Autophagy Modulators: Mechanistic Aspects and Drug Delivery Systems

 , , , , ,
, , , , , 
Abstract
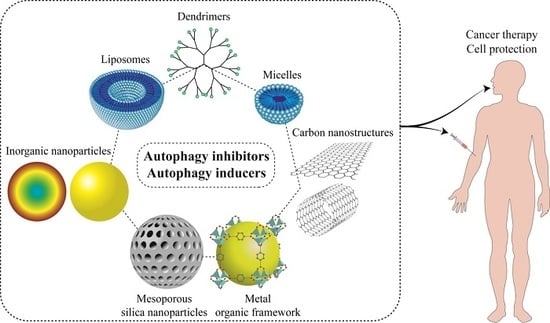
1. Introduction
2. mTOR Signaling Pathway
3. AMPK Signaling Pathway and Autophagy
4. MAPK Signaling Pathway and Autophagy
5. Autophagy Inducer Drugs
5.1. mTOR Inhibitors
5.1.1. Sirolimus and Its Analogues/Derivatives
5.1.2. Dactolisib
5.2. AMPK Activators
5.2.1. Metformin
5.2.2. Simvastatin
5.3. MAPK Activators
Carbamazepine
6. Autophagy Inhibitors: Main Agents and Mode of Action
6.1. Autophagosome Degradation Blockers
6.2. Class III PI3K Inhibitors
7. New Therapeutic Agents that Target Different Steps of the Autophagic Machinery
7.1. Targeting ULK1/2
7.2. Targeting Ubiquitin Specific Peptidases (USP)
7.3. Targeting Autophagy-Related Protein 4B (ATG4B)
7.4. Targeting p62
8. Autophagy Inhibition for Cancer Therapy
9. Nanostructures for Autophagy Modulator Delivery Systems
9.1. Liposomes
9.2. Micelles
9.3. Polymeric Nanoparticles
10. Co-Delivery of Autophagy Inducers/Inhibitors and Chemotherapeutics/siRNA
11. Nanocarriers as Autophagy Modulator: Challenging to Design Drug Delivery System
12. Conclusions and Remarks
Funding
Conflicts of Interest
References
- Ashrafizadeh, M.; Ahmadi, Z.; Mohammadinejad, R.; Kaviyani, N.; Tavakol, S. Monoterpenes modulating autophagy: A review study. Basic Clin. Pharmacol. Toxicol. 2019. [Google Scholar] [CrossRef]
- Mohammadinejad, R.; Ahmadi, Z.; Tavakol, S.; Ashrafizadeh, M. Berberine as a potential autophagy modulator. J. Cell. Physiol. 2019, 234, 14914–14926. [Google Scholar] [CrossRef] [PubMed]
- Ashrafizadeh, M.; Mohammadinejad, R.; Tavakol, S.; Ahmadi, Z.; Roomiani, S.; Katebi, M. Autophagy, anoikis, ferroptosis, necroptosis, and endoplasmic reticulum stress: Potential applications in melanoma therapy. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ashrafizadeh, M.; Yaribeygi, H.; Atkin, S.L.; Sahebkar, A. Effects of newly introduced antidiabetic drugs on autophagy. Diabetes Metab. Syndr. Clin. Res. Rev. 2019, 13, 2445–2449. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, Z.; Roomiani, S.; Bemani, N.; Ashrafizadeh, M. Targeting autophagy and endoplasmic reticulum stress mechanisms in Honokiol therapy. Rev. Clin. Med. 2019. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological functions of autophagy genes: A disease perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wang, Y.; An, H.-W.; Qi, B.; Wang, J.; Wang, L.; Shi, J.; Mei, L.; Wang, H. Peptide-based Autophagic Gene and Cisplatin Co-delivery Systems Enable Improve Chemotherapy Resistance. Nano Lett. 2019. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.; Mei, L.; Liu, J.; Tang, X.; Yin, S.; Xia, C.; Wei, J.; Wan, D.; Wang, X.; Wang, Y. Size-Adjustable Micelles Co-loaded with Chemotherapeutic Agent and Autophagy Inhibitor for Enhancing Cancer Treatment via Increasing Tumor Retention. Acta Biomater. 2019, 89, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Qiu, Y.; Grace, C.R.; Liu, X.; Klionsky, D.J.; Schulman, B.A. A switch element in the autophagy E2 Atg3 mediates allosteric regulation across the lipidation cascade. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Klionsky, D.J. UIM-UDS: A new interface between ATG8 and its interactors. Cell Res. 2019, 1, 507–508. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Green, D.R. Autophagy-independent functions of the autophagy machinery. Cell 2019, 177, 1682–1699. [Google Scholar] [CrossRef]
- Deng, S.; Shanmugam, M.K.; Kumar, A.P.; Yap, C.T.; Sethi, G.; Bishayee, A. Targeting autophagy using natural compounds for cancer prevention and therapy. Cancer 2019, 125, 1228–1246. [Google Scholar] [CrossRef] [PubMed]
- Gatica, D.; Damasio, A.; Pascual, C.; Klionsky, D.J.; Ragusa, M.J.; Popelka, H. The carboxy-terminus of yeast Atg13 binds phospholipid membrane via motifs that overlap with the Vac8-interacting domain. Autophagy 2019. [Google Scholar] [CrossRef]
- Bonam, S.R.; Wang, F.; Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discov. 2019, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.A.; Das, B.C.; Ray, S.K. Targeting autophagy for combating chemoresistance and radioresistance in glioblastoma. Apoptosis 2018, 23, 563–575. [Google Scholar] [CrossRef]
- Singh, S.S.; Vats, S.; Chia, A.Y.-Q.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jin, M.; Yao, Z.; Bernard, A.; Klionsky, D.J. Bidirectional roles of Dhh1 in regulating autophagy. Autophagy 2019. [Google Scholar] [CrossRef]
- Yang, M.; Chen, P.; Liu, J.; Zhu, S.; Kroemer, G.; Klionsky, D.J.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci. Adv. 2019, 5, eaaw2238. [Google Scholar] [CrossRef]
- Shakeri, A.; Cicero, A.F.; Panahi, Y.; Mohajeri, M.; Sahebkar, A. Curcumin: A naturally occurring autophagy modulator. J. Cell. Physiol. 2019, 234, 5643–5654. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Kroemer, G. Therapeutic modulation of autophagy: Which disease comes first? Cell Death Differ 2019. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, W.; Tang, G.; Wang, H.; Wu, M.; Yu, W.; Zhou, Z.; Mou, Y.; Liu, X. Targeted Co-delivery of Docetaxel and Atg7 siRNA for Autophagy Inhibition and Pancreatic Cancers Treatment. ACS Appl. Bio Mater. 2019. [Google Scholar] [CrossRef]
- Cai, J.; Qian, K.; Zuo, X.; Yue, W.; Bian, Y.; Yang, J.; Wei, J.; Zhao, W.; Qian, H.; Liu, B. PLGA nanoparticle-based docetaxel/LY294002 drug delivery system enhances antitumor activities against gastric cancer. J. Biomater. Appl. 2019, 33, 1394–1406. [Google Scholar] [CrossRef]
- Min-Wen, J.C.; Yan-Jiang, B.C.; Mishra, S.; Dai, X.; Magae, J.; Shyh-Chang, N.; Kumar, A.P.; Sethi, G. Molecular targets of ascochlorin and its derivatives for cancer therapy. In Advances in Protein Chemistry and Structural Biology; Elsevier: Amsterdam, The Neteherlands, 2017; Volume 108, pp. 199–225. [Google Scholar]
- Patel, S.M.; Venkata, K.C.N.; Bhattacharyya, P.; Sethi, G.; Bishayee, A. Potential of neem (Azadirachta indica L.) for prevention and treatment of oncologic diseases. Semin Cancer Biol. 2016, 40–41, 100–115. [Google Scholar] [CrossRef]
- Banik, K.; Ranaware, A.M.; Deshpande, V.; Nalawade, S.P.; Padmavathi, G.; Bordoloi, D.; Sailo, B.L.; Shanmugam, M.K.; Fan, L.; Arfuso, F.J.P.R. Honokiol for cancer therapeutics: A traditional medicine that can modulate multiple oncogenic targets. Pharmacol. Res. 2019, 144, 192–209. [Google Scholar] [CrossRef]
- Zhang, M.; Hagan IV, C.T.; Min, Y.; Foley, H.; Tian, X.; Yang, F.; Mi, Y.; Au, K.M.; Medik, Y.; Roche, K. Nanoparticle co-delivery of wortmannin and cisplatin synergistically enhances chemoradiotherapy and reverses platinum resistance in ovarian cancer models. Biomaterials 2018, 169, 1–10. [Google Scholar] [CrossRef]
- Feng, Y.; Gao, Y.; Wang, D.; Xu, Z.; Sun, W.; Ren, P. Autophagy Inhibitor (LY294002) and 5-fluorouracil (5-FU) Combination-Based Nanoliposome for Enhanced Efficacy Against Esophageal Squamous Cell Carcinoma. Nanoscale Res. Lett. 2018, 13, 325. [Google Scholar] [CrossRef]
- Lu, L.; Shen, X.; Tao, B.; Lin, C.; Li, K.; Luo, Z.; Cai, K. The nanoparticle-facilitated autophagy inhibition of cancer stem cells for improved chemotherapeutic effects on glioblastomas. J. Mater. Chem. B 2019, 7, 2054–2062. [Google Scholar] [CrossRef]
- Haas, N.B.; Appleman, L.J.; Stein, M.; Redlinger, M.; Wilks, M.; Xu, X.; Onorati, A.; Kalavacharla, A.; Kim, T.; Zhen, C.J. Autophagy inhibition to augment mTOR inhibition: A phase I/II trial of everolimus and hydroxychloroquine in patients with previously treated renal cell carcinoma. Clin. Cancer Res. 2019, 25, 2080–2087. [Google Scholar] [CrossRef]
- Shuhua, W.; Chenbo, S.; Yangyang, L.; Xiangqian, G.; Shuang, H.; Tangyue, L.; Dong, T. Autophagy-related genes Raptor, Rictor, and Beclin1 expression and relationship with multidrug resistance in colorectal carcinoma. Hum. Pathol. 2015, 46, 1752–1759. [Google Scholar] [CrossRef]
- Pan, B.; Chen, D.; Huang, J.; Wang, R.; Feng, B.; Song, H.; Chen, L. HMGB1-mediated autophagy promotes docetaxel resistance in human lung adenocarcinoma. Mol. Cancer 2014, 13, 165. [Google Scholar] [CrossRef]
- Xu, N.; Zhang, J.; Shen, C.; Luo, Y.; Xia, L.; Xue, F.; Xia, Q. Cisplatin-induced downregulation of miR-199a-5p increases drug resistance by activating autophagy in HCC cell. Biochem. Biophys. Res. Commun. 2012, 423, 826–831. [Google Scholar] [CrossRef]
- Faes, S.; Duval, A.P.; Planche, A.; Uldry, E.; Santoro, T.; Pythoud, C.; Stehle, J.-C.; Horlbeck, J.; Letovanec, I.; Riggi, N. Acidic tumor microenvironment abrogates the efficacy of mTORC1 inhibitors. Mol. Cancer 2016, 15, 78. [Google Scholar] [CrossRef]
- Wei, W.; Rosenkrans, Z.T.; Luo, Q.Y.; Lan, X.; Cai, W. Exploiting Nanomaterial-Mediated Autophagy for Cancer Therapy. Small Methods 2019, 3, 1800365. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, H.; Chen, S. Aptamer (AS1411)-Conjugated Liposome for Enhanced Therapeutic Efficacy of miRNA-29b in Ovarian Cancer. J. Nanosci. Nanotechnol. 2020, 20, 2025–2031. [Google Scholar] [CrossRef]
- Yoon, H.Y.; Chang, I.H.; Goo, Y.T.; Kim, C.H.; Kang, T.H.; Kim, S.-Y.; Lee, S.J.; Song, S.H.; Whang, Y.M.; Choi, Y.W. Intravesical delivery of rapamycin via folate-modified liposomes dispersed in thermo-reversible hydrogel. Int. J. Nanomed. 2019, 14, 6249. [Google Scholar] [CrossRef]
- Hajizadeh, M.R.; Parvaz, N.; Barani, M.; Khoshdel, A.; Fahmidehkar, M.A.; Mahmoodi, M.; Torkzadeh-Mahani, M. Diosgenin-loaded niosome as an effective phytochemical nanocarrier: Physicochemical characterization, loading efficiency, and cytotoxicity assay. Daru J. Fac. Pharm. Tehran Univ. Med Sci. 2019, 27, 329–339. [Google Scholar] [CrossRef]
- Cui, Y.; Yang, Y.; Ma, M.; Xu, Y.; Sui, J.; Li, H.; Liang, J.; Sun, Y.; Fan, Y.; Zhang, X. Reductive responsive micelle overcoming multidrug resistance of breast cancer by co-delivery of DOX and specific antibiotic. J. Mater. Chem. B 2019. [Google Scholar] [CrossRef]
- Wei, J.; Long, Y.; Guo, R.; Liu, X.; Tang, X.; Rao, J.; Yin, S.; Zhang, Z.; Li, M.; He, Q. Multifunctional polymeric micelle-based chemo-immunotherapy with immune checkpoint blockade for efficient treatment of orthotopic and metastatic breast cancer. Acta Pharm. Sin. B 2019, 9, 819–831. [Google Scholar] [CrossRef]
- Mohammadinejad, R.; Dadashzadeh, A.; Moghassemi, S.; Ashrafizadeh, M.; Dehshahri, A.; Pardakhty, A.; Sassan, H.A.; Sohrevardi, S.M.; Mandegary, A. Shedding light on gene therapy: Carbon dots for the minimally invasive image-guided delivery of plasmids and noncoding RNAs. J. Adv. Res. 2019. [Google Scholar] [CrossRef]
- Chung, H.J.; Kim, H.J.; Hong, S.T. Tumor-specific delivery of a paclitaxel-loading HSA-haemin nanoparticle for cancer treatment. Nanomed. Nanotechnol. Biol. Med. 2019. [Google Scholar] [CrossRef]
- Sala, R.; Sanchez-Garcia, L.; Serna, N.; Virtudes Cespedes, M.; Casanova, I.; Roldan, M.; Sanchez-Chardi, A.; Unzueta, U.; Vazquez, E.; Mangues, R.; et al. Collaborative membrane activity and receptor-dependent tumor cell targeting for precise nanoparticle delivery in CXCR4(+) colorectal cancer. Acta Biomater. 2019. [Google Scholar] [CrossRef]
- Sameiyan, E.; Hayes, A.W.; Karimi, G. The effect of medicinal plants on multiple drug resistance through autophagy: A review of in vitro studies. Eur. J. Pharmacol. 2019, 852, 244–253. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814. [Google Scholar] [CrossRef]
- Ghadimi, D.; Herrmann, J.; de Vrese, M.; Heller, K.J. Commensal lactic acid-producing bacteria affect host cellular lipid metabolism through various cellular metabolic pathways: Role of mTOR, FOXO1, and autophagy machinery system. PharmaNutrition 2018. [Google Scholar] [CrossRef]
- Qian, G.; Liu, D.; Hou, L.; Hamid, M.; Chen, X.; Gan, F.; Song, S.; Huang, K. Ochratoxin A induces cytoprotective autophagy via blocking AKT/mTOR signaling pathway in PK-15 cells. Food Chem. Toxicol. 2018, 122, 120–131. [Google Scholar] [CrossRef]
- Roohbakhsh, A.; Shamsizadeh, A.; Hayes, A.W.; Reiter, R.J.; Karimi, G. Melatonin as an endogenous regulator of diseases: The role of autophagy. Pharmacol. Res. 2018, 133, 265–276. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, J.; Kang, R.; Klionsky, D.J.; Kroemer, G.; Tang, D. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Hosseinpour-Moghaddam, K.; Caraglia, M.; Sahebkar, A. Autophagy induction by trehalose: Molecular mechanisms and therapeutic impacts. J. Cell. Physiol. 2018, 233, 6524–6543. [Google Scholar] [CrossRef]
- Faes, S.; Demartines, N.; Dormond, O. Resistance to mTORC1 inhibitors in cancer therapy: From kinase mutations to intratumoral heterogeneity of kinase activity. Oxidative Med. Cell. Longev. 2017, 2017, 1726078. [Google Scholar] [CrossRef]
- Faes, S.; Planche, A.; Uldry, E.; Santoro, T.; Pythoud, C.; Stehle, J.-C.; Horlbeck, J.; Letovanec, I.; Riggi, N.; Datta, D. Targeting carbonic anhydrase IX improves the anti-cancer efficacy of mTOR inhibitors. Oncotarget 2016, 7, 36666. [Google Scholar] [CrossRef]
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, M.; de Stanchina, E. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature 2016, 534, 272. [Google Scholar] [CrossRef]
- Habib, S.L.; Al-Obaidi, N.Y.; Nowacki, M.; Pietkun, K.; Zegarska, B.; Kloskowski, T.; Zegarski, W.; Drewa, T.; Medina, E.A.; Zhao, Z. Is mTOR inhibitor good enough for treatment all tumors in TSC patients? J. Cancer 2016, 7, 1621. [Google Scholar] [CrossRef]
- Krueger, D.A.; Care, M.M.; Holland, K.; Agricola, K.; Tudor, C.; Mangeshkar, P.; Wilson, K.A.; Byars, A.; Sahmoud, T.; Franz, D.N. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N. Engl. J. Med. 2010, 363, 1801–1811. [Google Scholar] [CrossRef]
- Pallet, N.; Legendre, C. Adverse events associated with mTOR inhibitors. Expert Opin. Drug Saf. 2013, 12, 177–186. [Google Scholar] [CrossRef]
- LoRusso, P.M. Inhibition of the PI3K/AKT/mTOR pathway in solid tumors. J. Clin. Oncol. 2016, 34, 3803–3815. [Google Scholar] [CrossRef]
- Hatakeyama, R.; Péli-Gulli, M.-P.; Hu, Z.; Jaquenoud, M.; Garcia Osuna, G.M.; Sardu, A.; Dengjel, J.; De Virgilio, C. Spatially Distinct Pools of TORC1 Balance Protein Homeostasis. Mol. Cell 2019, 73, 325–338.e8. [Google Scholar] [CrossRef]
- Lahiri, V.; Klionsky, D.J. Spatially distinct pools of TORC1 balance protein homeostasis. Autophagy 2019, 15, 561–564. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121. [Google Scholar] [CrossRef]
- Lahiri, V.; Hawkins, W.D.; Klionsky, D.J. Watch what you (self-) eat: Autophagic mechanisms that modulate metabolism. Cell Metab. 2019, 29, 803–826. [Google Scholar] [CrossRef]
- Kondratskyi, A.; Kondratska, K.; Skryma, R.; Klionsky, D.J.; Prevarskaya, N. Ion channels in the regulation of autophagy. Autophagy 2018, 14, 3–21. [Google Scholar] [CrossRef]
- Hardie, D.G. Keeping the home fires burning: AMP-activated protein kinase. J. R. Soc. Interface 2018, 15, 20170774. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132. [Google Scholar] [CrossRef]
- Shaw, R.J. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol. 2009, 196, 65–80. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated protein kinase: Maintaining energy homeostasis at the cellular and whole-body levels. Annu. Rev. Nutr. 2014, 34, 31–55. [Google Scholar] [CrossRef]
- Musi, N. AMP-activated protein kinase and type 2 diabetes. Curr. Med. Chem. 2006, 13, 583–589. [Google Scholar] [CrossRef]
- Rehman, G.; Shehzad, A.; Khan, A.L.; Hamayun, M. Role of AMP-activated protein kinase in cancer therapy. Arch. Der Pharm. 2014, 347, 457–468. [Google Scholar] [CrossRef]
- Alessi, D.R.; Sakamoto, K.; Bayascas, J.R. LKB1-dependent signaling pathways. Annu. Rev. Biochem. 2006, 75, 137–163. [Google Scholar] [CrossRef]
- Huang, X.; Wullschleger, S.; Shpiro, N.; McGuire, V.A.; Sakamoto, K.; Woods, Y.L.; Mcburnie, W.; Fleming, S.; Alessi, D.R. Important role of the LKB1–AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem. J. 2008, 412, 211–221. [Google Scholar] [CrossRef]
- Hemminki, A. The molecular basis and clinical aspects of Peutz-Jeghers syndrome. Cell. Mol. Life Sci. 1999, 55, 735–750. [Google Scholar] [CrossRef]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, S.; Bierhoff, H.; Cado, I.; Weber, A.; Tiebe, M.; Grummt, I.; Voit, R. AMP-activated protein kinase adapts rRNA synthesis to cellular energy supply. Proc. Natl. Acad. Sci. USA 2009. PNAS:0909873106. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Shao, S.H.; Xu, Z.-X.; Hennessy, B.; Ding, Z.; Larrea, M.; Kondo, S.; Dumont, D.J.; Gutterman, J.U.; Walker, C.L. The energy sensing LKB1–AMPK pathway regulates p27 kip1 phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 2007, 9, 218. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Levine, B.; Kroemer, G. Metabolic control of autophagy. Cell 2014, 159, 1263–1276. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Xiang, Y.; Wang, Y.; Baikati, K.; Cuervo, A.M.; Luu, Y.K.; Tang, Y.; Pessin, J.E.; Schwartz, G.J.; Czaja, M.J. Autophagy regulates adipose mass and differentiation in mice. J. Clin. Investig. 2009, 119, 3329–3339. [Google Scholar] [CrossRef]
- Kim, J.; Kim, Y.C.; Fang, C.; Russell, R.C.; Kim, J.H.; Fan, W.; Liu, R.; Zhong, Q.; Guan, K.-L. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 2013, 152, 290–303. [Google Scholar] [CrossRef]
- Bendell, J.C.; Kurkjian, C.; Infante, J.R.; Bauer, T.M.; Burris, H.A.; Greco, F.A.; Shih, K.C.; Thompson, D.S.; Lane, C.M.; Finney, L.H. A phase 1 study of the sachet formulation of the oral dual PI3K/mTOR inhibitor BEZ235 given twice daily (BID) in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Khavari, T.A.; Rinn, J.L. Ras/Erk MAPK signaling in epidermal homeostasis and neoplasia. Cell Cycl. 2007, 6, 2928–2931. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: A 10-year update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Sui, X.; Kong, N.; Ye, L.; Han, W.; Zhou, J.; Zhang, Q.; He, C.; Pan, H. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014, 344, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Luo, Q.; Yuan, L.; Miao, C.; Mu, X.; Xiao, W.; Li, J.; Sun, T.; Ma, E. JNK-dependent Atg4 upregulation mediates asperphenamate derivative BBP-induced autophagy in MCF-7 cells. Toxicol. Appl. Pharmacol. 2012, 263, 21–31. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; She, H.; Zhang, T.; Xu, H.; Cheng, L.; Yepes, M.; Zhao, Y.; Mao, Z. p38 MAPK inhibits autophagy and promotes microglial inflammatory responses by phosphorylating ULK1. J. Cell Biol. 2018, 217, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.-C.; Yu, L.; Wang, H.-J.; Tashiro, S.-i.; Onodera, S.; Ikejima, T. TNFα-induced necroptosis and autophagy via supression of the p38–NF-κB survival pathway in L929 cells. J. Pharmacol. Sci. 2011, 117, 160–169. [Google Scholar] [CrossRef]
- Li, C.; Johnson, D.E. Bortezomib induces autophagy in head and neck squamous cell carcinoma cells via JNK activation. Cancer Lett. 2012, 314, 102–107. [Google Scholar] [CrossRef]
- Sun, T.; Li, D.; Wang, L.; Xia, L.; Ma, J.; Guan, Z.; Feng, G.; Zhu, X. c-Jun NH2-terminal kinase activation is essential for up-regulation of LC3 during ceramide-induced autophagy in human nasopharyngeal carcinoma cells. J. Transl. Med. 2011, 9, 161. [Google Scholar] [CrossRef]
- Law, B.K. Rapamycin: An anti-cancer immunosuppressant? Crit. Rev. Oncol. Hematol. 2005, 56, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Canpolat, M.; Gumus, H.; Kumandas, S.; Coskun, A.; Per, H. The use of rapamycin in patients with tuberous sclerosis complex: Long-term results. Epilepsy Behav. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, D.; Silva, A.J. Rapamycin for treating Tuberous sclerosis and Autism spectrum disorders. Trends Mol. Med. 2011, 17, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Teachey, D.T.; Obzut, D.A.; Axsom, K.; Choi, J.K.; Goldsmith, K.C.; Hall, J.; Hulitt, J.; Manno, C.S.; Maris, J.M.; Rhodin, N. Rapamycin improves lymphoproliferative disease in murine autoimmune lymphoproliferative syndrome (ALPS). Blood 2006, 108, 1965–1971. [Google Scholar] [CrossRef] [PubMed]
- Bagherpour, B.; Salehi, M.; Jafari, R.; Bagheri, A.; Kiani-Esfahani, A.; Edalati, M.; Kardi, M.T.; Shaygannejad, V. Promising effect of rapamycin on multiple sclerosis. Mult. Scler. Relat. Disord. 2018, 26, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, K.; Venkatesan, S.; Low, E.S.H.; Hande, M.P. Effects of rapamycin on the mechanistic target of rapamycin (mTOR) pathway and telomerase in breast cancer cells. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2018. [Google Scholar] [CrossRef]
- Chen, W.; Zou, P.; Zhao, Z.; Chen, X.; Fan, X.; Vinothkumar, R.; Cui, R.; Wu, F.; Zhang, Q.; Liang, G. Synergistic antitumor activity of rapamycin and EF24 via increasing ROS for the treatment of gastric cancer. Redox Biol. 2016, 10, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Noh, W.-C.; Mondesire, W.H.; Peng, J.; Jian, W.; Zhang, H.; Dong, J.; Mills, G.B.; Hung, M.-C.; Meric-Bernstam, F. Determinants of rapamycin sensitivity in breast cancer cells. Clin. Cancer Res. 2004, 10, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Olsen, B.R. Rapamycin inhibits proliferation of hemangioma endothelial cells by reducing HIF-1-dependent expression of VEGF. PLoS ONE 2012, 7, e42913. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.H.; Samsel, P.; Shen, L.L.; Narov, K.; Yang, J.; Sampson, J.R. Assessment of Response of Kidney Tumors to Rapamycin and Atorvastatin in Tsc1+/− Mice. Transl. Oncol. 2017, 10, 793–799. [Google Scholar] [CrossRef]
- Karvelas, G.; Roumpi, A.; Komporozos, C.; Syrigos, K. Everolimus as cancer therapy: Cardiotoxic or an unexpected antiatherogenic agent? A narrative review. Hell. J. Cardiol. 2018, 59, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Morviducci, L.; Rota, F.; Rizza, L.; Di Giacinto, P.; Ramponi, S.; Nardone, M.; Tubili, C.; Lenzi, A.; Zuppi, P.; Baldelli, R. Everolimus is a new anti-cancer molecule: Metabolic side effects as lipid disorders and hyperglycemia. Diabetes Res. Clin. Pract. 2018. [Google Scholar] [CrossRef]
- Augustine, J.; Hricik, D. Experience with everolimus. Transplant. Proc. 2004, 36, S500–S503. [Google Scholar] [CrossRef] [PubMed]
- Klawitter, J.; Nashan, B.; Christians, U. Everolimus and sirolimus in transplantation-related but different. Expert Opin. Drug Saf. 2015, 14, 1055–1070. [Google Scholar] [CrossRef] [PubMed]
- Dantal, J.; Berthoux, F.; Moal, M.C.; Rostaing, L.; Legendre, C.; Genin, R.; Toupance, O.; Moulin, B.; Merville, P.; Rerolle, J.P. Efficacy and safety of de novo or early everolimus with low cyclosporine in deceased-donor kidney transplant recipients at specified risk of delayed graft function: 12-month results of a randomized, multicenter trial. Transpl. Int. 2010, 23, 1084–1093. [Google Scholar] [CrossRef] [PubMed]
- Montagnino, G.; Sandrini, S.; Iorio, B.; Schena, F.P.; Carmellini, M.; Rigotti, P.; Cossu, M.; Altieri, P.; Salvadori, M.; Stefoni, S. A randomized exploratory trial of steroid avoidance in renal transplant patients treated with everolimus and low-dose cyclosporine. Nephrol. Dial. Transplant. 2007, 23, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Levy, G.; Schmidli, H.; Punch, J.; Tuttle-Newhall, E.; Mayer, D.; Neuhaus, P.; Samuel, D.; Nashan, B.; Klempnauer, J.; Langnas, A. Safety, tolerability, and efficacy of everolimus in de novo liver transplant recipients: 12-and 36-month results. Liver Transplant. 2006, 12, 1640–1648. [Google Scholar] [CrossRef]
- Salman, J.; Jansson, K.; Siemeni, T.; Sommer, W.; Knoefel, A.-K.; Ahrens, L.; Nakagiri, T.; Ius, F.; Tudorache, I.; Kruse, B. Role for primary immunosuppression with everolimus after pulmonary transplantation. Transpl. Immunol. 2018. [Google Scholar] [CrossRef]
- Chan, E.Y.; Kir, S.; Tooze, S.A. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J. Biol. Chem. 2007, 282, 25464–25474. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef]
- Nakagawa, S.; Nishihara, K.; Inui, K.-i.; Masuda, S. Involvement of autophagy in the pharmacological effects of the mTOR inhibitor everolimus in acute kidney injury. Eur. J. Pharmacol. 2012, 696, 143–154. [Google Scholar] [CrossRef]
- Dhillon, S. Everolimus in combination with exemestane: A review of its use in the treatment of patients with postmenopausal hormone receptor-positive, HER2-negative advanced breast cancer. Drugs 2013, 73, 475–485. [Google Scholar] [CrossRef]
- Shtivelband, M.I. Everolimus in hormone receptor–positive advanced breast cancer: Targeting receptor-based mechanisms of resistance. Breast 2013, 22, 405–410. [Google Scholar] [CrossRef]
- Matter, M.S.; Decaens, T.; Andersen, J.B.; Thorgeirsson, S.S. Targeting the mTOR pathway in hepatocellular carcinoma: Current state and future trends. J. Hepatol. 2014, 60, 855–865. [Google Scholar] [CrossRef]
- Majewski, M.; Korecka, M.; Kossev, P.; Li, S.; Goldman, J.; Moore, J.; Silberstein, L.E.; Nowell, P.C.; Schuler, W.; Shaw, L.M. The immunosuppressive macrolide RAD inhibits growth of human Epstein–Barr virus-transformed B lymphocytes in vitro and in vivo: A potential approach to prevention and treatment of posttransplant lymphoproliferative disorders. Proc. Natl. Acad. Sci. USA 2000, 97, 4285–4290. [Google Scholar] [CrossRef]
- Bilbao, I.; Salcedo, M.; Gómez, M.A.; Jimenez, C.; Castroagudín, J.; Fabregat, J.; Almohalla, C.; Herrero, I.; Cuervas-Mons, V.; Otero, A. Renal function improvement in liver transplant recipients after early everolimus conversion: A clinical practice cohort study in Spain. Liver Transplant. 2015, 21, 1056–1065. [Google Scholar] [CrossRef]
- Bergmann, L.; Maute, L.; Guschmann, M. Temsirolimus for advanced renal cell carcinoma. Expert Rev. Anticancer Ther. 2014, 14, 9–21. [Google Scholar] [CrossRef]
- Kwitkowski, V.E.; Prowell, T.M.; Ibrahim, A.; Farrell, A.T.; Justice, R.; Mitchell, S.S.; Sridhara, R.; Pazdur, R. FDA approval summary: Temsirolimus as treatment for advanced renal cell carcinoma. Oncologist 2010, 15, 428–435. [Google Scholar] [CrossRef]
- Fingar, D.C.; Richardson, C.J.; Tee, A.R.; Cheatham, L.; Tsou, C.; Blenis, J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol. Cell. Biol. 2004, 24, 200–216. [Google Scholar] [CrossRef]
- Peralba, J.M.; Friedrichs, W.; Fulcher, L.; Grünwald, V.; Weiss, G.; Hidalgo, M. Pharmacodynamic evaluation of CCI-779, an inhibitor of mTOR, in cancer patients. Clin. Cancer Res. 2003, 9, 2887–2892. [Google Scholar]
- Schmelzle, T.; Hall, M.N. TOR, a central controller of cell growth. Cell 2000, 103, 253–262. [Google Scholar] [CrossRef]
- Dutcher, J.P.; de Souza, P.; McDermott, D.; Figlin, R.A.; Berkenblit, A.; Thiele, A.; Krygowski, M.; Strahs, A.; Feingold, J.; Hudes, G. Effect of temsirolimus versus interferon-α on outcome of patients with advanced renal cell carcinoma of different tumor histologies. Med Oncol. 2009, 26, 202–209. [Google Scholar] [CrossRef]
- Del Bufalo, D.; Ciuffreda, L.; Trisciuoglio, D.; Desideri, M.; Cognetti, F.; Zupi, G.; Milella, M. Antiangiogenic potential of the mammalian target of rapamycin inhibitor temsirolimus. Cancer Res. 2006, 66, 5549–5554. [Google Scholar] [CrossRef]
- Liu, W.; Huang, S.; Chen, Z.; Wang, H.; Wu, H.; Zhang, D. Temsirolimus, the mTOR inhibitor, induces autophagy in adenoid cystic carcinoma: In vitro and in vivo. Pathol. Res. Pract. 2014, 210, 764–769. [Google Scholar] [CrossRef]
- Younes, A.; Samad, N. Utility of mTOR inhibition in hematologic malignancies. Oncologist 2011, 16, 730–741. [Google Scholar] [CrossRef]
- Ito, D.; Fujimoto, K.; Mori, T.; Kami, K.; Koizumi, M.; Toyoda, E.; Kawaguchi, Y.; Doi, R. In vivo antitumor effect of the mTOR inhibitor CCI-779 and gemcitabine in xenograft models of human pancreatic cancer. Int. J. Cancer 2006, 118, 2337–2343. [Google Scholar] [CrossRef]
- Kang, H.-G.; Wang, B.-Z.; Zhang, J.; Liu, M.-R.; Li, Y.-X. Combination of temsirolimus and adriamycin exhibits an enhanced antitumor effect in hepatocellular carcinoma. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 197–203. [Google Scholar] [CrossRef]
- Galanis, E.; Buckner, J.C.; Maurer, M.J.; Kreisberg, J.I.; Ballman, K.; Boni, J.; Peralba, J.M.; Jenkins, R.B.; Dakhil, S.R.; Morton, R.F. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: A North Central Cancer Treatment Group Study. J. Clin. Oncol. 2005, 23, 5294–5304. [Google Scholar] [CrossRef]
- Lassen, U.; Sorensen, M.; Gaziel, T.B.; Hasselbalch, B.; Poulsen, H.S. Phase II study of bevacizumab and temsirolimus combination therapy for recurrent glioblastoma multiforme. Anticancer Res. 2013, 33, 1657–1660. [Google Scholar]
- Schiff, D.; Jaeckle, K.A.; Anderson, S.K.; Galanis, E.; Giannini, C.; Buckner, J.C.; Stella, P.; Flynn, P.J.; Erickson, B.J.; Schwerkoske, J.F. Phase 1/2 trial of temsirolimus and sorafenib in the treatment of patients with recurrent glioblastoma: North Central Cancer Treatment Group Study/Alliance N0572. Cancer 2018, 124, 1455–1463. [Google Scholar] [CrossRef]
- Baumann, P.; Mandl-Weber, S.; Oduncu, F.; Schmidmaier, R. The novel orally bioavailable inhibitor of phosphoinositol-3-kinase and mammalian target of rapamycin, NVP-BEZ235, inhibits growth and proliferation in multiple myeloma. Exp. Cell Res. 2009, 315, 485–497. [Google Scholar] [CrossRef]
- Liu, T.-J.; Koul, D.; LaFortune, T.; Tiao, N.; Shen, R.J.; Maira, S.-M.; Garcia-Echevrria, C.; Yung, W.A. NVP-BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Mol. Cancer Ther. 2009, 8, 2004–2010. [Google Scholar] [CrossRef]
- Maira, S.-M.; Stauffer, F.; Brueggen, J.; Furet, P.; Schnell, C.; Fritsch, C.; Brachmann, S.; Chene, P.; De Pover, A.; Schoemaker, K. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 2008, 7, 1851–1863. [Google Scholar] [CrossRef]
- Polivka Jr, J.; Janku, F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol. Ther. 2014, 142, 164–175. [Google Scholar] [CrossRef]
- Del Alcazar, C.R.G.; Hardebeck, M.C.; Mukherjee, B.; Tomimatsu, N.; Gao, X.; Yan, J.; Xie, X.-J.; Bachoo, R.; Li, L.; Habib, A.A. Inhibition of DNA double-strand break repair by the dual PI3K/mTOR inhibitor NVP-BEZ235 as a strategy for radiosensitization of glioblastoma. Clin. Cancer Res. 2014, 20, 1235–1248. [Google Scholar] [CrossRef]
- Lu, X.; Horner, J.W.; Paul, E.; Shang, X.; Troncoso, P.; Deng, P.; Jiang, S.; Chang, Q.; Spring, D.J.; Sharma, P. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature 2017, 543, 728. [Google Scholar] [CrossRef]
- Thomas, H.E.; Mercer, C.A.; Carnevalli, L.S.; Park, J.; Andersen, J.B.; Conner, E.A.; Tanaka, K.; Matsutani, T.; Iwanami, A.; Aronow, B.J. mTOR inhibitors synergize on regression, reversal of gene expression, and autophagy in hepatocellular carcinoma. Sci. Transl. Med. 2012, 4, 139ra84. [Google Scholar] [CrossRef]
- Falamarzian, A.; Montazeri Aliabadi, H.; Molavi, O.; Seubert, J.M.; Lai, R.; Uludağ, H.; Lavasanifar, A. Effective down-regulation of signal transducer and activator of transcription 3 (STAT3) by polyplexes of siRNA and lipid-substituted polyethyleneimine for sensitization of breast tumor cells to conventional chemotherapy. J. Biomed. Mater. Res. Part A 2014, 102, 3216–3228. [Google Scholar] [CrossRef]
- Shi, F.; Zhang, J.; Liu, H.; Wu, L.; Jiang, H.; Wu, Q.; Liu, T.; Lou, M.; Wu, H. The dual PI3K/mTOR inhibitor dactolisib elicits anti-tumor activity in vitro and in vivo. Oncotarget 2018, 9, 706. [Google Scholar]
- Wise-Draper, T.M.; Moorthy, G.; Salkeni, M.A.; Karim, N.A.; Thomas, H.E.; Mercer, C.A.; Beg, M.S.; O’Gara, S.; Olowokure, O.; Fathallah, H. A phase Ib study of the dual PI3K/mTOR inhibitor dactolisib (BEZ235) combined with everolimus in patients with advanced solid malignancies. Target. Oncol. 2017, 12, 323–332. [Google Scholar] [CrossRef]
- Massard, C.; Chi, K.N.; Castellano, D.; de Bono, J.; Gravis, G.; Dirix, L.; Machiels, J.-P.; Mita, A.; Mellado, B.; Turri, S. Phase Ib dose-finding study of abiraterone acetate plus buparlisib (BKM120) or dactolisib (BEZ235) in patients with castration-resistant prostate cancer. Eur. J. Cancer 2017, 76, 36–44. [Google Scholar] [CrossRef]
- Hardie, D.G. Regulation of AMP-activated protein kinase by natural and synthetic activators. Acta Pharm. Sin. B 2016, 6, 1–19. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef]
- Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar]
- Foretz, M.; Hébrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Investig. 2010, 120, 2355–2369. [Google Scholar] [CrossRef]
- Cha, J.-H.; Yang, W.-H.; Xia, W.; Wei, Y.; Chan, L.-C.; Lim, S.-O.; Li, C.-W.; Kim, T.; Chang, S.-S.; Lee, H.-H. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol. Cell 2018, 71, 606–620.e7. [Google Scholar] [CrossRef]
- Vancura, A.; Bu, P.; Bhagwat, M.; Zeng, J.; Vancurova, I. Metformin as an Anticancer Agent. Trends Pharmacol. Sci. 2018. [Google Scholar] [CrossRef]
- Pernicova, I.; Korbonits, M. Metformin—mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 2014, 10, 143. [Google Scholar] [CrossRef]
- Rabiee, S.; Tavakol, S.; Barati, M.; Joghataei, M.T. Autophagic, apoptotic, and necrotic cancer cell fates triggered by acidic pH microenvironment. J. Cell. Physiol. 2019, 234, 12061–12069. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Ahmadi, Z. Effects of statins on gut microbiota (microbiome). Rev. Clin. Med. 2019. [Google Scholar] [CrossRef]
- Zhong, S.; Zhang, X.; Chen, L.; Ma, T.; Tang, J.; Zhao, J. Statin use and mortality in cancer patients: Systematic review and meta-analysis of observational studies. Cancer Treat. Rev. 2015, 41, 554–567. [Google Scholar] [CrossRef]
- Zhao, H.; Ji, Z.; Tang, D.; Yan, C.; Zhao, W.; Gao, C. Role of autophagy in early brain injury after subarachnoid hemorrhage in rats. Mol. Biol. Rep. 2013, 40, 819–827. [Google Scholar] [CrossRef]
- Yin, Y.; Liu, L.; Zhao, Z.; Yin, L.; Bauer, N.; Nwaeburu, C.C.; Gladkich, J.; Gross, W.; Hackert, T.; Sticht, C. Simvastatin inhibits sonic hedgehog signaling and stemness features of pancreatic cancer. Cancer Lett. 2018, 426, 14–24. [Google Scholar] [CrossRef]
- Kamel, W.A.; Sugihara, E.; Nobusue, H.; Yamaguchi-Iwai, S.; Onishi, N.; Maki, K.; Fukuchi, Y.; Matsuo, K.; Muto, A.; Saya, H. Simvastatin-induced apoptosis in osteosarcoma cells: A key role of RhoA-AMPK/p38 MAPK signaling in antitumor activity. Mol. Cancer Ther. 2017, 16, 182–192. [Google Scholar] [CrossRef]
- Wei, Y.-M.; Li, X.; Xu, M.; Abais, J.M.; Chen, Y.; Riebling, C.R.; Boini, K.M.; Li, P.-L.; Zhang, Y. Enhancement of autophagy by simvastatin through inhibition of Rac1-mTOR signaling pathway in coronary arterial myocytes. Cell. Physiol. Biochem. 2013, 31, 925–937. [Google Scholar] [CrossRef]
- Yang, S.-H.; Sharrocks, A.D.; Whitmarsh, A.J. MAP kinase signalling cascades and transcriptional regulation. Gene 2013, 513, 1–13. [Google Scholar] [CrossRef]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487. [Google Scholar] [CrossRef]
- Rogawski, M.A.; Löscher, W.; Rho, J.M. Mechanisms of action of antiseizure drugs and the ketogenic diet. Cold Spring Harb. Perspect. Med. 2016. [Google Scholar] [CrossRef]
- Kroemer, G. Autophagy: A druggable process that is deregulated in aging and human disease. J. Clin. Investig. 2015, 125, 1–4. [Google Scholar] [CrossRef]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef]
- Murakami, N.; Oyama, F.; Gu, Y.; McLennan, I.S.; Nonaka, I.; Ihara, Y. Accumulation of tau in autophagic vacuoles in chloroquine myopathy. J. Neuropathol. Exp. Neurol. 1998, 57, 664–673. [Google Scholar] [CrossRef][Green Version]
- Briceno, E.; Reyes, S.; Sotelo, J. Therapy of glioblastoma multiforme improved by the antimutagenic chloroquine. Neurosurg. Focus 2003, 14, 1–6. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologyst 2014, 19, 637–638. [Google Scholar] [CrossRef]
- Rangwala, R.; Leone, R.; Chang, Y.C.; Fecher, L.A.; Schuchter, L.M.; Kramer, A.; Tan, K.-S.; Heitjan, D.F.; Rodgers, G.; Gallagher, M. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1369–1379. [Google Scholar] [CrossRef]
- Bhattacharjee, M.K. Chemistry of Antibiotics and Related Drugs; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Centers for Disease Control and Prevention. The History of Malaria, an Ancient Disease; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2010.
- McChesney, E.W. Animal toxicity and pharmacokinetics of hydroxychloroquine sulfate. Am. J. Med. 1983, 75, 11–18. [Google Scholar] [CrossRef]
- Surrey, A.R.; Hammer, H.F. The Preparation of 7-Chloro-4-(4-(N-ethyl-N-β-hydroxyethylamino)-1-methylbutylamino)-quinoline and Related Compounds. J. Am. Chem. Soc. 1950, 72, 1814–1815. [Google Scholar] [CrossRef]
- Fox, R.I. Mechanism of action of hydroxychloroquine as an antirheumatic drug. Semin Arthritis Rheum. 1993, 23, 82–91. [Google Scholar] [CrossRef]
- Lim, H.-S.; Im, J.-S.; Cho, J.-Y.; Bae, K.-S.; Klein, T.A.; Yeom, J.-S.; Kim, T.-S.; Choi, J.-S.; Jang, I.-J.; Park, J.-W. Pharmacokinetics of hydroxychloroquine and its clinical implications in chemoprophylaxis against malaria caused by Plasmodium vivax. Antimicrob. Agents Chemother. 2009, 53, 1468–1475. [Google Scholar] [CrossRef]
- Al-Bari, M.A.A. Chloroquine analogues in drug discovery: New directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J. Antimicrob. Chemother. 2015, 70, 1608–1621. [Google Scholar] [CrossRef]
- Kuma, A.; Mizushima, N. Physiological role of autophagy as an intracellular recycling system: With an emphasis on nutrient metabolism. Semin Cell Dev. Biol. 2010, 21, 683–690. [Google Scholar] [CrossRef]
- Manic, G.; Obrist, F.; Kroemer, G.; Vitale, I.; Galluzzi, L. Chloroquine and hydroxychloroquine for cancer therapy. Mol. Cell. Oncol. 2014, 1, e29911. [Google Scholar] [CrossRef]
- Choi, D.S.; Blanco, E.; Kim, Y.S.; Rodriguez, A.A.; Zhao, H.; Huang, T.H.M.; Chen, C.L.; Jin, G.; Landis, M.D.; Burey, L.A. Chloroquine eliminates cancer stem cells through deregulation of Jak2 and DNMT1. Stem Cells 2014, 32, 2309–2323. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondria: Master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 780. [Google Scholar] [CrossRef]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606. [Google Scholar] [CrossRef]
- Seglen, P.O.; Gordon, P.B. 3-Methyladenine: Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA 1982, 79, 1889–1892. [Google Scholar] [CrossRef]
- Blommaart, E.F.; Krause, U.; Schellens, J.P.; Vreeling-Sindelárová, H.; Meijer, A.J. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem. 1997, 243, 240–246. [Google Scholar] [CrossRef]
- Holen, I.; Gordon, P.; Seglen, P. Protein kinase-dependent effects of okadaic acid on hepatocytic autophagy and cytoskeletal integrity. Biochem. J. 1992, 284, 633–636. [Google Scholar] [CrossRef]
- Wu, Y.-T.; Tan, H.-L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.-N.; Codogno, P.; Shen, H.-M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 2010, 109, 080796. [Google Scholar] [CrossRef]
- Ui, M.; Okada, T.; Hazeki, K.; Hazeki, O. Wortmannin as a unique probe for an intracellular signalling protein, phosphoinositide 3-kinase. Trends Biochem. Sci. 1995, 20, 303–307. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, N.; Wu, J.; Dai, W.; Rosenblum, J.S. Polo-like kinases inhibited by wortmannin Labeling site and downstream effects. J. Biol. Chem. 2007, 282, 2505–2511. [Google Scholar] [CrossRef]
- Ferby, I.; Waga, I.; Kume, K.; Sakanaka, C.; Shimizu, T. PAF-induced MAPK activation is inhibited by wortmannin in neutrophils and macrophages. In Platelet-Activating Factor and Related Lipid Mediators 2; Springer: New York, NY, USA, 1996; pp. 321–326. [Google Scholar]
- Li, T.; Yue, J.; Huang, L.; Yang, M. Autophagy inhibitor Vacuolin-1 interferes with lipid-based small interference RNA delivery. Biochem. Biophys. Res. Commun. 2019, 510, 427–434. [Google Scholar] [CrossRef]
- Egan, D.F.; Chun, M.G.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.-C.; Sheffler, D.J. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol. Cell 2015, 59, 285–297. [Google Scholar] [CrossRef]
- Petherick, K.J.; Conway, O.J.; Mpamhanga, C.; Osborne, S.A.; Kamal, A.; Saxty, B.; Ganley, I.G. Pharmacological Inhibition of ULK1 Blocks mTOR-Dependent Autophagy. J. Biol. Chem. 2015, 114, 627778. [Google Scholar]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. In Autophagosome and Phagosome; Deretic, V., Ed.; Humana Press: Totowa, NJ, USA, 2008; pp. 77–88. [Google Scholar]
- Liu, J.; Xia, H.; Kim, M.; Xu, L.; Li, Y.; Zhang, L.; Cai, Y.; Norberg, H.V.; Zhang, T.; Furuya, T. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 2011, 147, 223–234. [Google Scholar] [CrossRef]
- Limpert, A.S.; Lambert, L.J.; Bakas, N.A.; Bata, N.; Brun, S.N.; Shaw, R.J.; Cosford, N.D. Autophagy in cancer: Regulation by small molecules. Trends Pharmacol. Sci. 2018, 39, 1021–1032. [Google Scholar] [CrossRef]
- Akin, D.; Wang, S.K.; Habibzadegah-Tari, P.; Law, B.; Ostrov, D.; Li, M.; Yin, X.-M.; Kim, J.-S.; Horenstein, N.; Dunn Jr, W.A. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy 2014, 10, 2021–2035. [Google Scholar] [CrossRef]
- Mohammadinejad, R.; Moosavi, M.A.; Tavakol, S.; Vardar, D.Ö.; Hosseini, A.; Rahmati, M.; Dini, L.; Hussain, S.; Mandegary, A.; Klionsky, D.J. Necrotic, apoptotic and autophagic cell fates triggered by nanoparticles. Autophagy 2019, 15, 4–33. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.-s.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.-i.; Ezaki, J.; Murata, S. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef]
- Shin, J. P62 and the sequestosome, a novel mechanism for protein metabolism. Arch. Pharmacal Res. 1998, 21, 629–633. [Google Scholar] [CrossRef]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiquitin-proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef]
- Donohue, E.; Tovey, A.; Vogl, A.W.; Arns, S.; Sternberg, E.; Young, R.N.; Roberge, M. Inhibition of autophagosome formation by the benzoporphyrin derivative verteporfin. J. Biol. Chem. 2011, 286, 7290–7300. [Google Scholar] [CrossRef]
- Donohue, E.; Thomas, A.; Maurer, N.; Manisali, I.; Zeisser-Labouebe, M.; Zisman, N.; Anderson, H.J.; Ng, S.S.; Webb, M.; Bally, M. The autophagy inhibitor verteporfin moderately enhances the antitumor activity of gemcitabine in a pancreatic ductal adenocarcinoma model. J. Cancer 2013, 4, 585. [Google Scholar] [CrossRef]
- Barangi, S.; Hayes, A.W.; Reiter, R.; Karimi, G. The therapeutic role of long non-coding RNAs in human diseases; a focus on the recent insights into autophagy. Pharmacol. Res. 2019. [Google Scholar] [CrossRef]
- Kong, C.; Li, Y.; Liu, Z.; Ye, J.; Wang, Z.; Zhang, L.; Kong, W.; Liu, H.; Liu, C.; Pang, H. Targeting the Oncogene KRAS Mutant Pancreatic Cancer by Synergistic Blocking of Lysosomal Acidification and Rapid Drug Release. ACS Nano 2019, 13, 4049–4063. [Google Scholar] [CrossRef]
- Mariño, G.; Kroemer, G. Ammonia: A diffusible factor released by proliferating cells that induces autophagy. Sci. Signal. 2010, 3, pe19. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296. [Google Scholar] [CrossRef]
- Iacobuzio-Donahue, C.A.; Herman, J.M. Autophagy, p53, and pancreatic cancer. N. Engl. J. Med. 2014, 370, 1352–1353. [Google Scholar] [CrossRef]
- Gewirtz, D.A. The four faces of autophagy: Implications for cancer therapy. Cancer Res. 2014, 74, 647–651. [Google Scholar] [CrossRef]
- Grácio, D.; Magro, F.; Lima, R.T.; Máximo, V. An overview on the role of autophagy in cancer therapy. Hematol. Med. Oncol. 2017. [Google Scholar] [CrossRef]
- Carew, J.S.; Kelly, K.R.; Nawrocki, S.T. Autophagy as a target for cancer therapy: New developments. Cancer Manag. Res. 2012, 4, 357. [Google Scholar]
- Ghidini, M.; Petrelli, F.; Ghidini, A.; Tomasello, G.; Hahne, J.C.; Passalacqua, R.; Barni, S. Clinical development of mTor inhibitors for renal cancer. Expert Opin. Investig. Drugs 2017, 26, 1229–1237. [Google Scholar] [CrossRef]
- Zhi, X.; Zhong, Q. Autophagy in cancer. F1000prime Rep. 2015, 7, 18. [Google Scholar] [CrossRef]
- Patel, S.; Hurez, V.; Nawrocki, S.T.; Goros, M.; Michalek, J.; Sarantopoulos, J.; Curiel, T.; Mahalingam, D. Vorinostat and hydroxychloroquine improve immunity and inhibit autophagy in metastatic colorectal cancer. Oncotarget 2016, 7, 59087. [Google Scholar] [CrossRef]
- Gorzalczany, Y.; Gilad, Y.; Amihai, D.; Hammel, I.; Sagi-Eisenberg, R.; Merimsky, O. Combining an EGFR directed tyrosine kinase inhibitor with autophagy-inducing drugs: A beneficial strategy to combat non-small cell lung cancer. Cancer Lett. 2011, 310, 207–215. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528. [Google Scholar] [CrossRef]
- Calabretta, B.; Salomoni, P. Inhibition of autophagy: A new strategy to enhance sensitivity of chronic myeloid leukemia stem cells to tyrosine kinase inhibitors. Leuk. Lymphoma 2011, 52, 54–59. [Google Scholar] [CrossRef]
- Tavakol, S.; Kiani, V.; Tavakol, B.; Derakhshan, M.A.; Joghataei, M.T.; Rezayat, S.M. Toxicity Concerns of Nanocarriers. In Nanotechnology-Based Approaches for Targeting and Delivery of Drugs and Genes; Elsevier: Amsterdam, The Netherlands, 2017; pp. 453–484. [Google Scholar]
- Simorgh, S.; Alizadeh, R.; Eftekharzadeh, M.; Haramshahi, S.M.A.; Milan, P.B.; Doshmanziari, M.; Ramezanpour, F.; Gholipourmalekabadi, M.; Seifi, M.; Moradi, F. Olfactory mucosa stem cells: An available candidate for the treatment of the Parkinson’s disease. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef]
- Lindvall, O.; Björklund, A. Cell therapy in Parkinson’s disease. NeuroRx 2004, 1, 382–393. [Google Scholar] [CrossRef]
- Salehi, M.; Bagher, Z.; Kamrava, S.K.; Ehterami, A.; Alizadeh, R.; Farhadi, M.; Falah, M.; Komeili, A. Alginate/chitosan hydrogel containing olfactory ectomesenchymal stem cells for sciatic nerve tissue engineering. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef]
- Alizadeh, R.; Kamrava, S.K.; Bagher, Z.; Farhadi, M.; Falah, M.; Moradi, F.; Boroujeni, M.E.; Soleimani, M.; Kamyab, A.; Komeili, A. Human olfactory stem cells: As a promising source of dopaminergic neuron-like cells for treatment of Parkinson’s disease. Neurosci. Lett. 2019, 696, 52–59. [Google Scholar] [CrossRef]
- Alizadeh, R.; Zarrintaj, P.; Kamrava, S.K.; Bagher, Z.; Farhadi, M.; Heidari, F.; Komeili, A.; Gutiérrez, T.J.; Saeb, M.R. Conductive hydrogels based on agarose/alginate/chitosan for neural disorder therapy. Carbohydr. Polym. 2019, 224, 115161. [Google Scholar] [CrossRef]
- Mehrpour, M.; Kyani, A.; Tafazzoli, M.; Fathi, F.; Joghataie, M.T. A metabonomics investigation of multiple sclerosis by nuclear magnetic resonance. Magn. Reson. Chem. 2013, 51, 102–109. [Google Scholar] [CrossRef]
- Abdolmaleky, H.M.; Pajouhanfar, S.; Faghankhani, M.; Joghataei, M.T.; Mostafavi, A.; Thiagalingam, S. Antipsychotic drugs attenuate aberrant DNA methylation of DTNBP1 (dysbindin) promoter in saliva and post-mortem brain of patients with schizophrenia and psychotic bipolar disorder. Am. J. Med Genet. Part B Neuropsychiatr. Genet. 2015, 168, 687–696. [Google Scholar] [CrossRef]
- Tavakol, S.; Jalili-Firoozinezhad, S.; Mashinchian, O.; Mahmoudi, M. Bioinspired Nanotechnologies for Skin Regeneration. In Nanoscience in Dermatology; Elsevier: Amsterdam, The Netherlands, 2016; pp. 337–352. [Google Scholar]
- Nadimi, A.E.; Ebrahimipour, S.Y.; Afshar, E.G.; Falahati-Pour, S.K.; Ahmadi, Z.; Mohammadinejad, R.; Mohamadi, M. Nano-scale drug delivery systems for antiarrhythmic agents. Eur. J. Med. Chem. 2018. [Google Scholar] [CrossRef]
- Tavakol, S.; Hoveizi, E.; Kharrazi, S.; Tavakol, B.; Karimi, S.; Rezayat Sorkhabadi, S.M. Organelles and chromatin fragmentation of human umbilical vein endothelial cell influence by the effects of zeta potential and size of silver nanoparticles in different manners. Artif. Cells Nanomed. Biotechnol. 2017, 45, 817–823. [Google Scholar] [CrossRef]
- Tavakol, S.; Nikpour, M.R.; Hoveizi, E.; Tavakol, B.; Rezayat, S.M.; Adabi, M.; Abokheili, S.S.; Jahanshahi, M. Investigating the effects of particle size and chemical structure on cytotoxicity and bacteriostatic potential of nano hydroxyapatite/chitosan/silica and nano hydroxyapatite/chitosan/silver; as antibacterial bone substitutes. J. Nanoparticle Res. 2014, 16, 2622. [Google Scholar] [CrossRef]
- Tavakol, S.; Kashani, I.R.; Azami, M.; Khoshzaban, A.; Tavakol, B.; Kharrazi, S.; Ebrahimi, S.; Sorkhabadi, S.M.R. In vitro and in vivo investigations on bone regeneration potential of laminated hydroxyapatite/gelatin nanocomposite scaffold along with DBM. J. Nanoparticle Res. 2012, 14, 1265. [Google Scholar] [CrossRef]
- Ajdary, M.; Moosavi, M.; Rahmati, M.; Falahati, M.; Mahboubi, M.; Mandegary, A.; Jangjoo, S.; Mohammadinejad, R.; Varma, R. Health concerns of various nanoparticles: A review of their in vitro and in vivo toxicity. Nanomaterials 2018, 8, 634. [Google Scholar] [CrossRef]
- Ghasemi Hamidabadi, H.; Rezvani, Z.; Nazm Bojnordi, M.; Shirinzadeh, H.; Seifalian, A.M.; Joghataei, M.T.; Razaghpour, M.; Alibakhshi, A.; Yazdanpanah, A.; Salimi, M. Chitosan-intercalated montmorillonite/poly (vinyl alcohol) nanofibers as a platform to guide neuronlike differentiation of human dental pulp stem cells. ACS Appl. Mater. Interfaces 2017, 9, 11392–11404. [Google Scholar] [CrossRef]
- Duo, Y.; Yang, M.; Du, Z.; Feng, C.; Xing, C.; Wu, Y.; Xie, Z.; Zhang, F.; Huang, L.; Zeng, X. CX-5461-loaded nucleolus-targeting nanoplatform for cancer therapy through induction of pro-death autophagy. Acta Biomater. 2018, 79, 317–330. [Google Scholar] [CrossRef]
- Meliani, A.; Boisgerault, F.; Hardet, R.; Marmier, S.; Collaud, F.; Ronzitti, G.; Leborgne, C.; Verdera, H.C.; Sola, M.S.; Charles, S. Antigen-selective modulation of AAV immunogenicity with tolerogenic rapamycin nanoparticles enables successful vector re-administration. Nat. Commun. 2018, 9, 4098. [Google Scholar] [CrossRef]
- Sabra, S.A.; Elzoghby, A.O.; Sheweita, S.A.; Haroun, M.; Helmy, M.W.; Eldemellawy, M.A.; Xia, Y.; Goodale, D.; Allan, A.L.; Rohani, S. Self-assembled amphiphilic zein-lactoferrin micelles for tumor targeted co-delivery of rapamycin and wogonin to breast cancer. Eur. J. Pharm. Biopharm. 2018, 128, 156–169. [Google Scholar] [CrossRef]
- Cao, L.; Jiang, Y.; Chen, Z. Hollow Fe3O4/Graphene Oxide Nanocomposites as Novel Rapamycin Carrier: Formulation Optimization and In Vitro Characterization. J. Nanosci. Nanotechnol. 2018, 18, 3067–3076. [Google Scholar] [CrossRef]
- Dhandhukia, J.P.; Li, Z.; Peddi, S.; Kakan, S.; Mehta, A.; Tyrpak, D.; Despanie, J.; MacKay, J.A. Berunda Polypeptides: Multi-Headed Fusion Proteins Promote Subcutaneous Administration of Rapamycin to Breast Cancer In Vivo. Theranostics 2017, 7, 3856. [Google Scholar] [CrossRef]
- Dhandhukia, J.P.; Shi, P.; Peddi, S.; Li, Z.; Aluri, S.; Ju, Y.; Brill, D.; Wang, W.; Janib, S.M.; Lin, Y.-A. Bifunctional Elastin-like Polypeptide Nanoparticles Bind Rapamycin and Integrins and Suppress Tumor Growth in Vivo. Bioconjugate Chem. 2017, 28, 2715–2728. [Google Scholar] [CrossRef]
- Gholizadeh, S.; Visweswaran, G.R.R.; Storm, G.; Hennink, W.E.; Kamps, J.A.; Kok, R.J. E-selectin targeted immunoliposomes for rapamycin delivery to activated endothelial cells. Int. J. Pharm. 2018, 548, 759–770. [Google Scholar] [CrossRef]
- Li, H.; Teng, Y.; Sun, J.; Liu, J. Inhibition of hemangioma growth using polymer–lipid hybrid nanoparticles for delivery of rapamycin. Biomed. Pharmacother. 2017, 95, 875–884. [Google Scholar] [CrossRef]
- Thapa, R.K.; Byeon, J.H.; Choi, H.-G.; Yong, C.S.; Kim, J.O. PEGylated lipid bilayer-wrapped nano-graphene oxides for synergistic co-delivery of doxorubicin and rapamycin to prevent drug resistance in cancers. Nanotechnology 2017, 28, 295101. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Tran, T.H.; Thapa, R.K.; Dai Phung, C.; Shin, B.S.; Jeong, J.-H.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Targeted co-delivery of polypyrrole and rapamycin by trastuzumab-conjugated liposomes for combined chemo-photothermal therapy. Int. J. Pharm. 2017, 527, 61–71. [Google Scholar] [CrossRef]
- Thapa, R.K.; Nguyen, H.T.; Jeong, J.-H.; Kim, J.R.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Progressive slowdown/prevention of cellular senescence by CD9-targeted delivery of rapamycin using lactose-wrapped calcium carbonate nanoparticles. Sci. Rep. 2017, 7, 43299. [Google Scholar] [CrossRef]
- Segura-Ibarra, V.; Amione-Guerra, J.; Cruz-Solbes, A.S.; Cara, F.E.; Iruegas-Nunez, D.A.; Wu, S.; Youker, K.A.; Bhimaraj, A.; Torre-Amione, G.; Ferrari, M. Rapamycin nanoparticles localize in diseased lung vasculature and prevent pulmonary arterial hypertension. Int. J. Pharm. 2017, 524, 257–267. [Google Scholar] [CrossRef]
- Eloy, J.O.; Petrilli, R.; Chesca, D.L.; Saggioro, F.P.; Lee, R.J.; Marchetti, J.M. Anti-HER2 immunoliposomes for co-delivery of paclitaxel and rapamycin for breast cancer therapy. Eur. J. Pharm. Biopharm. 2017, 115, 159–167. [Google Scholar] [CrossRef]
- Fan, Y.L.; Hou, H.W.; Tay, H.M.; Guo, W.M.; Berggren, P.-O.; Loo, S.C.J. Preservation of Anticancer and Immunosuppressive Properties of Rapamycin Achieved Through Controlled Releasing Particles. Aaps Pharmscitech. 2017, 18, 2648–2657. [Google Scholar] [CrossRef]
- Bai, H.; Lee, J.S.; Chen, E.; Wang, M.; Xing, Y.; Fahmy, T.M.; Dardik, A. Covalent modification of pericardial patches for sustained rapamycin delivery inhibits venous neointimal hyperplasia. Sci. Rep. 2017, 7, 40142. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Thapa, R.K.; Shin, B.S.; Jeong, J.-H.; Kim, J.-R.; Yong, C.S.; Kim, J.O. CD9 monoclonal antibody-conjugated PEGylated liposomes for targeted delivery of rapamycin in the treatment of cellular senescence. Nanotechnology 2017, 28, 095101. [Google Scholar] [CrossRef]
- Wang, B.; Li, H.; Yao, Q.; Zhang, Y.; Zhu, X.; Xia, T.; Wang, J.; Li, G.; Li, X.; Ni, S. Local in vitro delivery of rapamycin from electrospun PEO/PDLLA nanofibers for glioblastoma treatment. Biomed. Pharmacother. 2016, 83, 1345–1352. [Google Scholar] [CrossRef]
- Katiyar, S.S.; Muntimadugu, E.; Rafeeqi, T.A.; Domb, A.J.; Khan, W. Co-delivery of rapamycin-and piperine-loaded polymeric nanoparticles for breast cancer treatment. Drug Deliv. 2016, 23, 2608–2616. [Google Scholar] [CrossRef]
- Wang, J.; Guo, F.; Yu, M.; Liu, L.; Tan, F.; Yan, R.; Li, N. Rapamycin/DiR loaded lipid-polyaniline nanoparticles for dual-modal imaging guided enhanced photothermal and antiangiogenic combination therapy. J. Control. Release 2016, 237, 23–34. [Google Scholar] [CrossRef]
- Tomoda, K.; Tam, Y.T.; Cho, H.; Buehler, D.; Kozak, K.R.; Kwon, G.S. Triolimus: A Multi-Drug Loaded Polymeric Micelle Containing Paclitaxel, 17-AAG, and Rapamycin as a Novel Radiosensitizer. Macromol. Biosci. 2017, 17, 1600194. [Google Scholar] [CrossRef]
- Shirasu, T.; Koyama, H.; Miura, Y.; Hoshina, K.; Kataoka, K.; Watanabe, T. Nanoparticles effectively target rapamycin delivery to sites of experimental aortic aneurysm in rats. PLoS ONE 2016, 11, e0157813. [Google Scholar] [CrossRef]
- Dou, Y.; Guo, J.; Chen, Y.; Han, S.; Xu, X.; Shi, Q.; Jia, Y.; Liu, Y.; Deng, Y.; Wang, R. Sustained delivery by a cyclodextrin material-based nanocarrier potentiates antiatherosclerotic activity of rapamycin via selectively inhibiting mTORC1 in mice. J. Control. Release 2016, 235, 48–62. [Google Scholar] [CrossRef]
- Eloy, J.O.; Petrilli, R.; W Brueggemeier, R.; Maldonado Marchetti, J.; J Lee, R. Rapamycin-loaded immunoliposomes functionalized with trastuzumab: A strategy to enhance cytotoxicity to HER2-positive breast cancer cells. Anti-Cancer Agents Med. Chem. 2017, 17, 48–56. [Google Scholar] [CrossRef]
- Pang, X.; Wang, J.; Tan, X.; Guo, F.; Lei, M.; Ma, M.; Yu, M.; Tan, F.; Li, N. Dual-modal imaging-guided theranostic nanocarriers based on indocyanine green and mTOR inhibitor rapamycin. ACS Appl. Mater. Interfaces 2016, 8, 13819–13829. [Google Scholar] [CrossRef]
- Gupta, A.; Sharma, D.; Meena, J.; Pandya, S.; Sachan, M.; Kumar, S.; Singh, K.; Mitra, K.; Sharma, S.; Panda, A.K. Preparation and preclinical evaluation of inhalable particles containing rapamycin and anti-tuberculosis agents for induction of autophagy. Pharm. Res. 2016, 33, 1899–1912. [Google Scholar] [CrossRef]
- Eloy, J.O.; Petrilli, R.; Topan, J.F.; Antonio, H.M.R.; Barcellos, J.P.A.; Chesca, D.L.; Serafini, L.N.; Tiezzi, D.G.; Lee, R.J.; Marchetti, J.M. Co-loaded paclitaxel/rapamycin liposomes: Development, characterization and in vitro and in vivo evaluation for breast cancer therapy. Colloids Surf. B Biointerfaces 2016, 141, 74–82. [Google Scholar] [CrossRef]
- Wang, F.; Yang, K.; Wang, Z.; Ma, Y.; Gutkind, J.S.; Hida, N.; Niu, G.; Tian, J. Combined image guided monitoring the pharmacokinetics of rapamycin loaded human serum albumin nanoparticles with a split luciferase reporter. Nanoscale 2016, 8, 3991–4000. [Google Scholar] [CrossRef]
- Polchi, A.; Magini, A.; Mazuryk, J.; Tancini, B.; Gapiński, J.; Patkowski, A.; Giovagnoli, S.; Emiliani, C. Rapamycin loaded solid lipid nanoparticles as a new tool to deliver mTOR inhibitors: Formulation and in vitro characterization. Nanomaterials 2016, 6, 87. [Google Scholar] [CrossRef]
- Lance, K.D.; Good, S.D.; Mendes, T.S.; Ishikiriyama, M.; Chew, P.; Estes, L.S.; Yamada, K.; Mudumba, S.; Bhisitkul, R.B.; Desai, T.A. In vitro and in vivo sustained zero-order delivery of rapamycin (sirolimus) from a biodegradable intraocular device. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7331–7337. [Google Scholar] [CrossRef]
- Visweswaran, G.R.R.; Gholizadeh, S.; Ruiters, M.H.; Molema, G.; Kok, R.J.; Kamps, J.A. Targeting rapamycin to podocytes using a vascular cell adhesion molecule-1 (VCAM-1)-harnessed SAINT-based lipid carrier system. PLoS ONE 2015, 10, e0138870. [Google Scholar] [CrossRef]
- Sobhani, H.; Tarighi, P.; Ostad, S.N.; Shafaati, A.; Nafissi-Varcheh, N.; Aboofazeli, R. Formulation development and toxicity assessment of triacetin mediated nanoemulsions as novel delivery systems for rapamycin. Iran. J. Pharm. Res. 2015, 14, 3. [Google Scholar]
- Zandstra, J.; van Beuge, M.M.; Zuidema, J.; Petersen, A.H.; Staal, M.; Duque, L.F.; Rodriguez, S.; Lathuile, A.A.; Veldhuis, G.J.; Steendam, R. Microsphere-Based Rapamycin Delivery, Systemic Versus Local Administration in a Rat Model of Renal Ischemia/Reperfusion Injury. Pharm. Res. 2015, 32, 3238–3247. [Google Scholar] [CrossRef]
- Li, G.; Cao, L.; Zhou, Z.; Chen, Z.; Huang, Y.; Zhao, Y. Rapamycin loaded magnetic Fe3O4/carboxymethylchitosan nanoparticles as tumor-targeted drug delivery system: Synthesis and in vitro characterization. Colloids Surf. B Biointerfaces 2015, 128, 379–388. [Google Scholar] [CrossRef]
- Mendiburu-Eliçabe, M.; Gil-Ranedo, J. Combination therapy of intraperitoneal rapamycin and convection-enhanced delivery of nanoliposomal CPT-11 in rodent orthotopic brain tumor xenografts. Curr. Cancer Drug Targets 2015, 15, 352–362. [Google Scholar] [CrossRef]
- Miao, Z.L.; Deng, Y.J.; Du, H.Y.; Suo, X.B.; Wang, X.Y.; Wang, X.; Wang, L.; Cui, L.J.; Duan, N. Preparation of a liposomal delivery system and its in vitro release of rapamycin. Exp. Ther. Med. 2015, 9, 941–946. [Google Scholar] [CrossRef]
- Zhang, Q.; Pan, J.; Lubet, R.A.; Komas, S.M.; Kalyanaraman, B.; Wang, Y.; You, M. Enhanced antitumor activity of 3-bromopyruvate in combination with rapamycin in vivo and in vitro. Cancer Prev. Res. 2015. [Google Scholar] [CrossRef]
- Nieto, A.; Hou, H.; Moon, S.W.; Sailor, M.J.; Freeman, W.R.; Cheng, L. Surface engineering of porous silicon microparticles for intravitreal sustained delivery of rapamycin. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1070–1080. [Google Scholar] [CrossRef]
- Falke, L.L.; van Vuuren, S.H.; Kazazi-Hyseni, F.; Ramazani, F.; Nguyen, T.Q.; Veldhuis, G.J.; Maarseveen, E.M.; Zandstra, J.; Zuidema, J.; Duque, L.F. Local therapeutic efficacy with reduced systemic side effects by rapamycin-loaded subcapsular microspheres. Biomaterials 2015, 42, 151–160. [Google Scholar] [CrossRef]
- Chiesa, E.; Dorati, R.; Conti, B.; Modena, T.; Cova, E.; Meloni, F.; Genta, I. Hyaluronic acid-decorated chitosan nanoparticles for CD44-targeted delivery of everolimus. Int. J. Mol. Sci. 2018, 19, 2310. [Google Scholar] [CrossRef]
- Houdaihed, L.; Evans, J.; Allen, C. Co-Delivery of Paclitaxel and Everolimus at the Optimal Synergistic Ratio: A Promising Solution for the Treatment of Breast Cancer. Mol. Pharm. 2018. [Google Scholar] [CrossRef]
- Kasper, M.; Gabriel, D.; Möller, M.; Bauer, D.; Wildschütz, L.; Courthion, H.; Böhm, M.R.; Busch, M.; Loser, K.; Thanos, S. Novel everolimus-loaded nanocarriers for topical treatment of murine experimental autoimmune uveoretinitis (EAU). Exp. Eye Res. 2018, 168, 49–56. [Google Scholar] [CrossRef]
- Chen, X.; Tong, R.; Shi, Z.; Yang, B.; Liu, H.; Ding, S.; Wang, X.; Lei, Q.; Wu, J.; Fang, W. MOF Nanoparticles with Encapsulated Autophagy Inhibitor in Controlled Drug Delivery System for Antitumor. ACS Appl. Mater. Interfaces 2018, 10, 2328–2337. [Google Scholar] [CrossRef]
- Shi, Z.; Chen, X.; Zhang, L.; Ding, S.; Wang, X.; Lei, Q.; Fang, W. FA-PEG decorated MOF nanoparticles as a targeted drug delivery system for controlled release of an autophagy inhibitor. Biomater. Sci. 2018, 6, 2582–2590. [Google Scholar] [CrossRef]
- Gholizadeh, S.; Kamps, J.A.; Hennink, W.E.; Kok, R.J. PLGA-PEG nanoparticles for targeted delivery of the mTOR/PI3kinase inhibitor dactolisib to inflamed endothelium. Int. J. Pharm. 2018, 548, 747–758. [Google Scholar] [CrossRef]
- Ahmadi, Z.; Mohammadinejad, R.; Ashrafizadeh, M. Drug delivery systems for resveratrol, a non-flavonoid polyphenol: Emerging evidence in last decades. J. Drug Deliv. Sci. Technol. 2019, 51, 591–604. [Google Scholar] [CrossRef]
- Tavakol, S.; Mousavi, S.M.M.; Tavakol, B.; Hoveizi, E.; Ai, J.; Sorkhabadi, S.M.R. Mechano-transduction signals derived from self-assembling peptide nanofibers containing long motif of laminin influence neurogenesis in in-vitro and in-vivo. Mol. Neurobiol. 2017, 54, 2483–2496. [Google Scholar] [CrossRef]
- Tavakol, S.; Saber, R.; Hoveizi, E.; Tavakol, B.; Aligholi, H.; Ai, J.; Rezayat, S.M. Self-assembling peptide nanofiber containing long motif of laminin induces neural differentiation, tubulin polymerization, and neurogenesis: In vitro, ex vivo, and in vivo studies. Mol. Neurobiol. 2016, 53, 5288–5299. [Google Scholar] [CrossRef]
- Hoveizi, E.; Khodadadi, S.; Tavakol, S.; Karima, O.; Nasiri-Khalili, M.A. Small molecules differentiate definitive endoderm from human induced pluripotent stem cells on PCL scaffold. Appl. Biochem. Biotechnol. 2014, 173, 1727–1736. [Google Scholar] [CrossRef]
- Oloumi, H.; Mousavi, E.A.; Nejad, R.M. Multi-Wall Carbon Nanotubes Effects on Plant Seedlings Growth and Cadmium/Lead Uptake In Vitro. Russ. J. Plant Physiol. 2018, 65, 260–268. [Google Scholar] [CrossRef]
- Rezaee, M.; Behnam, B.; Banach, M.; Sahebkar, A. The Yin and Yang of carbon nanomaterials in atherosclerosis. Biotechnol. Adv. 2018. [Google Scholar] [CrossRef]
- Akhtartavan, S.; Karimi, M.; Karimian, K.; Azarpira, N.; Khatami, M.; Heli, H. Evaluation of a self-nanoemulsifying docetaxel delivery system. Biomed. Pharmacother. 2019, 109, 2427–2433. [Google Scholar] [CrossRef]
- Ranjbar, M.; Pardakhty, A.; Amanatfard, A.; Asadipour, A. Efficient drug delivery of β-estradiol encapsulated in Zn-metal–organic framework nanostructures by microwave-assisted coprecipitation method. Drug Des. Dev. Ther. 2018, 12, 2635. [Google Scholar] [CrossRef]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Petersen, G.H.; Alzghari, S.K.; Chee, W.; Sankari, S.S.; La-Beck, N.M. Meta-analysis of clinical and preclinical studies comparing the anticancer efficacy of liposomal versus conventional non-liposomal doxorubicin. J. Control. Release 2016, 232, 255–264. [Google Scholar] [CrossRef]
- Kim, J.-S. Liposomal drug delivery system. J. Pharm. Investig. 2016, 46, 387–392. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Tran, T.H.; Thapa, R.K.; Pham, T.T.; Jeong, J.-H.; Youn, Y.S.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Incorporation of chemotherapeutic agent and photosensitizer in a low temperature-sensitive liposome for effective chemo-hyperthermic anticancer activity. Expert Opin. Drug Deliv. 2017, 14, 155–164. [Google Scholar] [CrossRef]
- Sætern, A.M.; Flaten, G.E.; Brandl, M. A method to determine the incorporation capacity of camptothecin in liposomes. AAPS Pharmscitech 2004, 5, 30–37. [Google Scholar] [CrossRef]
- Ghanbarzadeh, S.; Arami, S.; Pourmoazzen, Z.; Khorrami, A. Improvement of the antiproliferative effect of rapamycin on tumor cell lines by poly (monomethylitaconate)-based pH-sensitive, plasma stable liposomes. Colloids Surf. B Biointerfaces 2014, 115, 323–330. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, T.; Wang, C.; Jiao, J.; Li, J.; Deng, Y. Coencapsulation of epirubicin and metformin in PEGylated liposomes inhibits the recurrence of murine sarcoma S180 existing CD133+ cancer stem-like cells. Eur. J. Pharm. Biopharm. 2014, 88, 737–745. [Google Scholar] [CrossRef]
- Alupei, M.C.; Licarete, E.; Patras, L.; Banciu, M. Liposomal simvastatin inhibits tumor growth via targeting tumor-associated macrophages-mediated oxidative stress. Cancer Lett. 2015, 356, 946–952. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Lo, C.-L.; Lin, Y.-F.; Hsiue, G.-H. Rapamycin encapsulated in dual-responsive micelles for cancer therapy. Biomaterials 2013, 34, 1115–1127. [Google Scholar] [CrossRef]
- Shaki, H.; Ganji, F.; Kempen, P.J.; Dolatshahi-Pirouz, A.; Vasheghani-Farahani, E. Self-assembled amphiphilic-dextran nanomicelles for delivery of rapamycin. J. Drug Deliv. Sci. Technol. 2018, 44, 333–341. [Google Scholar] [CrossRef]
- Liu, X.; Li, X.; Zhou, L.; Li, S.; Sun, J.; Wang, Z.; Gao, Y.; Jiang, Y.; Lu, H.; Wang, Q. Effects of simvastatin-loaded polymeric micelles on human osteoblast-like MG-63 cells. Colloids Surf. B Biointerfaces 2013, 102, 420–427. [Google Scholar] [CrossRef]
- Jia, Z.; Zhang, Y.; Chen, Y.H.; Dusad, A.; Yuan, H.; Ren, K.; Li, F.; Fehringer, E.V.; Purdue, P.E.; Goldring, S.R. Simvastatin prodrug micelles target fracture and improve healing. J. Control. Release 2015, 200, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, L.; Zhang, H.; Tian, B.; Li, R.; Hou, X.; Wei, F. Fluorescent magnetic PEI-PLGA nanoparticles loaded with paclitaxel for concurrent cell imaging, enhanced apoptosis and autophagy in human brain cancer. Colloids Surf. B Biointerfaces 2018, 172, 708–717. [Google Scholar] [CrossRef]
- Bala, I.; Hariharan, S.; Kumar, M.R. PLGA nanoparticles in drug delivery: The state of the art. Crit. Rev. Ther. Drug Carr. Syst. 2004, 21, 387–422. [Google Scholar] [CrossRef]
- Gao, J.; Chen, H.; Song, H.; Su, X.; Niu, F.; Li, W.; Li, B.; Dai, J.; Wang, H.; Guo, Y. Antibody-targeted immunoliposomes for cancer treatment. Mini Rev. Med. Chem. 2013, 13, 2026–2035. [Google Scholar] [CrossRef]
- Ding, L.; Wang, Q.; Shen, M.; Sun, Y.; Zhang, X.; Huang, C.; Chen, J.; Li, R.; Duan, Y. Thermoresponsive nanocomposite gel for local drug delivery to suppress the growth of glioma by inducing autophagy. Autophagy 2017, 13, 1176–1190. [Google Scholar] [CrossRef]
- Shi, Y.; Su, C.; Cui, W.; Li, H.; Liu, L.; Feng, B.; Liu, M.; Su, R.; Zhao, L. Gefitinib loaded folate decorated bovine serum albumin conjugated carboxymethyl-beta-cyclodextrin nanoparticles enhance drug delivery and attenuate autophagy in folate receptor-positive cancer cells. J. Nanobiotechnol. 2014, 12, 43. [Google Scholar] [CrossRef]
- Song, W.; Ma, Z.; Zhang, Y.; Yang, C. Autophagy plays a dual role during intracellular siRNA delivery by lipoplex and polyplex nanoparticles. Acta Biomater. 2017, 58, 196–204. [Google Scholar] [CrossRef]
- Zhang, X.; Dong, Y.; Zeng, X.; Liang, X.; Li, X.; Tao, W.; Chen, H.; Jiang, Y.; Mei, L.; Feng, S.-S. The effect of autophagy inhibitors on drug delivery using biodegradable polymer nanoparticles in cancer treatment. Biomaterials 2014, 35, 1932–1943. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Grabiner, B.C.; Van Allen, E.M.; Hodis, E.; Jacobus, S.; Supko, J.G.; Stewart, M.; Choueiri, T.K.; Gandhi, L.; Cleary, J.M. Activating mTOR mutations in a patient with an extraordinary response on a phase I trial of everolimus and pazopanib. Cancer Discov. 2014. [Google Scholar] [CrossRef] [PubMed]
- Taglieri, L.; De Iuliis, F.; Giuffrida, A.; Giantulli, S.; Silvestri, I.; Scarpa, S. Resistance to the mTOR inhibitor everolimus is reversed by the downregulation of survivin in breast cancer cells. Oncol. Lett. 2017, 14, 3832–3838. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Lo, Y.-L.; Hsu, C.; Wu, Y.-T.; Liao, Z.-X.; Wu, W.-J.; Chen, Y.-J.; Kao, C.; Chiu, C.-C.; Wang, L.-F. CS-PEI/Beclin-siRNA Downregulate Multidrug Resistance Proteins and Increase Paclitaxel Therapeutic Efficacy against NSCLC. Mol. Ther. Nucleic Acids 2019, 17, 477–490. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.-H.; Ye, C.; Bai, E.-H.; Zhang, L.-L.; Huo, S.-J.; Yu, H.-H.; Xiang, S.-Y.; Yu, S.-Q. Co-delivery nanoparticles of doxorubicin and chloroquine for improving the anti-cancer effect in vitro. Nanotechnology 2018, 30, 085101. [Google Scholar] [CrossRef] [PubMed]
- Tavakol, S. Acidic pH derived from cancer cells may induce failed reprogramming of normal differentiated cells adjacent tumor cells and turn them into cancer cells. Med Hypotheses 2014, 83, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, B.D.; Diering, G.H.; Bidinosti, M.A.; Dalal, K.; Alain, T.; Balgi, A.D.; Forestieri, R.; Nodwell, M.; Rajadurai, C.V.; Gunaratnam, C. Structure-activity analysis of niclosamide reveals potential role for cytoplasmic pH in control of mammalian target of rapamycin complex 1 (mTORC1) signaling. J. Biol. Chem. 2012, 287, 17530–17545. [Google Scholar] [CrossRef] [PubMed]
- Conciatori, F.; Ciuffreda, L.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M. mTOR cross-talk in cancer and potential for combination therapy. Cancers 2018, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Haagenson, K.K.; Wu, G.S. The role of MAP kinases and MAP kinase phosphatase-1 in resistance to breast cancer treatment. Cancer Metastasis Rev. 2010, 29, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Longo, V. Fasting vs dietary restriction in cellular protection and cancer treatment: From model organisms to patients. Oncogene 2011, 30, 3305. [Google Scholar] [CrossRef] [PubMed]
- Nencioni, A.; Caffa, I.; Cortellino, S.; Longo, V.D. Fasting and cancer: Molecular mechanisms and clinical application. Nat. Rev. Cancer 2018, 1, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Safdie, F.M.; Dorff, T.; Quinn, D.; Fontana, L.; Wei, M.; Lee, C.; Cohen, P.; Longo, V.D. Fasting and cancer treatment in humans: A case series report. Aging 2009, 1, 988. [Google Scholar] [CrossRef] [PubMed]
- Doyle, C.; Kushi, L.H.; Byers, T.; Courneya, K.S.; Demark-Wahnefried, W.; Grant, B.; McTiernan, A.; Rock, C.L.; Thompson, C.; Gansler, T. Nutrition and physical activity during and after cancer treatment: An American Cancer Society guide for informed choices. Ca. A Cancer J. Clin. 2006, 56, 323–353. [Google Scholar] [CrossRef] [PubMed]
- De Groot, S.; Vreeswijk, M.P.; Welters, M.J.; Gravesteijn, G.; Boei, J.J.; Jochems, A.; Houtsma, D.; Putter, H.; van der Hoeven, J.J.; Nortier, J.W. The effects of short-term fasting on tolerance to (neo) adjuvant chemotherapy in HER2-negative breast cancer patients: A randomized pilot study. BMC Cancer 2015, 15, 652. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, L.; Zhou, Y.; Ye, Q.; Yang, X.; Jiang, J.; Ye, Z.; Gao, F.; Tan, X.; Zhang, G. Hydroxychloroquine enhances the antitumor effects of BC001 in gastric cancer. Int. J. Oncol. 2019, 55, 405–414. [Google Scholar] [CrossRef]
- Pellegrini, P.; Strambi, A.; Zipoli, C.; Hägg-Olofsson, M.; Buoncervello, M.; Linder, S.; De Milito, A. Acidic extracellular pH neutralizes the autophagy-inhibiting activity of chloroquine: Implications for cancer therapies. Autophagy 2014, 10, 562–571. [Google Scholar] [CrossRef]
- Shi, T.-T.; Yu, X.-X.; Yan, L.-J.; Xiao, H.-T. Research progress of hydroxychloroquine and autophagy inhibitors on cancer. Cancer Chemother. Pharmacol. 2017, 79, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zeng, X.; Liang, X.; Yang, Y.; Li, X.; Chen, H.; Huang, L.; Mei, L.; Feng, S.-S. The chemotherapeutic potential of PEG-b-PLGA copolymer micelles that combine chloroquine as autophagy inhibitor and docetaxel as an anti-cancer drug. Biomaterials 2014, 35, 9144–9154. [Google Scholar] [CrossRef]
- Gong, C.; Hu, C.; Gu, F.; Xia, Q.; Yao, C.; Zhang, L.; Qiang, L.; Gao, S.; Gao, Y. Co-delivery of autophagy inhibitor ATG7 siRNA and docetaxel for breast cancer treatment. J. Control. Release 2017, 266, 272–286. [Google Scholar] [CrossRef]
- Zheng, Y.; Su, C.; Zhao, L.; Shi, Y. Chitosan nanoparticle-mediated co-delivery of shAtg-5 and gefitinib synergistically promoted the efficacy of chemotherapeutics through the modulation of autophagy. J. Nanobiotechnol. 2017, 15, 28. [Google Scholar] [CrossRef]
- Zhao, P.; Li, M.; Wang, Y.; Chen, Y.; He, C.; Zhang, X.; Yang, T.; Lu, Y.; You, J.; Lee, R.J. Enhancing anti-tumor efficiency in hepatocellular carcinoma through the autophagy inhibition by miR-375/sorafenib in lipid-coated calcium carbonate nanoparticles. Acta Biomater. 2018, 72, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Saiyin, W.; Wang, D.; Li, L.; Zhu, L.; Liu, B.; Sheng, L.; Li, Y.; Zhu, B.; Mao, L.; Li, G. Sequential release of autophagy inhibitor and chemotherapeutic drug with polymeric delivery system for oral squamous cell carcinoma therapy. Mol. Pharm. 2014, 11, 1662–1675. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Xia, C.; Wang, Y.; Wan, D.; Rao, J.; Tang, X.; Wei, J.; Wang, X.; Li, M.; Zhang, Z. Dual receptor recognizing liposomes containing paclitaxel and hydroxychloroquine for primary and metastatic melanoma treatment via autophagy-dependent and independent pathways. J. Control. Release 2018, 288, 148–160. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, M.H.; Gallagher, M.; Teitelbaum, U.R.; Giantonio, B.J.; Damjanov, N.; Loaiza-Bonilla, A.; Amaravadi, R.K.; Heitjan, D.F.; Vasilevskaya, I.; Von Hoff, D.D. Phase 1 trial of gemcitabine/nab-paclitaxel in combination with the autophagy inhibitor hydroxychloroquine in previously untreated patients with metastatic pancreatic adenocarcinoma. Am. Soc. Clinic. Oncol. 2015, 33, e15213. [Google Scholar] [CrossRef]
- Karasic, T.B.; O’Hara, M.H.; Loaiza-Bonilla, A.; Reiss-Binder, K.A.; Teitelbaum, U.R.; Borazanci, E.; De Jesus-Acosta, A.M.; Redlinger, C.; Burrell, J.A.; Von Hoff, D.D. Abstract CT085: Randomized phase II trial of hydroxychloroquine in combination with gemcitabine/nab-paclitaxel to inhibit autophagy in pancreatic cancer: A SU2C-funded trial. In Proceedings of the AACR Annual Meeting 2018, Chicago, IL, USA, 14–18 April 2018. [Google Scholar]
- Sotelo, J.; Briceno, E.; López-González, M.A. Adding chloroquine to conventional treatment for glioblastoma multiforme: A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 2006, 144, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Vogl, D.T.; Stadtmauer, E.A.; Tan, K.-S.; Heitjan, D.F.; Davis, L.E.; Pontiggia, L.; Rangwala, R.; Piao, S.; Chang, Y.C.; Scott, E.C. Combined autophagy and proteasome inhibition: A phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014, 10, 1380–1390. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Mita, M.; Sarantopoulos, J.; Wood, L.; Amaravadi, R.K.; Davis, L.E.; Mita, A.C.; Curiel, T.J.; Espitia, C.M.; Nawrocki, S.T. Combined autophagy and HDAC inhibition: A phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 2014, 10, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Poklepovic, A.; Gewirtz, D.A. Outcome of early clinical trials of the combination of hydroxychloroquine with chemotherapy in cancer. Autophagy 2014, 10, 1478–1480. [Google Scholar] [CrossRef]
- Wang, Y.; Qiu, Y.; Yin, S.; Zhang, L.; Shi, K.; Gao, H.; Zhang, Z.; He, Q. A functional nanocarrier that copenetrates extracellular matrix and multiple layers of tumor cells for sequential and deep tumor autophagy inhibitor and chemotherapeutic delivery. Autophagy 2017, 13, 359–370. [Google Scholar] [CrossRef]
- Lu, H.-Y.; Chang, Y.-J.; Fan, N.-C.; Wang, L.-S.; Lai, N.-C.; Yang, C.-M.; Wu, L.-C.; Ho, J.-a.A. Synergism through combination of chemotherapy and oxidative stress-induced autophagy in A549 lung cancer cells using redox-responsive nanohybrids: A new strategy for cancer therapy. Biomaterials 2015, 42, 30–41. [Google Scholar] [CrossRef]
- AbdElhamid, A.S.; Helmy, M.W.; Ebrahim, S.M.; Bahey-El-Din, M.; Zayed, D.G.; Zein El Dein, E.A.; El-Gizawy, S.A.; Elzoghby, A.O. Layer-by-layer gelatin/chondroitin quantum dots-based nanotheranostics: Combined rapamycin/celecoxib delivery and cancer imaging. Nanomed. Nanotechnol. Biol. Med. 2018. [Google Scholar] [CrossRef]
- Gao, X.; Yao, L.; Song, Q.; Zhu, L.; Xia, Z.; Xia, H.; Jiang, X.; Chen, J.; Chen, H. The association of autophagy with polyethylenimine-induced cytotoxity in nephritic and hepatic cell lines. Biomaterials 2011, 32, 8613–8625. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, H.; Sun, Y.; Wang, H.; Guo, F.; Rao, S.; Deng, J.; Zhang, Y.; Miao, Y.; Guo, C. PAMAM nanoparticles promote acute lung injury by inducing autophagic cell death through the Akt-TSC2-mTOR signaling pathway. J. Mol. Cell Biol. 2009, 1, 37–45. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Cong, C.; Li, X.; Zhu, R.; Li, A.; Zhao, S.; Li, X. Nano-drug System Based on Hierarchical Drug Release for Deep Localized/Systematic Cascade Tumor Therapy Stimulating Antitumor Immune Responses. Theranostics 2019, 9, 2897–2909. [Google Scholar] [CrossRef] [PubMed]
- Martinez Legaspi, S.; Segatori, L. Aggregation Behavior of Nanoparticle-Peptide Systems Affects Autophagy. Bioconjug Chem 2019, 30, 1986–1997. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kang, Y.; Yin, S.; Chen, A.; Wu, J.; Liang, H.; Shao, L. Key Role of Microtubule and Its Acetylation in a Zinc Oxide Nanoparticle-Mediated Lysosome-Autophagy System. Small 2019, 15, e1901073. [Google Scholar] [CrossRef]
- Jiang, L.; Li, Z.; Xie, Y.; Liu, L.; Cao, Y. Cyanidin chloride modestly protects Caco-2cells from ZnO nanoparticle exposure probably through the induction of autophagy. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2019, 127, 251–259. [Google Scholar] [CrossRef]
- Sun, X.; Cheng, C.; Zhang, J.; Jin, X.; Sun, S.; Mei, L.; Huang, L. Intracellular Trafficking Network and Autophagy of PHBHHx Nanoparticles and their Implications for Drug Delivery. Sci. Rep. 2019, 9, 9585. [Google Scholar] [CrossRef]





{kind=link}
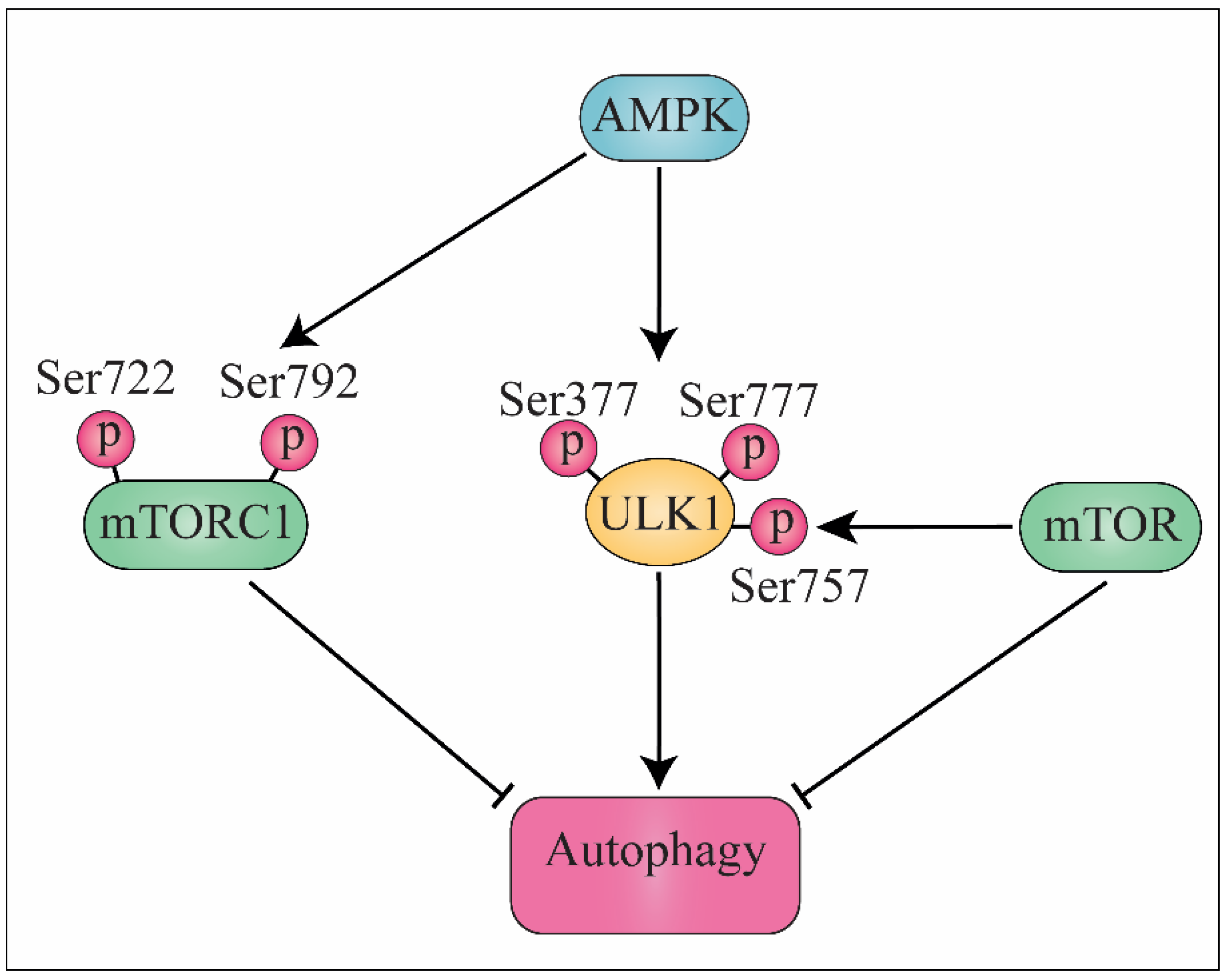{kind=link}
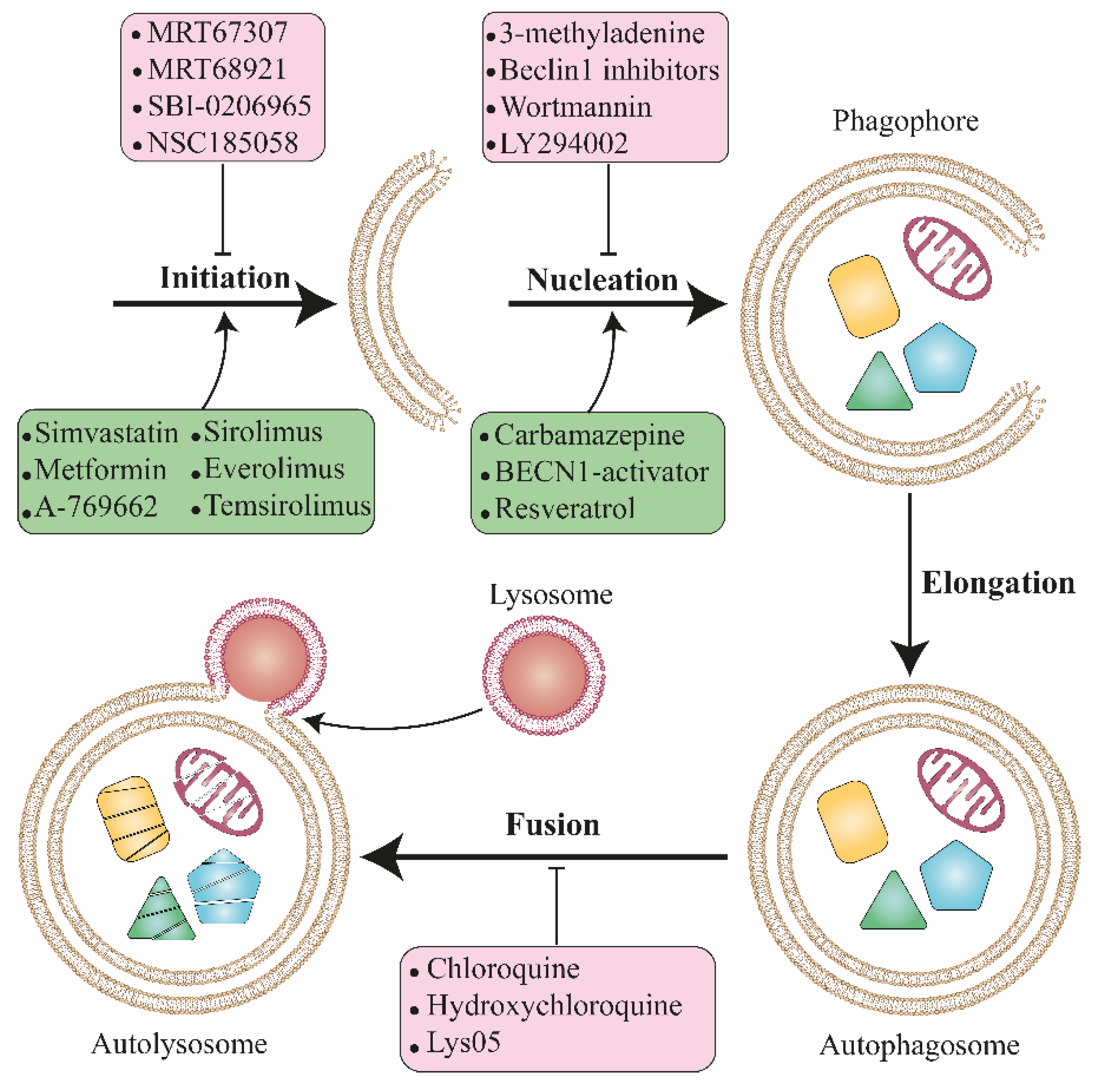{kind=link}
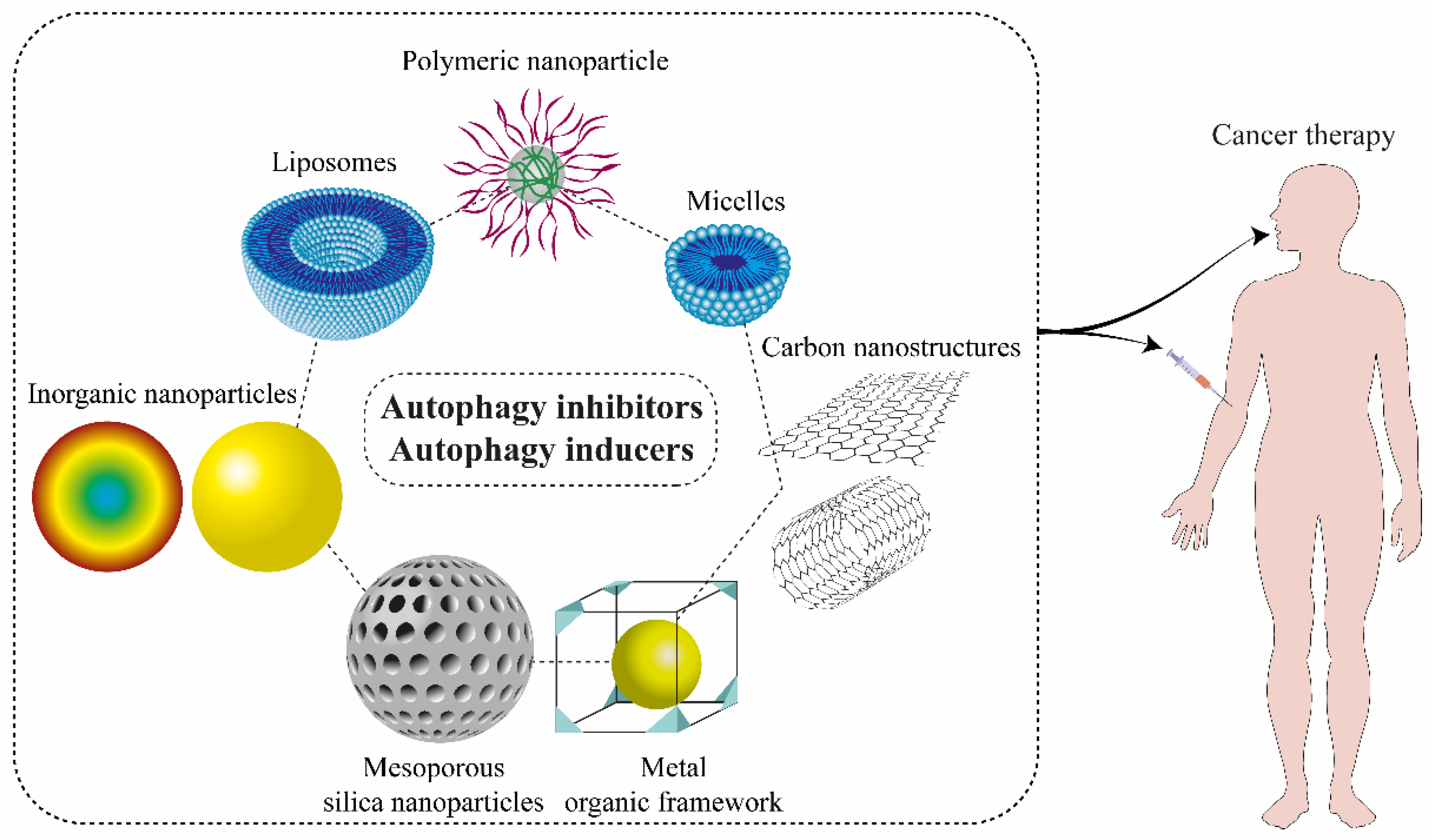{kind=link}
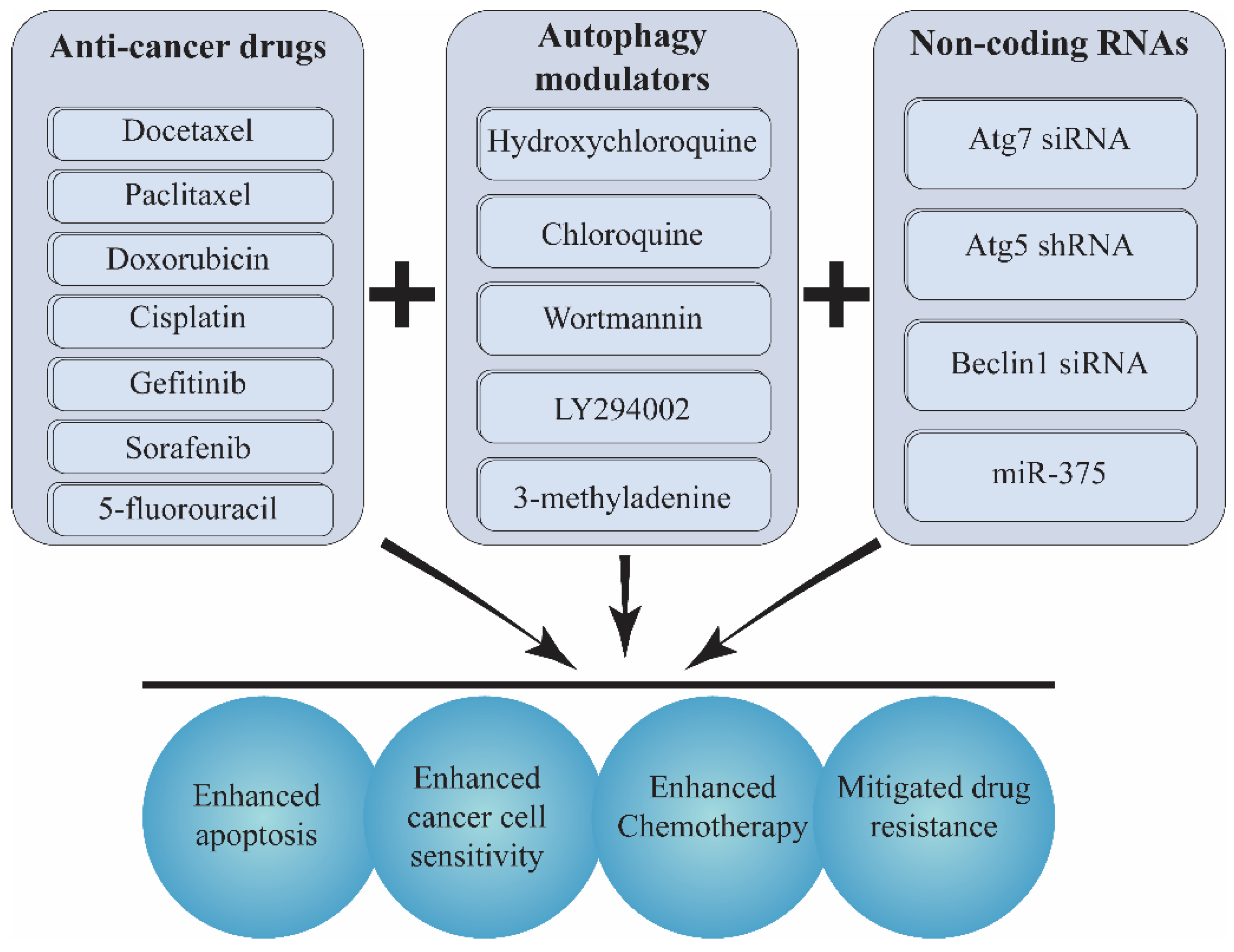{kind=link}
| Autophagy Inducers/Inhibitors | Nano/Micro-Carriers | Co-Delivered Drug/siRNA | Disease | Targeting Agent | In Vitro/In Vivo | Major Outcomes | Refs |
|---|---|---|---|---|---|---|---|
| Rapamycin | Zain-lactoferrin micelles | Wogonin | Breast cancer | - | In vitro (MCF-7 breast cancer cells) and in vivo (Ehrlich ascites tumor animal model) | Inhibition of tumor growth with minimized side effects | [230] |
| Rapamycin | Hollow Fe₃O₄/Graphene Oxide Nanocomposites | - | - | - | In vitro (HepG2 cells) | High EE (84.92%), good stability and great cytotoxicity against HepG2 cells | [231] |
| Rapamycin | Berunda Polypeptides | - | Breast Cancer | - | In vivo (human MDA-MB-468 orthotopic breast cancer xenografts) and in vitro | Suppression of tumor growth and decreased viability of tumor cells | [232] |
| Rapamycin | Elastin-like Polypeptide NPs | Integrins | Breast cancer | - | In vivo (MDA-MB-468 breast tumor) | Inhibition of tumor growth in a higher level (3 folds) | [233] |
| Rapamycin | Immunoliposomes | - | Inflammatory disorder | anti-E-selectin | In vitro (activated endothelial cells) | Inhibition of endothelial cells migration and proliferation as well as inflammatory cytokines expression | [234] |
| Rapamycin | Polymer-lipid hybrid NPs | - | Hemangioma | - | In vitro (human hemangioma endothelial cells) and in vivo (female Balb/c mice) | Effective binding with HemECs and remarkable proliferation inhibition and decreased expression of angiogenic factors as well as decreased hemangioma volume, weight and microvessel density in in vivo | [235] |
| Rapamycin | Graphene oxides wrapped with PEGylated lipid bilayer | DOX | - | - | In vitro (MCF-7, MDA-MB-221 and BT474 cells) | Treatment and prevention of resistant cancer cells by up-regulating Bax, P21, P53, and caspase-3 and apoptosis induction | [236] |
| Rapamycin | Liposomes | Polypyrrol | Breast cancer | trastuzumab | In vitro (BT-474 cells) | Overcoming against drug resistance in breast cancer and higher therapeutic efficacy in breast cancer cells | [237] |
| Rapamycin | Lactose-wrapped calcium carbonate NPs | - | Cellular senescence | CD9 | In vitro (senescent cells) | Prevention of cellular senescence and improved proliferation of aged cells | [238] |
| Rapamycin | PEG-PCL NPs | - | Pulmonary arterial hypertension (PAH) | - | In vivo (rat model of PAH) | High accumulation of nanoparticles in lung, attenuation PAH development and also decreased systemic side effects compared to the free rapamycin | [239] |
| Rapamycin | Immunoliposomes | Paclitaxel | Breast cancer | Anti-HER2 | In vitro (HER2(+) breast cancer cells and triple negative cancer cells) and in vivo (nude mice with HER2(+) breast cancer cells) | High cytotoxicity due to enhanced uptake through HER2 binding and decreased tumor volume in vivo | [240] |
| Rapamycin | PLGA-PCL NPs | - | Breast cancer | - | In vitro MCF-7 and human lymphocyte cell (Jurkat cells) | Inhibition of cell proliferation in MCF-7 cells, suppression of cell growth in Jurkat cells and simultaneously, maintaining the bioactivity of rapamycin | [241] |
| Rapamycin | PLGA NPs | - | Venous neointimal hyperplasia | Pericardial patches | In vitro (human smooth muscle cells) and in vivo (male Wistar rats) | Sustained rapamycin delivery and subsequently, less neointimal hyperplasia, less smooth muscle cells proliferation and lower infiltrating cells and simultaneously, maintaining endothelization | [242] |
| Rapamycin | PEGylated liposomes | - | Cellular senescence | CD9 monoclonal antibody | In vitro (CD9 receptor-overexpressing cells) | Promotion of cell proliferation and reduction in the number of cells that express the senescence-associated-galactosidase, showing higher anti-senescence activity of CD9-targeted liposome compared to the free rapamycin and conventional liposomes | [243] |
| Rapamycin | PEO/PDLLA nanofibers | - | Glioblastoma | - | In vitro (U251 and U87 human glioblastoma cell lines) | Local sustained delivery of rapamycin, showing potential targeted delivery systems for glioblastoma treatment | [244] |
| Rapamycin | Polymeric NPs | Piperine | Breast cancer | - | In vitro (breast cancer cells) | Increased cellular uptake and bioavailability of rapamycin as well as decreased viability of cancer cells | [245] |
| Rapamycin | Lipid-polyaniline NPs | 1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide (DiR) | Cancer | - | In vitro (HeLa cells) and in vivo (HeLa tumor bearing mice) | High antiangiogenic effect and great cytotoxicity as well as decreased tumor growth | [246] |
| Rapamycin | Polymeric Micelle | Paclitaxel and 17-allylamino-17-demethoxygeldaramycin (17-AAG) | Cancer | - | In vitro (A549 cells) | Inhibition of A549 tumor growth, increased cytotoxicity and enhanced radiosensitizing effect | [247] |
| Rapamycin | poly(ethylene glycol)-shelled NPs | - | Aortic Aneurysm | - | In vivo (experimental aortic aneurysm in rat) | Remarkable inhibition of activities of matrix metalloproteinase and expression of inflammatory cytokines, showing their potential in targeting aortic aneurysm | [248] |
| Rapamycin | Acetalated b-CD (Ac-bCDs)-based NPs | - | Atherosclerosis | - | In vitro (smooth muscle cells) and in vivo (apoliproprotein E-deficient (ApoE) mice) | Decreased formation of atherosclerotic lesions, increased stability of plaques, decreased level of pro-inflammatory factors and suppression of mTORC1 | [249] |
| Rapamycin | Immunoliposomes | - | Breast cancer | Trastuzumab | In vitro (triple negative MDA-MB-231 and SKBR3cell lines) | High cytotoxicity against breast cancer cells | [250] |
| Rapamycin | Thermal sensitive liposomes | Indocyanine Green | - | - | In vitro (HeLa and HUVEC cells) and in vivo (HeLa cell bearing mice) | Great drug accumulation and cytotoxicity in vitro experiment and inhibited tumor growth in vivo with minimal side effects | [251] |
| Rapamycin | PLGA particles | Isoniazid and rifabutin | - | - | In vivo (infected mice) and in vitro (THP-1 human monocytes) | Stimulating more autophagy in infected macrophages, decreased bacterial burden in lung and spleen and inducing phagosome-lysosome fusion | [252] |
| Rapamycin | Liposomes | Paclitaxel | Breast cancer | - | In vitro (cancer 4T1 breast cancer cell line) and in vivo (4T1-tumor bearing mice) | Higher cytotoxicity against 4T1 cells and decreased tumor growth and viability in mice | [253] |
| Rapamycin | Human serum albumin NPs | Split luciferase reporter | Combined image guided monitoring the pharmacokinetics | [254] | |||
| Rapamycin | Solid Lipid NPs | - | - | - | In vitro (SH-SY5Y neuroblastoma cells) | High cellular uptake, sustained release and higher mTORC1 inhibition | [255] |
| Rapamycin | Biodegradable intraocular device | - | - | - | In vivo (New Zeland white rabbits) | Prolonged release, good stability and good ocular compatibility | [256] |
| Rapamycin | Lipid SAINT-O-Somes | - | - | Anti-VCAM-1 | In vitro (ABN12 and MPC-5 cell lines) | Remarkable inhibition of AB8/cell migration in targeted nanocarriers | [257] |
| Rapamycin | Nanoemulsions | - | - | - | In vitro (SKBR3 and Caco-2 cell lines) | Great cytotoxicity again SKBR-3 cell and good uptake by Caco-2 cells | [258] |
| Rapamycin | Microsphere | - | Kidney disease | - | In vivo (rat model of renal ischemic/reperfusion injury) | Lack of adverse effects, decreased macrophage infiltration and lower amount of myofibroblasts in kidney | [259] |
| Rapamycin | magnetic Fe3O4/carboxymethylchitosan NPs | - | Cancer | - | In vitro (liver cell line LO2 and human hepatocarcinoma cell line HepG2) | Sustained release of rapamycin, higher cytotoxicity against LO2 and HepG2 cells, increased cellular uptake and decreased damage to normal cells | [260] |
| Rapamycin | Nanoliposomal | CPT-11 | Brain tumor | - | In vivo (rodent orthotopic brain tumor xenografts) | Significant efficacy in increasing survival with minimal side effects | [261] |
| Rapamycin | Liposome | - | - | - | In vitro | High rapamycin encapsulation rate, good reproducibility and sustained release | [262] |
| Rapamycin | Aerosol treatment | 3-bromopyruvate | Lung cancer | - | In vitro (human non-small cell lung cancer (NSCLC)) and in vivo (mice with lung cancer0 | Remarkable inhibition of cell proliferation, decreased glycolytic activity, resulting in antitumor effect | [263] |
| Rapamycin | Porous silicon microparticles | - | - | - | In vivo (rabbit) | Great rapamycin loading, increased bioavailability and simultaneously, maintaining clear optical media and normal histology of retina | [264] |
| Rapamycin | Subcapsular microspheres | - | Chronic kidney disease | - | In vivo (ureter-obstructed rats) | Decreased intrarenal mTOR activity, over-expression of fibrotic genes, myofibroblast accumulation and T-lymphocyte infiltration and subsequently, successful inhibition of local fibrotic response | [265] |
| Everolimus | Chitosan NPs | - | Bronchiolitis obliterans syndrome (BOS) | - | In vitro (CD44-overexpressing mesenchymal cells) | Great properties in terms of average size (≤200 nm), good zeta potential (-30.9mV) and sustained release behavior as well as good uptake by mesenchymal cells | [266] |
| Everolimus | Polymeric NPs | Paclitaxel | Breast cancer | - | In vitro (MCF-7 and SKBR3 cells) | Synergistic effect on inhibiting the growth and decreasing viability of breast cancer cells | [267] |
| Everolimus | mPEGhexPLA nanocarriers | - | Autoimmune uveoretinitis (EAU) | - | In vivo (B10,RIII mice) | Significant decrease in EAU severity in both eyes and decreased secretion of IL-10 and CD4+ CD25+ FoxP3+ | [268] |
| 3-methyladenine | Metal-organic framework NPs | - | Cancer | - | In vitro (HeLa cells) and in vivo (nude mice) | Significant inhibition of autophagosome formation in HeLa cells and higher anti-tumor activity and inhibition of Beclin-1 and LC3 in mice | [269] |
| Cholorquine diphosphate | Metal-organic framework NPs | - | Cancer | Methoxy poly (ethylene glycol)-folate (FA-PEG) | In vitro (HeLa cells) | Remarkable inhibition of autophagosome formation and autophagy flux | [270] |
| Dactolisib | PLGA-PEG NPs | - | - | Anti-human E-selectin antibody | In vitro (TNF-activated endothelial cells) | High cellular uptake and great anti-inflammatory effects | [271] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tavakol, S.; Ashrafizadeh, M.; Deng, S.; Azarian, M.; Abdoli, A.; Motavaf, M.; Poormoghadam, D.; Khanbabaei, H.; Ghasemipour Afshar, E.; Mandegary, A.; et al. Autophagy Modulators: Mechanistic Aspects and Drug Delivery Systems. Biomolecules 2019, 9, 530. https://doi.org/10.3390/biom9100530
Tavakol S, Ashrafizadeh M, Deng S, Azarian M, Abdoli A, Motavaf M, Poormoghadam D, Khanbabaei H, Ghasemipour Afshar E, Mandegary A, et al. Autophagy Modulators: Mechanistic Aspects and Drug Delivery Systems. Biomolecules. 2019; 9(10):530. https://doi.org/10.3390/biom9100530
Chicago/Turabian StyleTavakol, Shima, Milad Ashrafizadeh, Shuo Deng, Maryam Azarian, Asghar Abdoli, Mahsa Motavaf, Delaram Poormoghadam, Hashem Khanbabaei, Elham Ghasemipour Afshar, Ali Mandegary, and et al. 2019. "Autophagy Modulators: Mechanistic Aspects and Drug Delivery Systems" Biomolecules 9, no. 10: 530. https://doi.org/10.3390/biom9100530
APA StyleTavakol, S., Ashrafizadeh, M., Deng, S., Azarian, M., Abdoli, A., Motavaf, M., Poormoghadam, D., Khanbabaei, H., Ghasemipour Afshar, E., Mandegary, A., Pardakhty, A., Yap, C. T., Mohammadinejad, R., & Kumar, A. P. (2019). Autophagy Modulators: Mechanistic Aspects and Drug Delivery Systems. Biomolecules, 9(10), 530. https://doi.org/10.3390/biom9100530