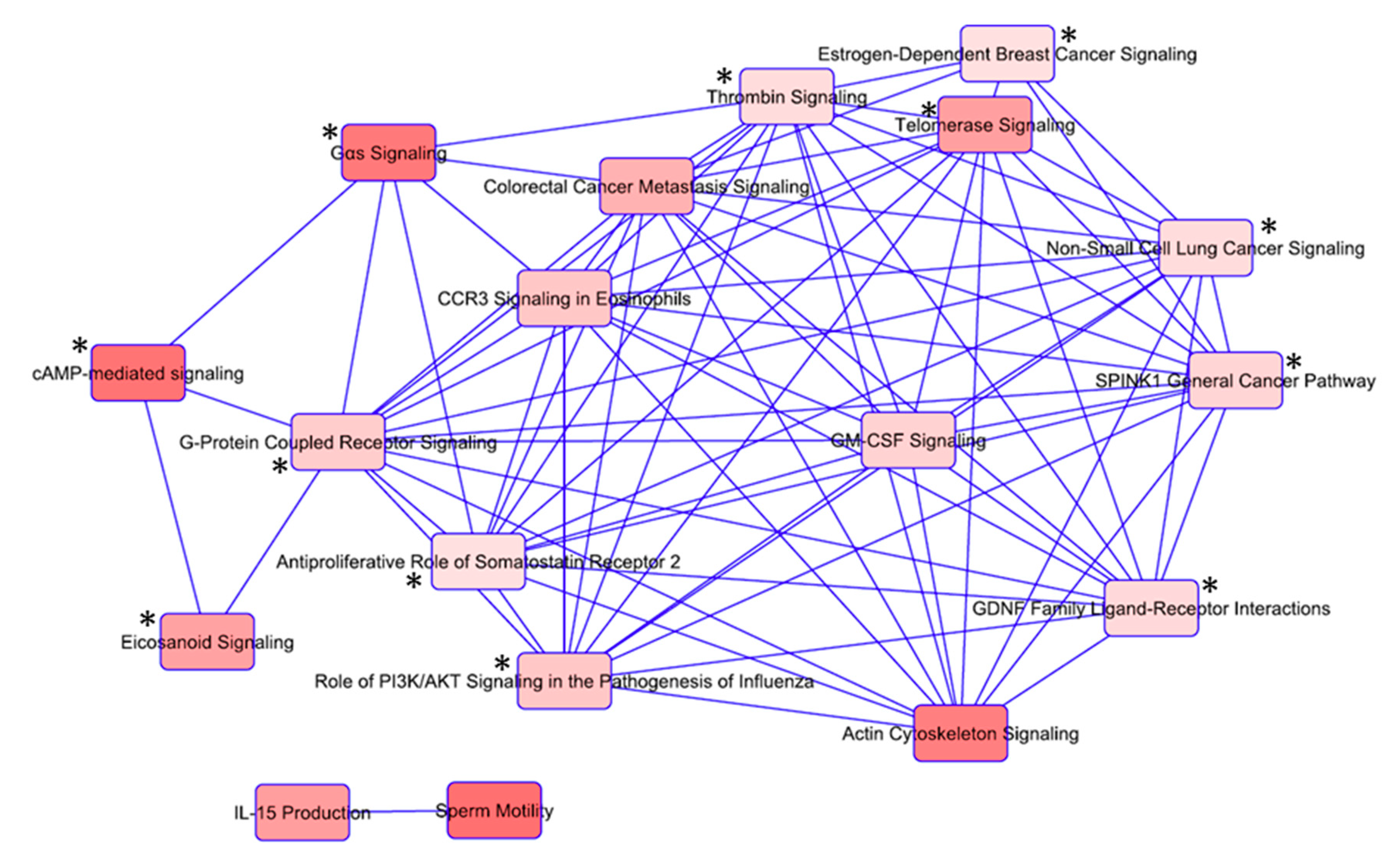
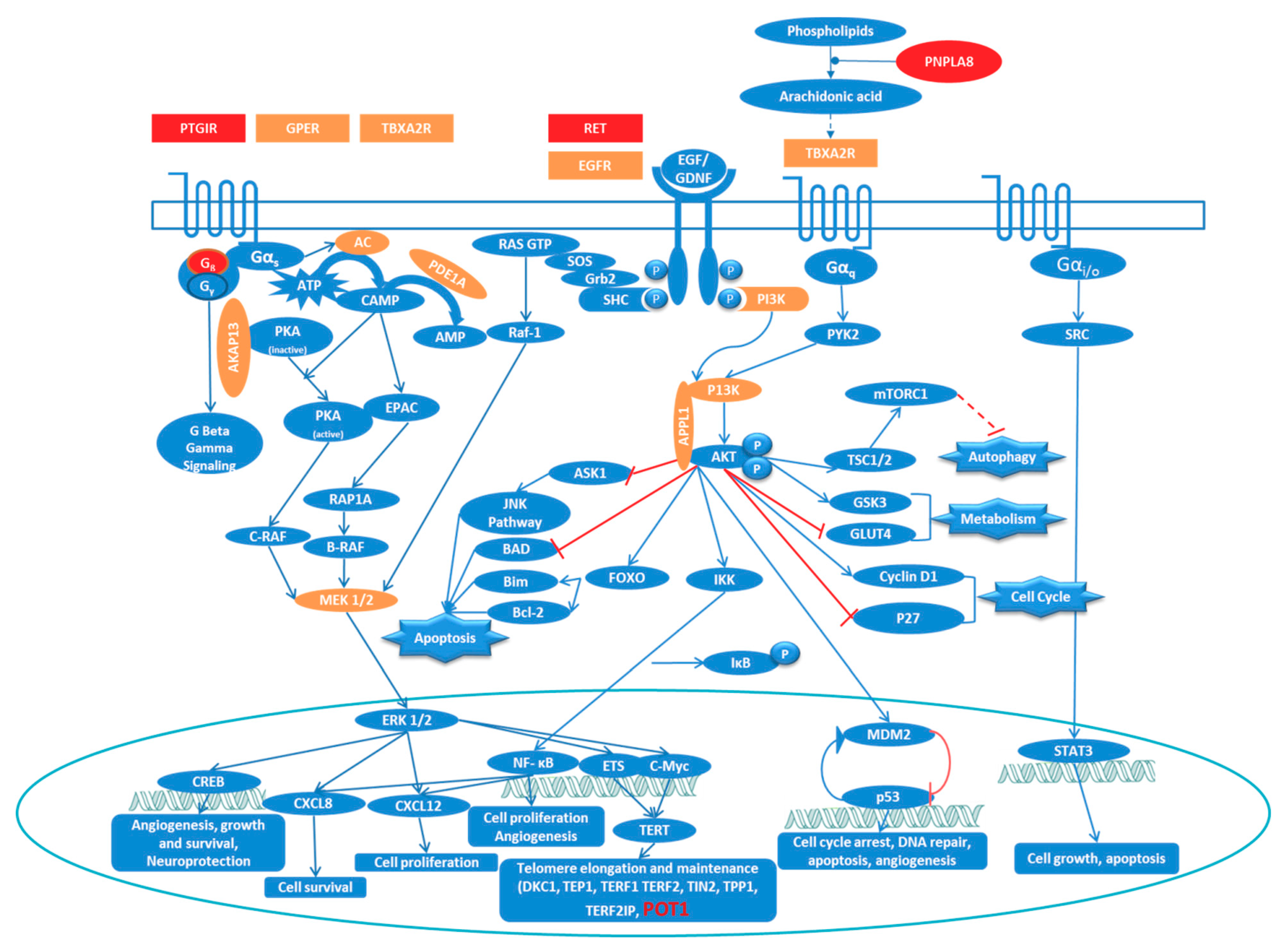
3.3. Ingenuity Pathway Analysis (IPA) Shows Enrichment of GPCR and RTK Mediated Pathways
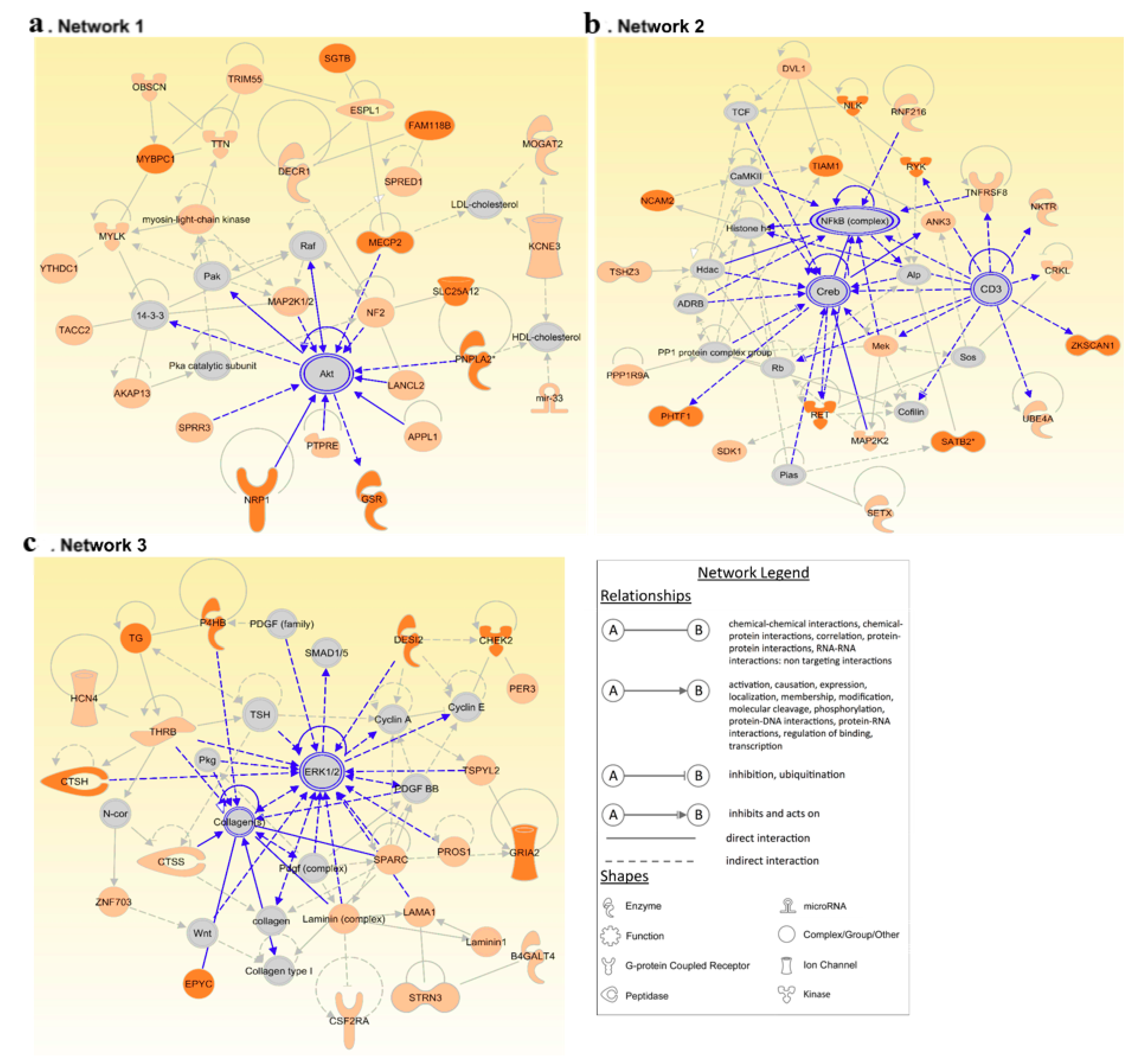
3.4. Network Analysis Reinforces the Central Role of PI3K/AKT and MAPK/ERK Signaling in FNMTC
3.5. Overlapping Pathways in Familial Non-Medullary Thyroid Cancer
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Lubina, A.; Cohen, O.; Barchana, M.; Liphshiz, I.; Vered, I.; Sadetzki, S.; Karasik, A. Time trends of incidence rates of thyroid cancer in israel: What might explain the sharp increase. Thyroid 2006, 16, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Malandrino, P.; Scollo, C.; Marturano, I.; Russo, M.; Tavarelli, M.; Attard, M.; Richiusa, P.; Violi, M.A.; Dardanoni, G.; Vigneri, R.; et al. Descriptive epidemiology of human thyroid cancer: Experience from a regional registry and the “volcanic factor”. Front. Endocrinol. 2013, 4, 65. [Google Scholar] [CrossRef] [PubMed]
- Frank, C.; Sundquist, J.; Yu, H.; Hemminki, A.; Hemminki, K. Concordant and discordant familial cancer: Familial risks, proportions and population impact. Int. J. Cancer 2017, 140, 1510–1516. [Google Scholar] [CrossRef] [PubMed]
- Grossman, R.F.; Tu, S.H.; Duh, Q.Y.; Siperstein, A.E.; Novosolov, F.; Clark, O.H. Familial nonmedullary thyroid cancer. An emerging entity that warrants aggressive treatment. Arch. Surg. 1995, 130, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Peiling Yang, S.; Ngeow, J. Familial non-medullary thyroid cancer: Unraveling the genetic maze. Endocrine-related cancer 2016, 23, R577–r595. [Google Scholar] [CrossRef]
- Gara, S.K.; Jia, L.; Merino, M.J.; Agarwal, S.K.; Zhang, L.; Cam, M.; Patel, D.; Kebebew, E. Germline habp2 mutation causing familial nonmedullary thyroid cancer. N. Engl. J. Med. 2015, 373, 448–455. [Google Scholar] [CrossRef]
- He, H.; Bronisz, A.; Liyanarachchi, S.; Nagy, R.; Li, W.; Huang, Y.; Akagi, K.; Saji, M.; Kula, D.; Wojcicka, A.; et al. Srgap1 is a candidate gene for papillary thyroid carcinoma susceptibility. J. Clin. Endocrinol. Metab. 2013, 98, E973–E980. [Google Scholar] [CrossRef]
- Ngan, E.S.; Lang, B.H.; Liu, T.; Shum, C.K.; So, M.T.; Lau, D.K.; Leon, T.Y.; Cherny, S.S.; Tsai, S.Y.; Lo, C.Y.; et al. A germline mutation (a339v) in thyroid transcription factor-1 (titf-1/nkx2.1) in patients with multinodular goiter and papillary thyroid carcinoma. J. Natl. Cancer Inst. 2009, 101, 162–175. [Google Scholar] [CrossRef]
- Pereira, J.S.; da Silva, J.G.; Tomaz, R.A.; Pinto, A.E.; Bugalho, M.J.; Leite, V.; Cavaco, B.M. Identification of a novel germline foxe1 variant in patients with familial non-medullary thyroid carcinoma (fnmtc). Endocrine 2015, 49, 204–214. [Google Scholar] [CrossRef]
- Tomsic, J.; He, H.; Akagi, K.; Liyanarachchi, S.; Pan, Q.; Bertani, B.; Nagy, R.; Symer, D.E.; Blencowe, B.J.; Chapelle, A.d.l. A germline mutation in srrm2, a splicing factor gene, is implicated in papillary thyroid carcinoma predisposition. Sci. Reports 2015, 5, 10566. [Google Scholar] [CrossRef] [PubMed]
- Capezzone, M.; Cantara, S.; Marchisotta, S.; Filetti, S.; De Santi, M.M.; Rossi, B.; Ronga, G.; Durante, C.; Pacini, F. Short telomeres, telomerase reverse transcriptase gene amplification, and increased telomerase activity in the blood of familial papillary thyroid cancer patients. J. Clin. Endocrinol. Metab. 2008, 93, 3950–3957. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.J. Clinical behavior and genetics of nonsyndromic, familial nonmedullary thyroid cancer. Front. Horm. Res. 2013, 41, 141–148. [Google Scholar] [PubMed]
- Kumar, A.; Bandapalli, O.R.; Paramasivam, N.; Giangiobbe, S.; Diquigiovanni, C.; Bonora, E.; Eils, R.; Schlesner, M.; Hemminki, K.; Forsti, A. Familial cancer variant prioritization pipeline version 2 (fcvppv2) applied to a papillary thyroid cancer family. Sci. Rep. 2018, 8, 11635. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. Dbnsfp v3.0: A one-stop database of functional predictions and annotations for human nonsynonymous and splice-site snvs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microrna target sites in mammalian mrnas. eLife 2015, 4. [Google Scholar] [CrossRef]
- Dayem Ullah, A.Z.; Oscanoa, J.; Wang, J.; Nagano, A.; Lemoine, N.R.; Chelala, C. Snpnexus: Assessing the functional relevance of genetic variation to facilitate the promise of precision medicine. Nucleic Acids Res. 2018, 46, W109–W113. [Google Scholar] [CrossRef]
- Andersson, R.; Gebhard, C.; Miguel-Escalada, I.; Hoof, I.; Bornholdt, J.; Boyd, M.; Chen, Y.; Zhao, X.; Schmidl, C.; Suzuki, T.; et al. An atlas of active enhancers across human cell types and tissues. Nature 2014, 507, 455. [Google Scholar] [CrossRef]
- Lizio, M.; Harshbarger, J.; Shimoji, H.; Severin, J.; Kasukawa, T.; Sahin, S.; Abugessaisa, I.; Fukuda, S.; Hori, F.; Ishikawa-Kato, S.; et al. Gateways to the fantom5 promoter level mammalian expression atlas. Genome Biol. 2015, 16, 22. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, F.; Su, J.; Shang, S.; Zhang, S.; Li, S.; Wang, X.; Wei, Y.; Liu, H.; Zhang, Y.; et al. Sea: A super-enhancer archive. Nucleic Acids Res. 2015, 44, D172–D179. [Google Scholar]
- Khan, A.; Zhang, X. Dbsuper: A database of super-enhancers in mouse and human genome. Nucleic Acids Res. 2016, 44, D164–D171. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Kellis, M. Chromhmm: Automating chromatin-state discovery and characterization. Nat. Methods 2012, 9, 215. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M.M.; Buske, O.J.; Wang, J.; Weng, Z.; Bilmes, J.A.; Noble, W.S. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat. Methods 2012, 9, 473. [Google Scholar] [CrossRef] [PubMed]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microrna.Org resource: Targets and expression. Nucleic Acids Res. 2008, 36, D149–D153. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdottir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant review with the integrative genomics viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef]
- Zannini, L.; Delia, D.; Buscemi, G. Chk2 kinase in the DNA damage response and beyond. J. Mol. Cell Biol. 2014, 6, 442–457. [Google Scholar] [CrossRef]
- Noujaim, J.; Jones, R.L.; Swansbury, J.; Gonzalez, D.; Benson, C.; Judson, I.; Fisher, C.; Thway, K. The spectrum of ewsr1-rearranged neoplasms at a tertiary sarcoma centre; assessing 772 tumour specimens and the value of current ancillary molecular diagnostic modalities. Br. J. Cancer 2017, 116, 669. [Google Scholar] [CrossRef]
- Hu, C.; Jiang, X. Role of nrp-1 in vegf-vegfr2-independent tumorigenesis. Target. Oncol. 2016, 11, 501–505. [Google Scholar] [CrossRef]
- Robles-Espinoza, C.D.; Harland, M.; Ramsay, A.J.; Aoude, L.G.; Quesada, V.; Ding, Z.; Pooley, K.A.; Pritchard, A.L.; Tiffen, J.C.; Petljak, M.; et al. Pot1 loss-of-function variants predispose to familial melanoma. Nat. Genet. 2014, 46, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Effraimidis, G.; Wiersinga, W.M. Mechanisms in endocrinology: Autoimmune thyroid disease: Old and new players. Eur. J. Endocrinol. 2014, 170, R241–R252. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (refseq) database at ncbi: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Mezghrani, A.; Courageot, J.; Mani, J.C.; Pugniere, M.; Bastiani, P.; Miquelis, R. Protein-disulfide isomerase (pdi) in frtl5 cells. Ph-dependent thyroglobulin/pdi interactions determine a novel pdi function in the post-endoplasmic reticulum of thyrocytes. J. Biol. Chem. 2000, 275, 1920–1929. [Google Scholar] [CrossRef] [PubMed]
- Su, G.H.; Bansal, R.; Murphy, K.M.; Montgomery, E.; Yeo, C.J.; Hruban, R.H.; Kern, S.E. Acvr1b (alk4, activin receptor type 1b) gene mutations in pancreatic carcinoma. Proc. Natl. Acad. Sci. USA 2001, 98, 3254–3257. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; He, J.; Du, Y.; Gao, X.-H.; Liu, Y.; Liu, Q.-Z.; Chang, W.-J.; Cao, G.-W.; Fu, C.-G. Upregulation of nemo-like kinase is an independent prognostic factor in colorectal cancer. World J. Gastroenterol. 2015, 21, 8836–8847. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.Y.; El-Gebali, S.; Fraser, M.I.; et al. Interpro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019, 47, D351–D360. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The genecards suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Biol. 2016, 54, 1.30.31–31.30.33. [Google Scholar]
- Wong, K.; Robles-Espinoza, C.D.; Rodriguez, D.; Rudat, S.S.; Puig, S.; Potrony, M.; Wong, C.C.; Hewinson, J.; Aguilera, P.; Puig-Butille, J.A.; et al. Association of the pot1 germline missense variant p.I78t with familial melanoma. JAMA Dermatol. 2019, 155, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Palles, C.; Cazier, J.B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of pole and pold1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Ngeow, J.; Eng, C. Habp2 in familial non-medullary thyroid cancer: Will the real mutation please stand up? J. Natl. Cancer Inst. 2016, 108, djw013. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tu, D.G.; Chang, W.W.; Jan, M.S.; Tu, C.W.; Lu, Y.C.; Tai, C.K. Promotion of metastasis of thyroid cancer cells via nrp-2-mediated induction. Oncol. Lett. 2016, 12, 4224–4230. [Google Scholar] [CrossRef] [PubMed]
- Chiappetta, G.; Valentino, T.; Vitiello, M.; Pasquinelli, R.; Monaco, M.; Palma, G.; Sepe, R.; Luciano, A.; Pallante, P.; Palmieri, D.; et al. Patz1 acts as a tumor suppressor in thyroid cancer via targeting p53-dependent genes involved in emt and cell migration. Oncotarget 2015, 6, 5310–5323. [Google Scholar] [CrossRef] [PubMed]
- Iesato, A.; Nakamura, T.; Izumi, H.; Uehara, T.; Ito, K.I. Patz1 knockdown enhances malignant phenotype in thyroid epithelial follicular cells and thyroid cancer cells. Oncotarget 2017, 8, 82754–82772. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.K. Map kinase pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef] [PubMed]
- Chambard, J.C.; Lefloch, R.; Pouyssegur, J.; Lenormand, P. Erk implication in cell cycle regulation. Biochim. Biophys. Acta 2007, 1773, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Yang, X.; Geng, M.; Huang, M. Targeting erk, an achilles’ heel of the mapk pathway, in cancer therapy. Acta Pharm. Sin. B 2018, 8, 552–562. [Google Scholar] [CrossRef]
- Burotto, M.; Chiou, V.L.; Lee, J.M.; Kohn, E.C. The mapk pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef]
- Fedorenko, I.V.; Paraiso, K.H.T.; Smalley, K.S.M. Acquired and intrinsic braf inhibitor resistance in braf v600e mutant melanoma. Biochem. Pharmacol. 2011, 82, 201–209. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the braf gene in human cancer. Nature 2002, 417, 949. [Google Scholar] [CrossRef]
- Tiacci, E.; Trifonov, V.; Schiavoni, G.; Holmes, A.; Kern, W.; Martelli, M.P.; Pucciarini, A.; Bigerna, B.; Pacini, R.; Wells, V.A.; et al. Braf mutations in hairy-cell leukemia. N. Engl. J. Med. 2011, 364, 2305–2315. [Google Scholar] [CrossRef] [PubMed]
- Benvenga, S.; Koch, C.A. Molecular pathways associated with aggressiveness of papillary thyroid cancer. Curr. Genom. 2014, 15, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Du, Y.; Zhang, X.; Li, M.; Li, X. Downregulation of bancr promotes aggressiveness in papillary thyroid cancer via the mapk and pi3k pathways. J. Cancer 2018, 9, 1318–1328. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, J.; Gao, J.; Li, Y. Microrna-4728 mediated regulation of mapk oncogenic signaling in papillary thyroid carcinoma. Saudi J. Biol. Sci. 2018, 25, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.T.; Zhong, Q.; Chen, R.H.; Han, P.; Li, S.B.; Zhang, H.; Yuan, L.; Xia, T.L.; Zeng, M.S.; Huang, X.M. Crlf1 promotes malignant phenotypes of papillary thyroid carcinoma by activating the mapk/erk and pi3k/akt pathways. Cell Death Dis. 2018, 9, 371. [Google Scholar] [CrossRef]
- Xing, M. Genetic alterations in the phosphatidylinositol-3 kinase/akt pathway in thyroid cancer. Thyroid 2010, 20, 697–706. [Google Scholar] [CrossRef]
- Xue, Y.; Martelotto, L.; Baslan, T.; Vides, A.; Solomon, M.; Mai, T.T.; Chaudhary, N.; Riely, G.J.; Li, B.T.; Scott, K.; et al. An approach to suppress the evolution of resistance in brafv600e-mutant cancer. Nat. Med. 2017, 23, 929. [Google Scholar] [CrossRef]
- Bandapalli, O.R.; Paramasivam, N.; Giangiobbe, S.; Kumar, A.; Benisch, W.; Engert, A.; Witzens-Harig, M.; Schlesner, M.; Hemminki, K.; Forsti, A. Whole genome sequencing reveals dicer1 as a candidate predisposing gene in familial hodgkin lymphoma. Int. J. Cancer 2018, 143, 2076–2078. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Gene | Chrom_Pos_Ref_Alt | Exonic Classification | CADD_PHRED Score | Int (%) | Del (%) | Interactions (STRING) | Domain | Function |
|---|---|---|---|---|---|---|---|---|---|
| 1 | CHEK2 | 22_29107974_C_T | nonsynonymous SNV | 24.8 | 75 | 42 | ATM, ATR, CDC25C, CDC25A, TP53BP1, TP53, MRE11A, BRCA1, RAD50, H2AFX | Serine/ threonine-protein kinase-like domain | DNA repair, cell cycle arrest or apoptosis in response to DNA damage; tumor suppressor gene |
| 1 | SLC35A4 | 5_139947647_T_C | nonsynonymous SNV | 26.5 | 50 | 75 | SCAMP3, PRKAA1, SLC35B2, SLC35D2, ABCB10 | Nucleotide-sugar transporter | Pyrimidine nucleotide-sugar transmembrane transporter activity, sialic acid transmembrane transporter activity |
| 1 | ANXA3 | 4_79531211_C_T | nonsynonymous SNV | 27.9 | 50 | 75 | STX4, SNAP23, STXBP2, ANXA11, ANXA4, FPR1, CACNA1B, NLRP3, SUMF1, FPR2 | Annexin repeat, conserved site | Phospoholipase A2 inhibition, anti-coagulant properties, formation of inositol 1-phosphate from inositol 1,2-cyclic phosphate |
| 1 | EWSR1 | 22_29687556_C_A | nonsynonymous SNV | 22.7 | 100 | 75 | BARD1, ETV1, TAF5, TAF5L, FUS, TAF12, DHX9, TP53, PIOK2, POLR2G | NA | Gene expression, cell signaling, RNA processing and transport; oncogene |
| 1 | RTTN | 18_67776873_G_A | nonsynonymous SNV | 26.7 | 25 | 83 | INVS, LEFTY2, DNAH11, CCDC102B, EN1, CCDC178, L3MBTL4, CHML, CHM, DLL1 | NA | Involved in the genetic cascade that governs left-right specification and in the maintenance of a normal ciliary structure. |
| 1 | TIAM1 | 21_32526579_G_A | nonsynonymous SNV | 35 | 100 | 92 | CDC42, SRC, RAC1, EFNB1, RAC2, NME1, EPHA2, RHOA, PARD6A, ARF6 | Dbl homology (DH) domain | Modulates the activity of Rho GTP-binding proteins, connects extracellular signals to cytoskeletal activities, activates Rac1, CDC42, and to a lesser extent RhoA. |
| 1 | MAN2B2 | 4_6612617_C_T | nonsynonymous SNV | 34 | 25 | 100 | MAN2C1, NAAA, SIAE, GLB1L3, GLB1, PYGB, PYGL, PYGM, NAGA | Glycosyl hydrolase family 38, C-terminal | carbohydrate binding, alpha-mannosidase activity, involved in metabolism and other glycan degradation |
| 2 | CLEC18B | 16_74446758_G_A | nonsynonymous SNV | 23.3 | 50 | 67 | FRAS1, LEO1, FREM2 | Epidermal growth factor-like domain | Ca2+ independent binding of polysaccharides |
| 2 | PTGIR | 19_47124811_C_T | nonsynonymous SNV | 35 | 100 | 67 | HTR7, NPS, AVP, VIP, ADM, AVPR2, ADRB2, PTH, ADCY6, GNB1 | NA | Member of GPCR family 1, receptor for prostacyclin, elicits potent vasodilation and inhibition of platelet aggregation |
| 2 | UBN1 | 16_4911084_G_A | nonsynonymous SNV | 34 | 75 | 67 | ASF1A, HIRA, CABIN1, RB1, TP53, EP400, HMGA1, HMGA2, H1F0, HIST1H1C | Ubinuclein middle domain | Novel regulator of senescence, involved in DNA damage/telomere stress induced senescence and cellular senescence, required for replication independent chromatin assembly |
| 2 | GALNT10 | 5_153789322_G_C | nonsynonymous SNV | 24.6 | 100 | 67 | MUC7, MUC1, C1GALT1, MUC5AC, GCNT1, ST6GALNAC1, B3GNT6, MUC2, MUC16, C1GALT1C1 | Ricin B-related lectin | Catalyzes the initial reaction in O-linked oligosaccharide biosynthesis |
| 2 | OSGIN2 | 8_90921899_A_T | nonsynonymous SNV | 23.7 | 100 | 67 | CALB1, CA7, DECR1, DECR2, CALB2, NBN, SLC39A3 | NA | Possibly involved in meiosis or the maturation of germ cells, associated with retinitis pigmentosa |
| 2 | TG | 8_133900661_A_C | nonsynonymous SNV | 25 | 0 | 75 | TPO, LRP2, TSHR, ASGR1, NKX2-1, INS, SLC5A5, PAX8, ASGR2, ALB | Thyroglobulin type-1 domain | Precursors of iodinated thyroid hormones (T4) and triiodothyronine (T3), associated with susceptibility to autoimmune thyroid diseases (AITD) |
| 2 | GSR | 8_30585111_C_T | nonsynonymous SNV | 34 | 100 | 75 | GPX1, GPX3, GPX2, CAT, GPX4, GSS, GPX7, HPGDS, TXN, ACLY | Pyridine nucleotide-disulphide oxidoreductase, FAD/NAD(P)-binding domain | Oxidoreductase activity and flavin adenine dinucleotide binding |
| 2 | KCNT1 | 9_138676399_A_G | nonsynonymous SNV | 11.1 | 100 | 75 | GPR55, C11orf40, ASRGL1, SLC11A1 | NA | Sodium/Chloride/Calcium-activated potassium channel subunit, activated upon stimulation of GPCRs |
| 2 | KLHL18 | 3_47385160_A_G | nonsynonymous SNV | 27.4 | 100 | 75 | COPS5, GPKOW, CNIH4, COPS6, PDE7A, CNIH3, PDE/B, PDE6D, EEF1G, CNIH2 | Galactose oxidase, beta-propeller | Involved in the ubiquination process, specific role has yet to be elucidated |
| 2 | CDRT1, RP11-385D13.1 | 17_15501921_G_A | nonsynonymous SNV | 25.3 | - | 83 | - | WD40/YVTN repeat-like-containing domain | CDRT1: a protein-ubiquitin ligase; RP11: a component of the spliceosome complex, one of several retinitis pigmentosa-causing genes |
| 2 | RET | 10_43600559_T_C | nonsynonymous SNV | 26.3 | 75 | 83 | GDNF, GFRA1, NRTN, SHC1, PSPN, PIK3CA, GFRA2, PIK3CD, PIK3CB, GRB2 | Cadherin-like domain | Proto-oncogene, receptor tyrosine kinase; involved in cell differentiation, growth, migration and survival |
| 2 | SCN10A | 3_38755465_C_A | nonsynonymous SNV | 35 | 50 | 92 | SCN5A, CALM2, SCN8A, SCN2A, SCN11A, SCH3A, SCN1A, SCN9A, SCN4A, SCN1B | Ion transport | Tetrodotoxin-resistant channel that mediates the voltage-dependent sodium ion permeability of excitable membranes, plays a role in neuropathic pain mechanisms |
| 3 | C1orf27 | 1_186355211_G_A | nonsynonymous SNV | 25.1 | 0 | 67 | DRAM1, PID1, TXLNG | ODR-4-like domain | Possible involvement in the trafficking of a subset of GPCRs |
| 3 | CPXM1 | 20_2776248_C_T | nonsynonymous SNV | 32 | 100 | 75 | FAM196A, PPP2R2B, SEC13 | Peptidase M14, carboxypeptidase A | Binds collagen, involved in adipogenesis through extracellular matrix remodeling, may act as a TSG in breast cancer |
| 3 | ZBTB41 | 1_197128680_C_T | nonsynonymous SNV | 23.1 | 100 | 75 | POTEE, POTEI, POTEJ, POTEF, SKIV2L, CFHR4, RIPK4, PHLPP2, PHLPP1, C7orf73 | NA | May be involved in transcriptional regulation |
| 3 | AR | X_66765158_T_TGCAGCAGCA | nonframeshift insertion | 12.8 | 67 | - | NCOA2, NCOA4, KLK3, KDM1A, FOXA1, SRC, HSP90AA1, FKBP5, NCOA1, CCND1 | Androgen receptor domain | Steroid-hormone activated transcription factor. Stimulates transcription of androgen responsive genes. |
| 4 | PKHD1L1 | 8_110477162_G_A | nonsynonymous SNV | 27.5 | 0 | 100 | TMEM2, CUEDC1, PKHD1, PKD1P1, C2orf74, RAD21-AS1, FAM135B, CSMD3, MUM1L1, HSPA12B | NA | Signaling receptor activity, immune response |
| 4 | ECE2 | 3_184008594_G_C | nonsynonymous SNV | 32 | 75 | 100 | RPS6KA2, EDN3, EDNRA, DHX40, MYSM1. EDNRB, EDN1, EZR, LARP6, PRKCE | Peptidase M13, neprilysin, C-terminal/Metallopeptidase, catalytic domain | Metalloprotease involved in the generation of functionally pleiotropic members of the endothelin vasoactive family, possibly involved in amyloid-beta processing |
| 5 | EPYC | 12_91365726_C_G | nonsynonymous SNV | 27 | 25 | 67 | RIPK4, PPIE, POTEI, POTEE, POTEJ, POTEF, PRKAR1B, PRKAR1A, CNBD2, PRKAR2B | Leucine-rich repeat | Regulates fibrillogenesis by interacting with collagen fibrils and other extracellular matrix proteins |
| 5 | SPOCK1 | 5_136448179_G_A | nonsynonymous SNV | 25.7 | 100 | 67 | SPARC, MMP16, FST, MMP14, SPARCL1, MMP2, CITED2, CHD1L, CFTR, HMCN1 | Proteinase inhibitor I1, Kazal | Calcium ion binding, cysteine-type endopeptidase inhibitor activity, cell-cell interactions, may contribute to various neuronal mechanisms |
| 5 | MYBPC1 | 12_102046527_A_G | nonsynonymous SNV | 25.9 | 100 | 67 | MYH3, TTN, TNNT3, NEB, TNNI2, DMD, MYL1, TMOD4, TNNI1, MYL3 | Immunoglobulin subtype | Member of the myosin-binding protein C family, binds actin and titin, modulates muscle contraction |
| 5 | ACSS3 | 12_81593172_T_G | nonsynonymous SNV | 32 | 100 | 83 | ALDH2, ALDH3A2, EHHADH, ACLY, ECHDC1, ACADM, ALDH6A1, ALDH9A1, ALDH1B1 | AMP-dependent synthetase/ligase | Activates acetate for use in lipid synthesis or energy generation |
| 5 | NRP1 | 10_33469205_G_C | nonsynonymous SNV | 24.2 | 75 | 83 | SEMA3A, KDR, FLT1, PLXNA1, PLXNA2, SEMA3C, PLXNA4, SEMA3F, PLXNA3, SEMA3E | Neuropilin-1, C-terminal | Membrane-bound coreceptor to a tyrosine kinase receptor for both VEGF and semaphorin family members; plays roles in angiogenesis, axon guidance, cell survival, migration and invasion |
| 5 | POT1 | 7_124532359_C_A | nonsynonymous SNV | 32 | 50 | 92 | TERF1, TINF2, ACD, TERF2, TERF2IP, RAD50, MRE11A, H2AFx, DCLRE1B, BRCA1 | Nucleic acid-binding, OB-fold | Member of the shelterin complex; involved in regulating telomere length and protecting chromosome ends from illegitimate recombination, catastrophic instability and abnormal segregation |
| Variant Details | FANTOM5, SEA | Annotations From CADD | SNPnexus | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FI | Gene | Variation_ Annotation | Chrom_Pos_Ref_Alt | Promoter/Enhancer_ Start..End, Strand | CADD_PHRED Score | Chromatin StateII | TFBS | TFs | In a CpG Island? | Non-coding scores (%)III | |||
| Chrom-Hmm State | Score | Segway | TFBS | TFBS PeaksI | |||||||||
| 1 | PCM1 | SNV_UTR5 | 8_17780410_G_A | − | 17.2 | TssA | 0.95 | TSS | 50 | 92 | Egr-3, AP-2alphaA, AP-2gamma | Yes | 67 |
| 1 | STAP1 | SNV_UTR5 | 4_68424468_A_G | Promoter_68424462..68424469,+ | 15.4 | Quies | 0.71 | GM0 | 18 | 24 | − | No | 71 |
| 1 | DAPL1 | SNV_ Upstream | 2_159651789_C_T | − | 13.1 | − | − | TF0 | 1 | 2 | MAZR, Sp1 | No | 50 |
| 2 | LRRC48 | SNV_UTR5 | 17_17876279_G_T | − | 10.8 | TssA | 0.803 | GS | 28 | 56 | − | No | 50 |
| 2 | P4HB | SNV_UTR5 | 17_79818442_T_G | − | 11.4 | TssA | 0.945 | TSS | 51 | 78 | − | Yes | 60 |
| 2 | FAM118B | SNV_UTR5 | 11_126081608_C_T | − | 10.5 | TssA | 0.969 | TSS | 60 | 129 | − | Yes | 33 |
| 2 | AZIN1 | SNV_UTR5 | 8_103876327_G_A | − | 12.4 | TssA | 0.929 | TSS | 47 | 76 | − | No | 50 |
| 2 | RPS3A | SNV_UTR5 | 4_152020778_C_G | Promoter_152020736..152020788,+ | 16.0 | TssA | 0.961 | TSS | 81 | 184 | − | Yes | 86 |
| 3 | C20orf194 | SNV_ Upstream | 20_3388577_C_A | − | 13.4 | TssA | 0.921 | TSS | 17 | 26 | Egr-2, Egr-3 | Yes | 50 |
| 4 | DNAI1 | SNV_UTR5 | 9_34458888_T_C | Promoter_34458851..34458908,+ | 14.4 | TssAFlnk | 0.575 | GM1 | 10 | 13 | − | Yes | 71 |
| 4 | PNPLA2 | SNV_UTR5 | 11_819602_G_C | Promoter_819601..819612,+ | 10.6 | TssA | 0.945 | TSS | 38 | 65 | − | Yes | 50 |
| 4 | GNB2 | Indel_UTR5 | 7_100271438_G_GCGCCGCCGCCGC | − | 17.5 | TssA | 0.992 | TSS | 65 | 115 | CUTL-1 | Yes | 25 |
| 4 | PHTF1 | SNV_UTR5 | 1_114301745_G_T | − | 16.2 | TssA | 0.961 | TSS | 20 | 28 | CREB, delta CREB | Yes | 50 |
| 4 | ZKSCAN1 | SNV_UTR5 | 7_99613211_C_G | − | 21.4 | TssA | 0.937 | TSS | 65 | 140 | Elk-1, LCR-F1 | Yes | 67 |
| Variant Details | TargetScan | Annotations from CADD | SNP-nexus | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FI | Gene | Variation_ Annotation | Chrom_Pos_Ref_Alt | miRNA Target Sites | Context Score ++ PercentileII | Site Type | mirSVR-Score | CADD_ PHRED Score | Chromatin StateIII | Non-Coding Scores IV (%) | ||
| Chrom-Hmm | Score | Segway | ||||||||||
| 1 | DESI2 | SNV_UTR3 | 1_244872281_A_G | DESI2:miR-3651 | 94 | 7mer-m8 | -0.84 | 15.6 | TxWk | 0.73 | R2 | 60 |
| 1 | DPYSL3 | SNV_UTR3 | 5_146770537_A_T | DPYSL3:miR-4693-5p, DPYSL3:miR-4768-3p, DPYSL3:miR-6888-5p | 20, 52, 59 | 7mer-1a, 7mer-m8, 7mer-m8 | -0.24 | 11.1 | − | − | L1 | 40 |
| 1 | MECP2 | SNV_UTR3 | X_153295452_G_A | MECP2:miR-6812-3p | 72 | 7mer-1a | NA | 10.4 | Tx | 0.46 | TF2 | 25 |
| 1 | RYK | SNV_UTR3 | 3_133876591_C_T | RYK:miR-548aq-3p/548am-3p/548aj-3p/548ah-3p/548ae-3p/548j-3p/548x-3p; RYK:miR-5582-3p | 93, 95 | 7mer-m8, 7mer-m8 | −1.25 | 12.7 | TxWk | 0.50 | F1 | 80 |
| 1 | SGTB | SNV_UTR3 | 5_64965337_A_C | SGTB:miR-3187-3p, SGTB:miR-4529-5p | 84, 46 | 7mer-m8, 7mer-1a | −0.75 | 16.8 | TxWk | 0.68 | GE0 | 67 |
| 1 | SLC25A12 | SNV_UTR3 | 2_172641178_G_A | SLC25A12:miR-3622b-5p | 62 | 7mer-1a | −0.31 | 15.1 | − | − | GE1 | 60 |
| 2 | ACVR1B | SNV_UTR3 | 12_52388057_A_G | ACVR1B:miR-6871-5p | 53 | 7mer-m8 | 14.8 | − | − | − | 60 | |
| 2 | NCAM2 | Indel_UTR3 | 21_22913891_AT_A | NCAM2:miR-6885-3p | 46 | 7mer-m8 | NA | 11.3 | Quies | 0.99 | F0 | 50 |
| 2 | NOP2 | SNV_UTR3 | 12_6666047_A_T | NOP2:miR-3662 | 98 | 7mer-1a | −1.29 | 14.2 | Tx | 0.48 | GE0 | 50 |
| 2 | NUPL1 | SNV_UTR3 | 13_25909315_T_C | NUPL1:miR-3145-3p | 69 | 8mer | − | 11.3 | − | − | − | 80 |
| 2 | PNPLA8 | SNV_UTR3 | 7_108112453_A_G | PNPLA8:miR-3163, PNPLA8:miR-4668-3p, PNPLA8:miR-551b-5p | 65, 56, 62 | 7mer-m8, 7mer-1, 7mer-m8 | −1.25 | 13.3 | TxWk | 0.53 | F0 | 80 |
| 2 | STK32A | SNV_UTR3 | 5_146763869_G_A | STK32A:miR-4484, STK32A:miR-548an, STK32A:miR-6768-3p | 99, 80, 74 | 8mer, 7mer-1a, 7mer-1a | NA | 11.8 | Quies | 0.48 | F1 | 40 |
| 2 | SVEP1 | SNV_UTR3 | 9_113128472_T_C | SVEP1:miR-1468-3p | 96 | 7mer-m8 | −1.32 | 17.0 | Quies | 0.77 | F1 | 60 |
| 2 | TFCP2 | Indel_UTR3 | 12_51487616_A_AACAC | TFCP2:miR-8485 | 95 | 7mer-m8 | NA | 10.2 | Tx, TxWk | 0.47, 0.52 | GE0 | 67 |
| 2 | MRPL51 | SNV_ downstream | 12_6600160_C_T | MRPL51: miR-6802-3p | 90 | 7mer-m8 | NA | 13.4 | TxWk | 0.63 | H3K9 me1 | 50 |
| 2 | ZNF45 | SNV_ncRNA_UTR3 | 19_44417402_A_G | ZNF45: miR-6777-3p | 96 | 8mer | −0.39 | 11.3 | ZnfRpts | 0.78 | GE1 | 60 |
| 3 | NLK | Indel_UTR3 | 17_26522009_T_TCACA | NLK:miR-6818-5p, NLK:miR-6867-5p | 88, 79 | 7mer-m8, 8mer | −0.62 | 11.7 | TxWk | 0.63 | TF1 | 100 |
| 4 | ADAMTS1 | SNV_UTR3 | 21_28208629_T_C | ADAMTS1:miR-325, ADAMTS1:miR-628-3p | 88, 97 | 7mer-1a, 8mer | −1.31 | 16.0 | TxWk | 0.58 | F1 | 60 |
| 4 | GRIA2 | SNV_UTR3 | 4_158284635_G_A | GRIA2:miR-486-5p, GRIA2:miR-7152-5p | 88, 84 | 7mer-1a, 7mer-m8 | −0.87 | 22.3 | Quies | 0.84 | L1 | 60 |
| 4 | IGSF9 | Indel_UTR3 | 1_159896866_TCACA_T | IGSF9:miR-377-3p, IGSF9:miR-5582-3p, IGSF9:miR-8485 | 98, 82, -1 | 8mer,7mer-m8, 3’compensatory | −0.92 | 17.0 | − | - | TF1 | 50 |
| 4 | MPP6 | SNV_UTR3 | 7_24727611_A_G | MPP6:miR-138-2-3p, MPP6:miR-205-3p, MPP6:miR-498 | 93, 50, 48 | 7mer-m8, 7mer-1a, 7mer-1a | NA | 15.5 | TxWk, Quies | 0.50, 0.45 | GE0 | 60 |
| 4 | ZNF532 | SNV_UTR3 | 18_56651809_T_C | ZNF532: miR-1277-5p | 53 | 7mer-m8 | −0.86 | 15.2 | TxWk | 0.73 | R0 | 80 |
| 5 | KLF7 | Indel_UTR3 | 2_207945783_ATATGTG_A | KLF7:miR-511-3p, KLF7:miR-223-5p | 82, 59 | 7mer-1a, 7mer-1a | −1.10 | 11.9 | Tx | 0.73 | F1 | 50 |
| 5 | SATB2 | SNV_UTR3 | 2_200134548_A_G | SATB2:miR-3156-5p, SATB2:miR-3126-3p, SATB2:miR-4720-5p/4799-3p/5588-5p, SATB2:miR-3128, SATB2:miR-6868-5p | 37, 86, 74, 76, 83 | 7mer-m8, 7mer-m8, 7mer-1a, 8mer, 7mer-1a | -1.22 | 15.2 | Quies | 0.74 | F1 | 60 |
| 5 | ZNF608 | SNV_ downstream | 5_123972606_C_A | ZNF608: miR-4786-3p | 87 | 7mer-m8 | NA | 16.8 | TxWk | 0.69 | D | 60 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srivastava, A.; Kumar, A.; Giangiobbe, S.; Bonora, E.; Hemminki, K.; Försti, A.; Bandapalli, O.R. Whole Genome Sequencing of Familial Non-Medullary Thyroid Cancer Identifies Germline Alterations in MAPK/ERK and PI3K/AKT Signaling Pathways. Biomolecules 2019, 9, 605. https://doi.org/10.3390/biom9100605
Srivastava A, Kumar A, Giangiobbe S, Bonora E, Hemminki K, Försti A, Bandapalli OR. Whole Genome Sequencing of Familial Non-Medullary Thyroid Cancer Identifies Germline Alterations in MAPK/ERK and PI3K/AKT Signaling Pathways. Biomolecules. 2019; 9(10):605. https://doi.org/10.3390/biom9100605
Chicago/Turabian StyleSrivastava, Aayushi, Abhishek Kumar, Sara Giangiobbe, Elena Bonora, Kari Hemminki, Asta Försti, and Obul Reddy Bandapalli. 2019. "Whole Genome Sequencing of Familial Non-Medullary Thyroid Cancer Identifies Germline Alterations in MAPK/ERK and PI3K/AKT Signaling Pathways" Biomolecules 9, no. 10: 605. https://doi.org/10.3390/biom9100605
APA StyleSrivastava, A., Kumar, A., Giangiobbe, S., Bonora, E., Hemminki, K., Försti, A., & Bandapalli, O. R. (2019). Whole Genome Sequencing of Familial Non-Medullary Thyroid Cancer Identifies Germline Alterations in MAPK/ERK and PI3K/AKT Signaling Pathways. Biomolecules, 9(10), 605. https://doi.org/10.3390/biom9100605