Epibatidine: A Promising Natural Alkaloid in Health

 ,
, 
 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Pharmacokinetics of Epibatidine and Its Synthetic Derivatives
3. Role of Epibatidine in Health
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Spande, T.F.; Garraffo, H.M.; Yeh, H.J.; Pu, Q.L.; Pannell, L.K.; Daly, J.W. A new class of alkaloids from a dendrobatid poison frog: A structure for alkaloid 251f. J. Nat. Prod. 1992, 55, 707. [Google Scholar] [CrossRef] [PubMed]
- Spande, T.F.; Garraffo, H.M.; Edwards, M.W.; Yeh, H.J.C.; Pannell, L.; Daly, J.W. Epibatidine: A novel (chloropyridyl)azabicycloheptane with potent analgesic activity from an Ecuadoran poison frog. J. Am. Chem. Soc. 1992, 3475. [Google Scholar] [CrossRef]
- Posadas, I.; Lopez-Hernandez, B.; Cena, V. Nicotinic receptors in neurodegeneration. Curr. Neuropharmacol. 2013, 11, 298–314. [Google Scholar] [CrossRef] [PubMed]
- Boyle, J. Molecular biology of the cell, by b. Alberts, a. Johnson, j. Lewis, m. Raff, k. Roberts, and p. Walter. Biochem. Mol. Biol. Educ. 2008, 36, 317–318. [Google Scholar] [CrossRef]
- Lloyd, G.K.; Williams, M. Neuronal nicotinic acetylcholine receptors as novel drug targets. J. Pharmacol. Exp. Ther. 2000, 292, 461–467. [Google Scholar] [PubMed]
- Qian, C.; Li, T.; Shen, T.Y.; Libertine-Garahan, L.; Eckman, J.; Biftu, T. Epibatidine is a nicotinic analgesic. Eur. J. Pharmacol. 1993, 250, 13–14. [Google Scholar] [CrossRef]
- Sullivan, J.P.; Decker, M.W.; Brioni, J.D.; Donnelly-Roberts, D.; Anderson, D.J.; Bannon, A.W.; Kang, C.H.; Adams, P.; Piattoni-Kaplan, M.; Buckley, M.J. (+/−)-epibatidine elicits a diversity of in vitro and in vivo effects mediated by nicotinic acetylcholine receptors. J. Pharmacol. Exp. Ther. 1994, 271, 624–631. [Google Scholar] [PubMed]
- Thompson, A.J.; Metzger, S.; Lochner, M.; Ruepp, M.-D. The binding orientation of epibatidine at α7 nach receptors. Neuropharmacology 2017, 116, 421–428. [Google Scholar] [CrossRef]
- Baranowska, U.; Wiśniewska, R.J. The α7-nach nicotinic receptor and its role in memory and selected diseases of the central nervous system. Postepy. Hig. Med. Dosw. 2017, 30, 633–648. [Google Scholar] [CrossRef]
- Donnelly-Roberts, D.L.; Puttfarcken, P.S.; Kuntzweiler, T.A.; Briggs, C.A.; Anderson, D.J.; Campbell, J.E.; Piattoni-Kaplan, M.; McKenna, D.G.; Wasicak, J.T.; Holladay, M.W.; et al. Abt-594 [(r)-5-(2-azetidinylmethoxy)-2-chloropyridine]: A novel, orally effective analgesic acting via neuronal nicotinic acetylcholine receptors: I. In vitro characterization. J. Pharmacol. Exp. Ther. 1998, 285, 777–786. [Google Scholar]
- Shimizu, T.; Tanaka, K.; Hasegawa, T.; Yokotani, K. Brain α4β2 nicotinic acetylcholine receptors are involved in the secretion of noradrenaline and adrenaline from adrenal medulla in rats. Eur. J. Pharmacol. 2011, 654, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Green, B.T.; Lee, S.T.; Keele, J.W.; Welch, K.D.; Cook, D.; Pfister, J.A.; Kem, W.R. Complete inhibition of fetal movement in the day 40 pregnant goat model by the piperidine alkaloid anabasine but not related alkaloids. Toxicon 2018, 144, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Traynor, J.R. Epibatidine and pain. Br. J. Anaesth. 1998, 81, 69–76. [Google Scholar] [CrossRef] [PubMed]
- London, E.D.; Scheffel, U.; Kimes, A.S.; Kellar, K.J. In vivo labeling of nicotinic acetylcholine receptors in brain with [3h] epibatidine. Eur. J. Pharmacol. 1995, 278, R1–R2. [Google Scholar] [CrossRef]
- Javors, M.A.; Sanchez, J.J.; King, T.S.; Rohde, A.R.; Wilson, S.G.; Flores, C.M. Extraction and quantification of epibatidine in plasma. J. Chromatogr. B Biomed. Sci. Appl. 2001, 755, 379–382. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Ogawa, T.; Suzuki, T. Simultaneous quantification of batrachotoxin and epibatidine in plasma by ultra-performance liquid chromatography/tandem mass spectrometry. Legal. Med. 2017, 25, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Watt, A.P.; Hitzel, L.; Morrison, D.; Locker, K.L. Determination of the in vitro metabolism of (+)- and (−)-epibatidine. J. Chromatogr. A 2000, 896, 229–238. [Google Scholar] [CrossRef]
- Heugebaert, T.S.A.; Van Overtveldt, M.; De Blieck, A. Synthesis of 1-substituted epibatidine analogues and their in vitro and in vivo evaluation as a4ß2 nicotinic acetylcholine receptor ligands. RSC Adv. 2014, 4, 2226–2234. [Google Scholar] [CrossRef]
- Patt, M.; Becker, G.A.; Grossmann, U. Evaluation of metabolism, plasma protein binding and other biological parameters after administration of (−)-[18f]flubatine in humans. Nucl. Med. Biol. 2014, 41, 489–494. [Google Scholar] [CrossRef]
- Ludwig, F.; Fischer, S.; Smits, R. Exploring the metabolism of (+)-[18f] flubatine in vitro and in vivo: Lc-ms/ms aided identification of radiometabolites in a clinical pet study. Molecules 2018, 23, 464. [Google Scholar] [CrossRef]
- Bunnelle, W.H.; MJ Dart, M.; Schrimpf, M.R. Design of ligands for the nicotinic acetylcholine receptors: The quest for selectivity. Curr. Top. Med. Chem. 2004, 4, 299–334. [Google Scholar] [CrossRef] [PubMed]
- Yogeeswari, P.; Sriram, D.; Bal, T.R.; Thirumurugan, R. Epibatidine and its analogues as nicotinic acetylcholine receptor agonist: An update. Nat. Prod. Res. 2006, 20, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Carroll, F.I. Epibatidine structure-activity relationships. Bioorganic Med. Chem. Lett. 2004, 14, 5713. [Google Scholar]
- Seerden, J.G.; Tulp, M.T.; Scheeren, H.W.; Kruse, C.G. Synthesis and structure-activity data of some new epibatidine analogues. Bioorganic Med. Chem. Lett. 1998, 6, 2103–2110. [Google Scholar] [CrossRef]
- Badio, B.; Garraffo, H.M.; Plummer, C.V.; Padgett, W.L.; Daly, J.W. Synthesis and nicotinic activity of epiboxidine: An isoxazole analogue of epibatidine. Eur. J. Pharmacol. 1997, 321, 189–194. [Google Scholar] [CrossRef]
- Rizzi, L.; Dallanoce, C.; Matera, C.; Magrone, P.; Pucci, L.; Gotti, C.; Clementi, F.; De Amici, M. Epiboxidine and novel-related analogues: A convenient synthetic approach and estimation of their affinity at neuronal nicotinic acetylcholine receptor subtypes. Bioorg. Med. Chem. Lett. 2008, 18, 4651–4654. [Google Scholar] [CrossRef] [PubMed]
- Holladay, M.W.; Wasicak, J.T.; Lin, N.-H.; He, Y.; Ryther, K.B.; Bannon, A.W.; Buckley, M.J.; Kim, D.J.; Decker, M.W.; Anderson, D.J. Identification and initial structure—Activity relationships of (r)-5-(2-azetidinylmethoxy)-2-chloropyridine (abt-594), a potent, orally active, non-opiate analgesic agent acting via neuronal nicotinic acetylcholine receptors. J. Med. Chem. 1998, 41, 407–412. [Google Scholar] [CrossRef]
- Meyer, M.D.; Anderson, D.J.; Campbell, J.E.; Carroll, S.; Marsh, K.C.; Rodrigues, A.D.; Decker, M.W. Preclinical pharmacology of abt-594: A nicotinic acetylcholine receptor agonist for the treatment of pain. CNS Drug Rev. 2000, 6, 183–194. [Google Scholar] [CrossRef]
- Rowbotham, M.C.; Duan, W.R.; Thomas, J.; Nothaft, W.; Backonja, M.-M. A randomized, double-blind, placebo-controlled trial evaluating the efficacy and safety of abt-594 in patients with diabetic peripheral neuropathic pain. Pain 2009, 146, 245–252. [Google Scholar] [CrossRef]
- Boyce, S.; Webb, J.K.; Shepheard, S.L.; Russell, M.G.; Hill, R.G.; Rupniak, N.M. Analgesic and toxic effects of abt-594 resemble epibatidine and nicotine in rats. Pain 2000, 85, 443–450. [Google Scholar] [CrossRef]
- Bannon, A.W.; Decker, M.W.; Curzon, P.; Buckley, M.J.; Kim, D.J.; Radek, R.J.; Lynch, J.K.; Wasicak, J.T.; Lin, N.-H.; Arnold, W.H. Abt-594 [(r)-5-(2-azetidinylmethoxy)-2-chloropyridine]: A novel, orally effective antinociceptive agent acting vianeuronal nicotinic acetylcholine receptors: Ii. In vivocharacterization. J. Pharmacol. Exp. Ther. 1998, 285, 787–794. [Google Scholar] [PubMed]
- Munro, G.; Dyhr, H.; Grunnet, M. Selective potentiation of gabapentin-mediated antinociception in the rat formalin test by the nicotinic acetylcholine receptor agonist abt-594. Neuropharmacology 2010, 59, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.P. Mechanisms of action of gabapentin. Rev. Neurol. 1997, 153 (Suppl. 1), S39–S45. [Google Scholar] [PubMed]
- Bitner, R.S.; Nikkel, A.L.; Curzon, P.; Arneric, S.P.; Bannon, A.W.; Decker, M.W. Role of the nucleus raphe magnus in antinociception produced by abt-594: Immediate early gene responses possibly linked to neuronal nicotinic acetylcholine receptors on serotonergic neurons. J. Neurosci. 1998, 18, 5426–5432. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Katsuyama, S.; Orito, T.; Suzuki, T.; Sakurada, S. Antinociceptive effect of tebanicline for various noxious stimuli-induced behaviours in mice. Neurosci. Lett. 2017, 638, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Hone, A.J.; McIntosh, J.M. Nicotinic acetylcholine receptors in neuropathic and inflammatory pain. FEBS Lett. 2018, 592, 1045–1062. [Google Scholar] [CrossRef] [PubMed]
- Sihver, W.; Nordberg, A.; Långström, B.; Mukhin, A.G.; Koren, A.O.; Kimes, A.S.; London, E.D. Development of ligands for in vivo imaging of cerebral nicotinic receptors. Behav. Brain Res. 2000, 113, 143–157. [Google Scholar] [CrossRef]
- Sihver, W.; Långström, B.; Nordberg, A. Ligands for in vivo imaging of nicotinic receptor subtypes in alzheimer brain. Acta Neurol. Scand. 2000, 102, 27–33. [Google Scholar] [CrossRef]
- Belluardo, N.; Mudò, G.; Blum, M.; Cheng, Q.; Caniglia, G.; Dell’Albani, P.; Fuxe, K. The nicotinic acetylcholine receptor agonist (±)-epibatidine increases fgf-2 mrna and protein levels in the rat brain. Mol. Brain Res. 1999, 74, 98–110. [Google Scholar] [CrossRef]
- Egea, J.; Rosa, A.O.; Cuadrado, A.; García, A.G.; López, M.G. Nicotinic receptor activation by epibatidine induces heme oxygenase-1 and protects chromaffin cells against oxidative stress. J. Neurochem. 2007, 102, 1842–1852. [Google Scholar] [CrossRef]
- Potter, A.; Corwin, J.; Lang, J.; Piasecki, M.; Lenox, R.; Newhouse, P.A. Acute effects of the selective cholinergic channel activator (nicotinic agonist) abt-418 in alzheimer’s disease. Psychopharmacology 1999, 142, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Wilens, T.E.; Biederman, J.; Spencer, T.J.; Bostic, J.; Prince, J.; Monuteaux, M.C.; Soriano, J.; Fine, C.; Abrams, A.; Rater, M. A pilot controlled clinical trial of abt-418, a cholinergic agonist, in the treatment of adults with attention deficit hyperactivity disorder. Am. J. Psychiatry 1999, 156, 1931–1937. [Google Scholar] [PubMed]
- Perry, D.C.; Kellar, K.J. [3h] epibatidine labels nicotinic receptors in rat brain: An autoradiographic study. J. Pharmacol. Exp. Ther. 1995, 275, 1030–1034. [Google Scholar] [PubMed]



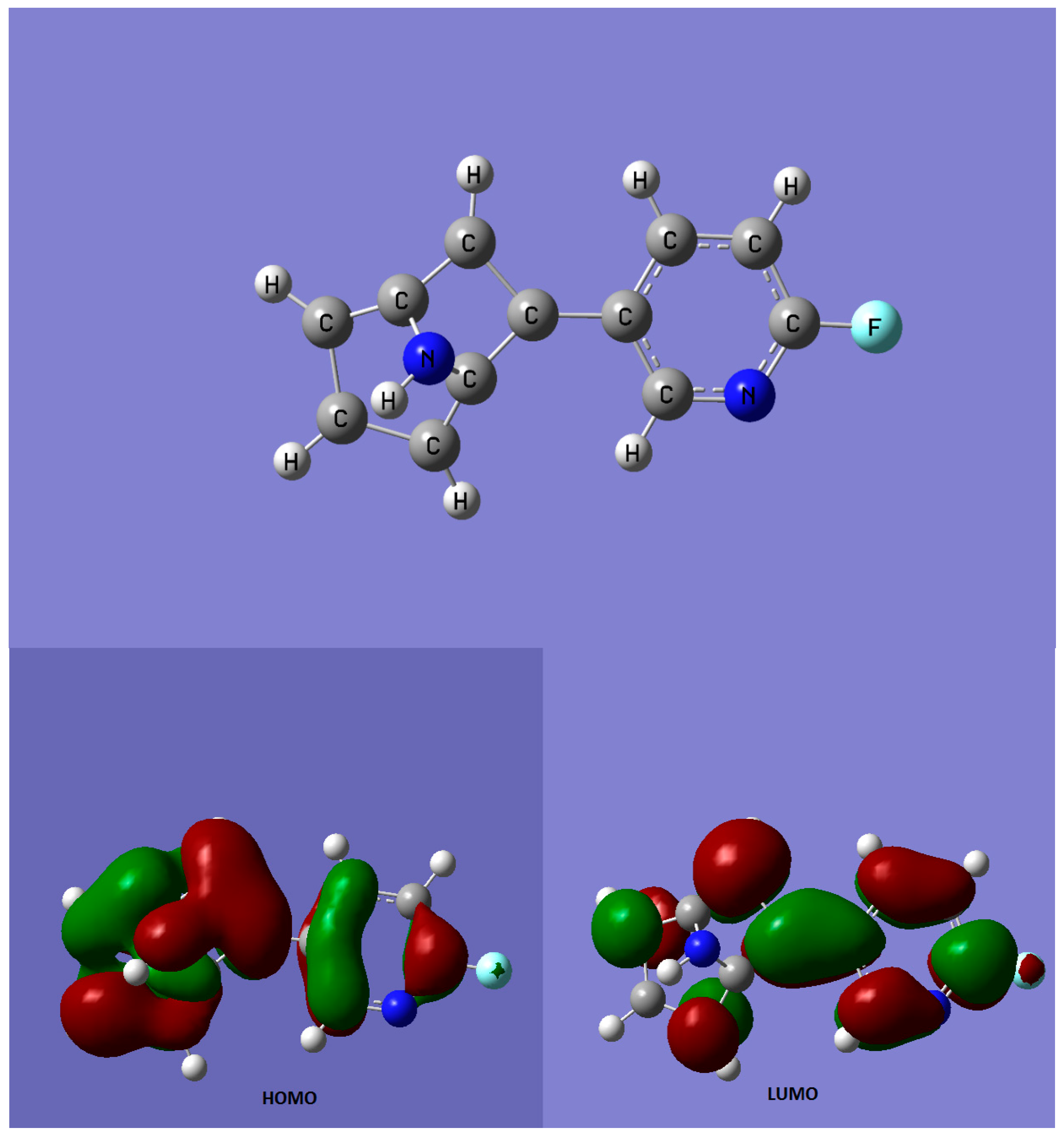{kind=link}
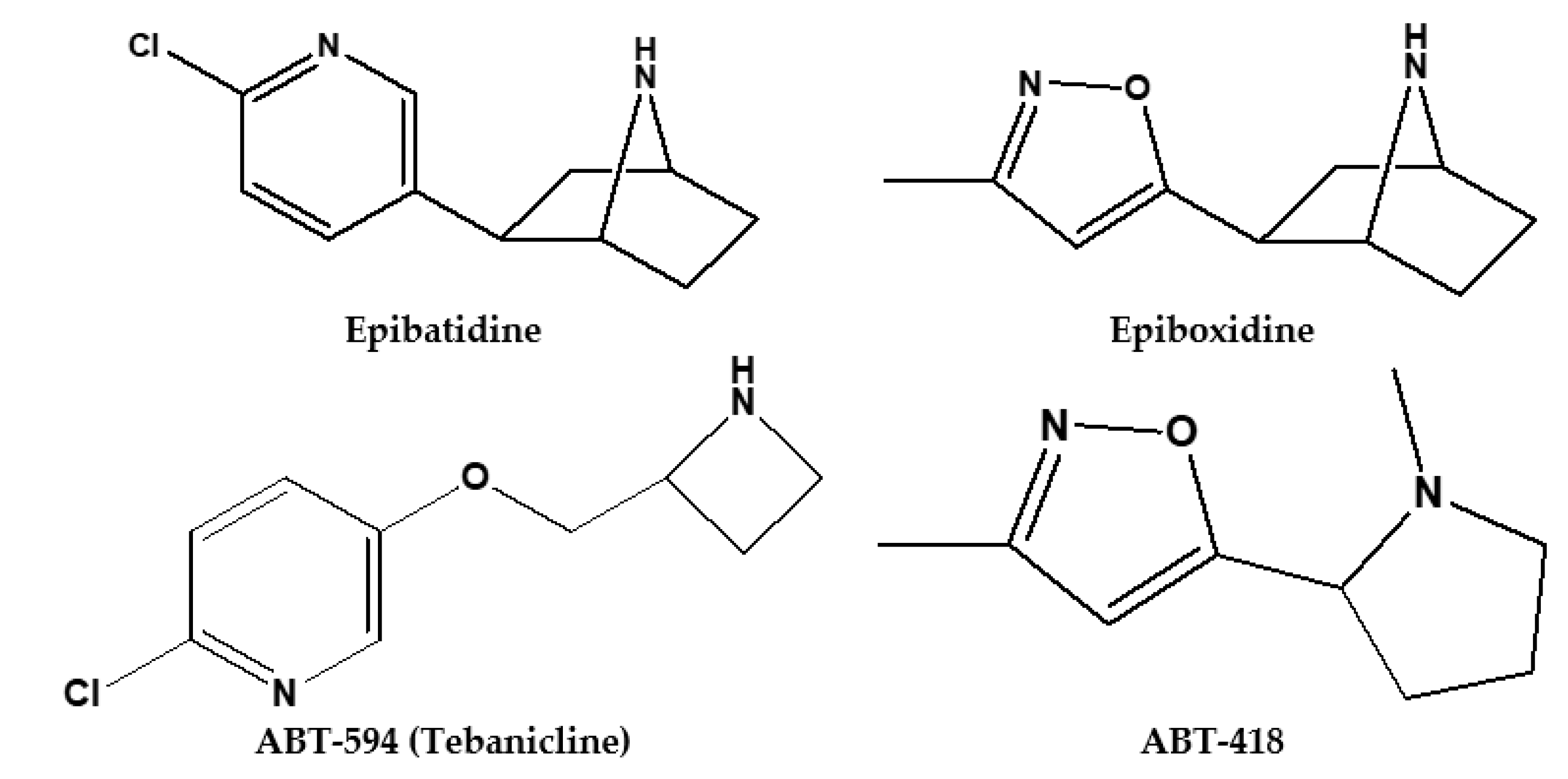{kind=link}
| Compound | Type of Study | References | |
|---|---|---|---|
| In Vivo Studies Using Animal Models | |||
| Epibatidine | Animal model | Primary outcomes | |
| Mice and rats | Antinociceptive | [6] | |
| Rats | Increased adrenaline and noradrenaline neuromediators | [12] | |
| Pregnant goats | Lack of completely inhibition of fetal movement | [11] | |
| Rats | Neuroprotective | [39] | |
| ABT-594 tebanicline | Rodent pain models (rats, mice) | Antinociceptive | [27,28,30,31,35] |
| Rat formalin test | Analgesic (multimodal analgesia) | [32] | |
| Epiboxidine | Rats | Cognitive disfunction treatment | [26] |
| Mice | Antinociceptive | ||
| In vitro studies using cell lines | |||
| Epibatidine | Bovine chromaffin cells | Antioxidant, antiapoptotic | [40] |
| Human clinical studies | |||
| ABT-594tebanicline | Study design | Primary outcomes | [29] |
| Randomized, multicenter, double-blind, placebo-controlled study (phase 2) | Analgesic in diabetic patients with neuropathic pain | ||
| ABT-418Epiboxidine | Double-blind, placebo-controlled study | Cognitive enhancement in moderate Alzheimer’s disease | [41] |
| Double-blind, randomized, placebo-controlled, crossover trial | Increased attention in deficit hyperactivity disorder (ADHD) in adults | [42] | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salehi, B.; Sestito, S.; Rapposelli, S.; Peron, G.; Calina, D.; Sharifi-Rad, M.; Sharopov, F.; Martins, N.; Sharifi-Rad, J. Epibatidine: A Promising Natural Alkaloid in Health. Biomolecules 2019, 9, 6. https://doi.org/10.3390/biom9010006
Salehi B, Sestito S, Rapposelli S, Peron G, Calina D, Sharifi-Rad M, Sharopov F, Martins N, Sharifi-Rad J. Epibatidine: A Promising Natural Alkaloid in Health. Biomolecules. 2019; 9(1):6. https://doi.org/10.3390/biom9010006
Chicago/Turabian StyleSalehi, Bahare, Simona Sestito, Simona Rapposelli, Gregorio Peron, Daniela Calina, Mehdi Sharifi-Rad, Farukh Sharopov, Natália Martins, and Javad Sharifi-Rad. 2019. "Epibatidine: A Promising Natural Alkaloid in Health" Biomolecules 9, no. 1: 6. https://doi.org/10.3390/biom9010006
APA StyleSalehi, B., Sestito, S., Rapposelli, S., Peron, G., Calina, D., Sharifi-Rad, M., Sharopov, F., Martins, N., & Sharifi-Rad, J. (2019). Epibatidine: A Promising Natural Alkaloid in Health. Biomolecules, 9(1), 6. https://doi.org/10.3390/biom9010006