Crosstalk between PPARγ Ligands and Inflammatory-Related Pathways in Natural T-Regulatory Cells from Type 1 Diabetes Mouse Model


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Section
2.1. Mice
2.2. Antibodies and Reagents
2.3. Flow Cytometry Analysis
2.4. Total RNA Isolation, cDNA Synthesis and Real-Time PCR for the Detection of PPARγ and Foxp3
2.5. PPARγ-PPRE Binding Activity
2.6. Signaling Pathways Modulation by PCR Array
2.7. Statistical Analyses
3. Results
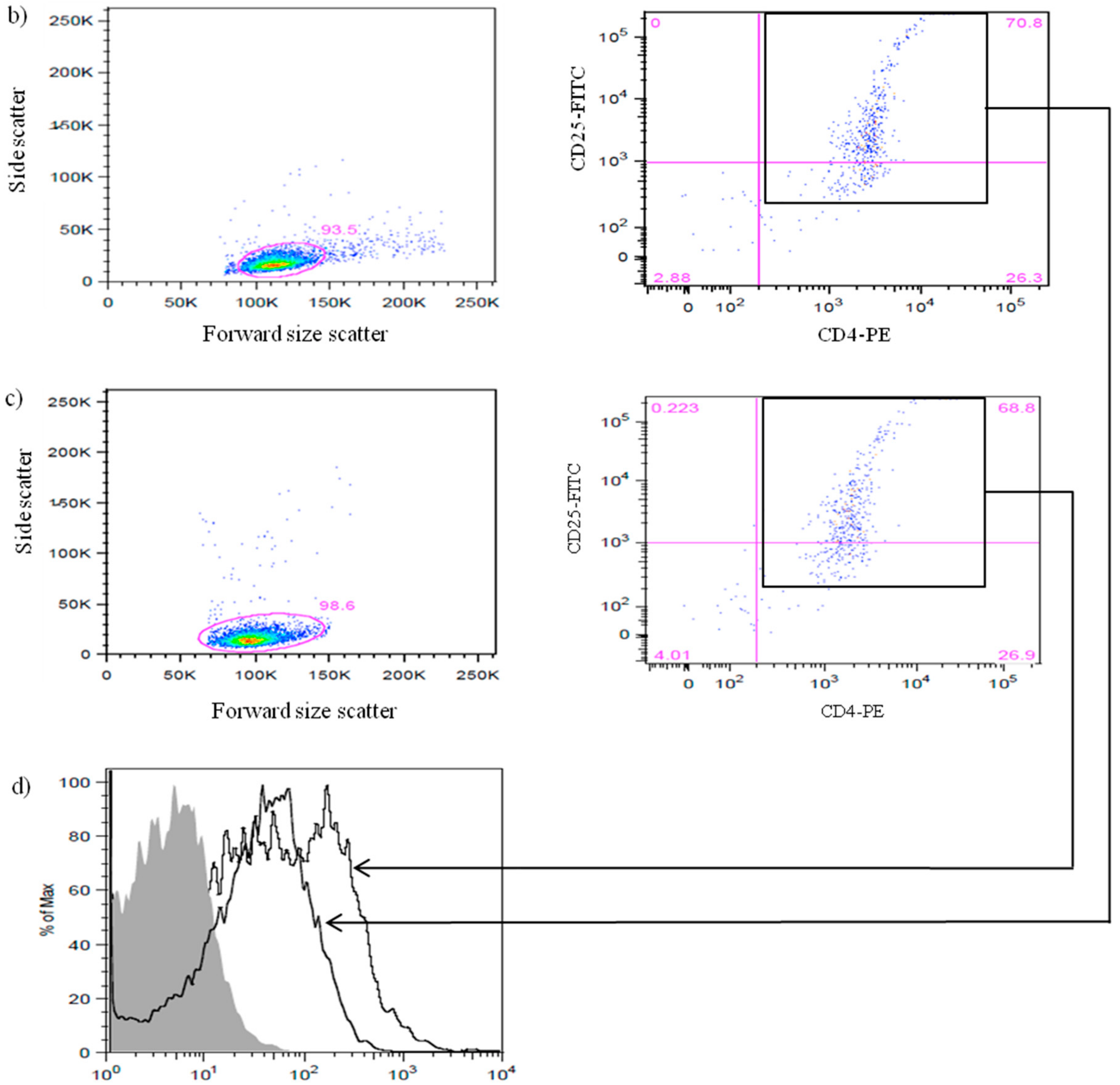
3.1. Efficiency of CD4+CD25+Foxp3+ nTreg Cells Isolation from NOD and NOR Mice
3.2. Expression of Foxp3 in nTreg Cells of NOD and NOR Mice
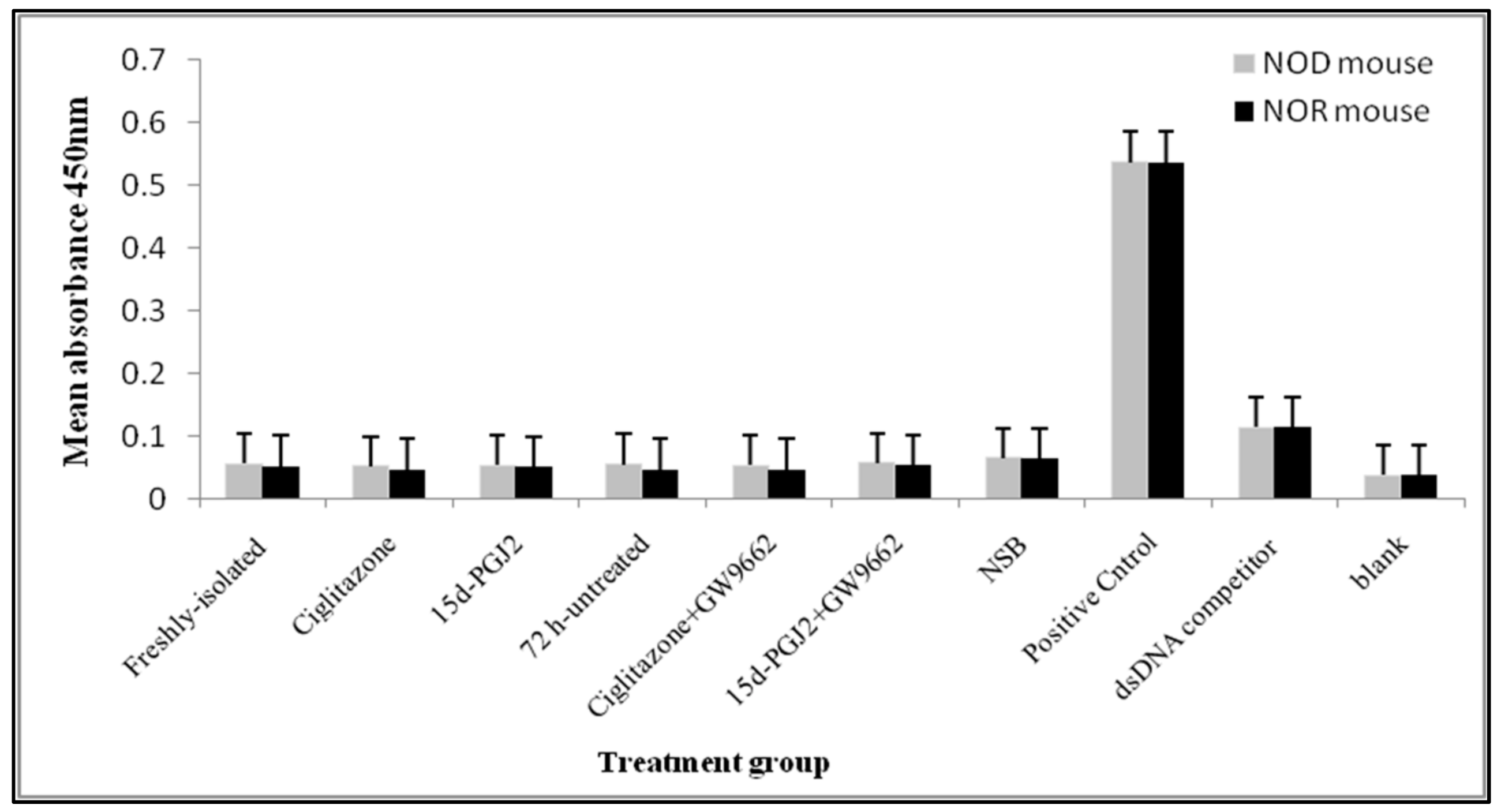
3.3. Binding Activity between PPARγ and PPRE in nTreg Cells of NOD and NOR Mice
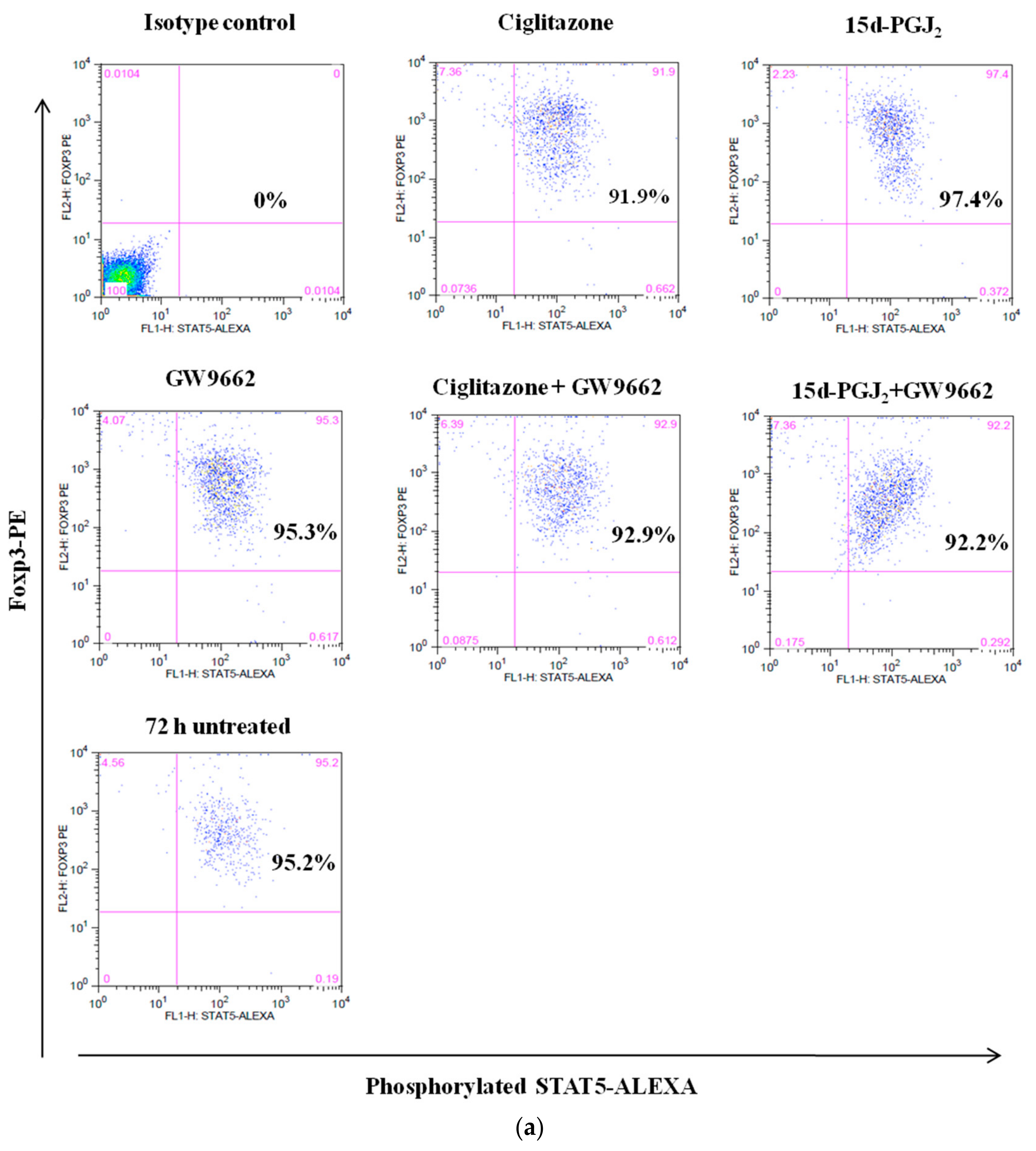
3.4. Phosphorylation Levels of ZAP-70 & STAT-5 Transduction Proteins
3.5. Differential Gene Expression Involved in Signal Transduction Pathways in nTreg Cells of NOD and NOR Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bettini, M.L.; Vignali, D.A.A. Development of thymically derived natural regulatory T cells. Ann. N. Y. Acad. Sci. 2010, 1183, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programmes the development and function of CD4+CD25+ Regulatory T-Cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Fontenot, J.D.; Rasmussen, J.P.; Williams, L.M.; Dooley, J.L.; Farr, A.G.; Rudensky, A.Y. Regulatory T cell lineage specification by the Forkhead Transcription Factor Foxp3. Immunity 2005, 22, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Pascual, G.; Glass, C.K. Peroxisome Proliferator-Activated Receptor γ-dependent repression of the inducible nitric oxide synthase gene. Mol. Cell. Biol. 2000, 20, 4699–4707. [Google Scholar] [CrossRef] [PubMed]
- Rotondo, D.; Davidson, J. Prostaglandin and PPAR control of immune cell function. Immunology 2002, 105, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Von Knethen, A.; Soller, M.; Tzieply, N.; Weigert, A.; Johann, A.M.; Jennewein, C.; Kohl, R.; Brune, B. PPARγ1 attenuates cytosol to membrane translocation of PKCα to desensitize monocytes/macrophages. J. Cell. Biol. 2007, 176, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Sauter, M.; Kastenmuller, K.; Belling, F.; Wornle, M.; Ladurner, R.; Mussack, T.; Sitte, T. Activation of Peroxisome Proliferator-Activated Receptor-Gamma by glitazones reduces the expression and release of monocyte chemoattractant protein-1 in human mesothelial cells. Mediat. Inflamm. 2012, 2012, 2176960. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.B.; Bishop, B.D.; Estrada, H.D.; Hla, T.; Puddington, L.; Padula, S.J. The nuclear receptor PPARγ and immunoregulation: PPARγ mediates inhibition of helper T cell responses. J. Immunol. 2000, 164, 1364–1371. [Google Scholar] [CrossRef] [PubMed]
- Hammad, H.; de Heer, H.J.; Soullie, T.; Angeli, V.; Trottein, F.; Hoogsteden, H.C.; Lambrecht, B.N. Activation of Peroxisome Proliferator-Activated Receptor-γ in Dendritic Cells inhibits the development of eosinophilic airway inflammation in a mouse model of asthma. Am. J. Pathol. 2004, 164, 263–271. [Google Scholar] [CrossRef]
- Hontecillas, R.; Horne, W.T.; Climent, M.; Guri, A.J.; Evans, C.; Zhang, Y.; Sobral, B.W.; Bassaganya-Riera, J. Immunoregulatory mechanisms of macrophage PPAR-γ in mice with experimental inflammatory bowel disease. Mucosal Immunol. 2011, 4, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.; Schmidt, M.; Giese, T.; Sastre, M.; Knolle, P.; Klockgether, T.; Heneka, M.T. Proinflammatory stimulation and pioglitazone treatment regulate Peroxisome Proliferator-Activated Receptor γ levels in peripheral blood mononuclear cells from healthy controls and multiple sclerosis patients. J. Immunol. 2005, 175, 4948–4955. [Google Scholar] [CrossRef] [PubMed]
- Racke, M.K.; Gocke, A.R.; Muir, M.; Diab, A.; Drew, P.D.; Lovett-Racke, A.E. Nuclear Receptors and Autoimmune disease: The Potential of PPAR Agonists to Treat Multiple Sclerosis. J. Nutr. 2006, 136, 700–703. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M. PPARα/γ independent Effects of PPARα/γ Ligands on Cysteinyl Leukotriene production in mast cells. PPAR Res. 2008, 2008, 293538. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhu, Z.; Zhu, B.; Ma, Z. Peroxisome Proliferator-Activated Receptor–γ Agonist Induces Regulatory T Cells in a Murine Model of Allergic Rhinitis. Otolaryngol. Head Neck Surg. 2011, 144, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Nencioni, A.; Lauber, K.; Grunebach, F.; Van Parijs, L.; Denzlinger, C.; Wesselborg, S.; Brossart, P. Cyclopentenone prostaglandins induce lymphocyte apoptosis by activating the mitochondrial apoptosis pathway independent of external death receptor signaling. J. Immunol. 2003, 171, 5148–5156. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.-H.; Park, S.-M.; Lee, Y.H.; Cha, J.Y.; Lee, J.-Y.; Shin, E.K.; Park, J.-S.; Park, B.-L.; Shin, H.D.; Park, C.-S. Association of peroxisome proliferator-activated receptor-gamma gene polymorphisms with the development of asthma. Resp. Med. 2009, 103, 1020–1024. [Google Scholar] [CrossRef] [PubMed]
- IJpenberg, A.; Jeannin, E.; Wahli, W.; Desvergne, B. Polarity and specific sequence requirements of Peroxisome Proliferator-activated Receptor (PPAR)/retinoid X receptor heterodimer binding to DNA: A functional analysis of the malic enzyme gene PPAR response element. J. Biol. Chem. 1997, 272, 20108–20117. [Google Scholar] [CrossRef] [PubMed]
- Gilmour, K.C.; Pine, R.; Reich, N. Interleukin 2 activates STAT5 transcription factor (mammary gland factor) and specific gene expression in T lymphocytes. Proc. Natl. Acad. Sci. USA 1995, 92, 10772–10776. [Google Scholar] [CrossRef] [PubMed]
- Burchill, M.A.; Goetz, C.A.; Prylic, M.; O’Neil, J.J.; Harmon, I.R.; Bensinger, S.J.; Turk, L.A.; Brennan, P.; Jameson, S.C.; Farrar, M.A. Distinct effects of STAT5 activation on CD4+ and CD8+ T cell homeostasis: Development of CD4+CD25+ Regulatory T cells versus CD8+ Memory T cells. J. Immunol. 2003, 171, 5853–5864. [Google Scholar] [CrossRef] [PubMed]
- Passerini, L.; Allan, S.E.; Battaglia, M.; Di Nunzio, S.; Alstad, A.N.; Levings, M.K.; Roncarolo, M.G.; Bacchetta, R. STAT5-signaling cytokines regulate the expression of FOXP3 in CD4+CD25+ regulatory T cells and CD4+CD25− effector T cells. Int. Immunol. 2008, 20, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, D.L.; Galea, E.; Gavrilyuk, V.; Brosnan, C.F.; Whitacre, C.C.; Dumitrescu-Ozimek, L.; Landreth, G.E.; Pershadsingh, H.A.; Weinberg, G.; Heneka, M.T. Peroxisome proliferator-activated receptor-γ agonists prevent experimental autoimmune encephalomyelitis. Ann. Neurol. 2002, 51, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.S.; Ricote, M.; Akiyama, T.E.; Gonzalez, F.J.; Glass, C. KPPARγ and PPARδ negatively regulate specific subsets of lipopolysaccharide and IFN-γ target genes in macrophages. Proc. Natl. Acad. Sci. USA 2003, 100, 6712–6717. [Google Scholar] [CrossRef] [PubMed]
- Diab, A.; Deng, C.; Smith, J.D.; Hussain, R.Z.; Phanavanh, B.; Lovett-Racke, A.E.; Drew, P.D.; Racke, M.K. Ligands for the peroxisome proliferator-activated receptor-γ and the retinoid X receptor exert additive anti-inflammatory effects on experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2004, 148, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Hitoshi, H.; Takuya, M.; Masaki, Y. Peroxisome Proliferator-Activated Receptor α and γ agonist together with TGF-β convert human CD4+CD25− T cells into functional Foxp3+ regulatory cells. J. Immunol. 2010, 201, 7186–7198. [Google Scholar] [CrossRef] [PubMed]
- Seargent, J.M.; Yates, E.A.; Gill, J. HGW9662, a potent antagonist of PPARγ, inhibits growth of breast tumour cells and promotes the anticancer effects of the PPARγ agonist rosiglitazone, independently of PPARγ activation. Br. J. Pharmacol. 2004, 143, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Jaudszus, A.; Lorkowski, S.; Gruen, M.; Roth, A.; Jahreis, G. Limited Applicability of GW9662 to Elucidate PPARg-mediated Fatty Acid Effects in Primary Human T-helper Cells. Int. J. Inflamm. 2014, 2014, 149628. [Google Scholar] [CrossRef] [PubMed]
- Raman, P.; Kaplan, B.L.F.; Kaminski, N. E15-Deoxy-∆12,14-Prostaglandin J2-Glycerol, a Putative Metabolite of 2-Arachidonyl Glycerol and a Peroxisome Proliferator-Activated Receptor γ Ligand, Modulates Nuclear Factor of Activated T Cells. J. Pharm. Exp. Ther. 2012, 342, 816–926. [Google Scholar] [CrossRef] [PubMed]
- Hontecillas, R.; Bassaganya-Riera, J. Peroxisome proliferator-Activated Receptor γ is required for regulatory CD4+ T cell mediated Protection against colitis. J. Immunol. 2007, 178, 2940–2949. [Google Scholar] [CrossRef] [PubMed]
- Wohlfert, E.A.; Nichols, F.C.; Nevius, E.; Clark, R.B. Peroxisome Proliferator-activated Receptor-γ is and immunoregulation: Enhancement of regulatory T-cells through PPARγ-dependant and independant mechanism. J. Immunol. 2007, 178, 4129–4135. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.Y.; Wang, L.H.; Chen, T.; Hodge, D.R.; Resau, J.H.; DaSilva, L.; Farrar, W.L. Activation of human T lymphocytes is inhibited by Peroxisome Proliferator-activated Receptor γ (PPARγ) Agonists: PPARγ co-association with transcription factor NFAT. J. Biol. Chem. 2000, 275, 4541–4544. [Google Scholar] [CrossRef] [PubMed]
- Delerive, P.; Fruchart, J.C.; Staels, B. Peroxisome proliferator-activated receptors in inflammation control. J. Endocrinol. 2001, 169, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Cunard, R.; Ricote, M.; DiCampli, D.; Archer, D.; Clay, K.; Daniel, A.; Glass, C.K.; Kelly, C.J. Regulation of cytokine expression by ligands of Peroxisome Proliferator Activated Receptors. J. Immunol. 2002, 168, 2795–2802. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Anderson, P.O.; Chen, S.; Paulsson, K.M.; Sjogren, H.-O.; Li, S. Inhibition of the transcription factors AP-1 and NF-κB in CD4 T cells by peroxisome proliferator-activated receptor γ ligands. Int. Immunopharmacol. 2001, 1, 803–812. [Google Scholar] [CrossRef]
- Mantel, P.-Y.; Ouaked, N.; Ruckert, B.; Karagiannidis, C.; Welz, R.; Blaser, K.; Schmidt-Weber, C.B. Molecular mechanisms underlying FOXP3 induction in human T cells. J. Immunol. 2006, 176, 3593–3602. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Borde, M.; Heissmeyer, V.; Feuerer, M.; Lapan, A.D.; Stroud, J.C.; Bates, D.L.; Guo, L.; Han, A.; Ziegler, S.F.; et al. FOXP3 controls Regulatory T cell function through cooperation with NFAT. Cell 2006, 126, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Long, M.; Park, S.-G.; Strickland, I.; Hayden, M.S.; Ghosh, S. Nuclear Factor-kB Modulates Regulatory T Cell Development by Directly Regulating Expression of Foxp3 Transcription Factor. Immunity 2009, 31, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Fong, A.L.; Ogawa, S.; Gamliel, A.; Li, A.C.; Perissi, V.; Rose, D.W.; Willson, T.M.; Rosenfeld, M.G.; Glass, C.K. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPARγ. Nature 2005, 437, 759. [Google Scholar] [CrossRef] [PubMed]
- Ricote, M.; Glass, C.K. PPARs and molecular mechanisms of transrepression. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2007, 1771, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Weil, R.; Israel, A. Deciphering the pathway from the TCR to NF-κB. Cell Death Differ. 2006, 13, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Carson, B.D.; Ziegler, S.F. Impaired T cell receptor signaling in Foxp3+ CD4+ T Cells. Ann. N. Y. Acad. Sci. 2007, 1103, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Shipley, J.M.; Waxman, D.J. Down-Regulation of STAT5b Transcriptional Activity by Ligand-Activated Peroxisome Proliferator-Activated Receptor-α and -γ. Mol. Pharmacol. 2003, 64, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, S.R.; Lee, W.M.F.; Williams, P.W.; Giels, G.M.; Williams, L.T. c-myc gene expression is stimulated by agents that activate protein kinase C and does not account for the mitogenic effect of PDGF. Cell 1985, 43, 243–251. [Google Scholar] [CrossRef]
- Pendeville, H.; Carpino, N.; Marine, J.-C.; Takahashi, Y.; Muller, M.; Martial, J.A.; Cleveland, J.L. The ornithine decarboxylase gene is essential for cell survival during early murine development. Mol. Cell. Biol. 2001, 21, 6549–6558. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.P.; Yang, J.; Thomas, R.M.; Wells, A.D.; Turka, L.A. Defective activation of Protein Kinase C and Ras-ERK pathways limits IL-2 production and proliferation by CD4+CD25+ Regulatory T Cells. J. Immunol. 2006, 177, 2186–2194. [Google Scholar] [CrossRef] [PubMed]
- Angelin, A.; Gil-de-Gomez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J., III; Kopinski, P.K.; Wang, L.; Akimova, T. Foxp3 reprograms T cell metabolism to function in low-glucose, high lactate environments. Cell Metab. 2017, 25, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Cordenonsi, M.; Dupont, S.; Maretto, S.; Insinga, A.; Imbriano, C.; Piccolo, S. Links between tumor suppressors. Cell 2003, 113, 301–314. [Google Scholar] [CrossRef]
- Elston, R.; Inman, G.J. Crosstalk between p53 and TGF-β Signaling. J. Signal Transduct. 2012, 2012, 294097. [Google Scholar] [CrossRef] [PubMed]
- Dasu, T.; Qualls, J.E.; Tuna, H.; Raman, C.; Cohen, D.A.; Bondada, S. CD5 plays an inhibitory role in the suppressive function of murine CD4+CD25+Treg cells. Immunol. Lett. 2008, 119, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.; Lutz, M.A.; Zarek, P.E.; Anders, R.A.; Kersh, G.J.; Powell, J.D. Opposing regulation of T cell function by Egr-1/NAB2 and Egr-2/Egr-3. Eur. J. Immunol. 2008, 38, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Gardner, O.S.; Shiau, C.-W.; Chen, C.-S.; Graves, L.M. Peroxisome Proliferator-activated Receptor γ-independent Activation of p38 MAPK by Thiazolidinediones involves calcium/calmodulin-dependent Protein Kinase II and Protein Kinase R: Correlation with endoplasmic reticulum stress. J. Biol. Chem. 2005, 280, 10109–10118. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Suzuki, M.; Yamada, H.; Horita, S.; Kunimi, M.; Yamazaki, O.; Shirai, A.; Nakamura, M.; Iso-O, N.; Li, Y.; et al. Thiazolidinediones enhance sodium-coupled bicarbonate absorption from renal proximal tubules via PPARγ-dependent non-genomic signaling. Cell Metab. 2011, 13, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, S.A.; Frigola, X.; Bonne-Annee, S.; Mercader, M.; Kuntz, S.M.; Krambeck, A.E.; Sengupta, S.; Dong, H.; Cheville, J.C.; Lohse, C.M.; et al. Tumor-infiltrating Foxp3-CD4+CD25+ T cells predict poor survival in renal cell carcinoma. Clin. Cancer Res. 2007, 13, 2075–2081. [Google Scholar] [CrossRef] [PubMed]
- Ladanyi, A.; Mohos, A.; Somlai, B.; Liszkay, G.; Gilde, K.; Fejos, Z.; Gaudi, I.; Timar, J. FOXP3+ cell density in primary tumor has no prognostic impact in patients with cutaneous malignant melanoma. Pathol. Oncol. Res. 2010, 16, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Mougiakakos, D.; Johansson, C.C.; Trocme, E.; All-Ericsson, C.; Economou, M.A.; Larsson, O.; Seregard, S.; Kiessling, R. Intratumoral forkhead box P3-positive regulatory T cells predict poor survival in cyclooxygenase-2-positive uveal melanoma. Cancer 2010, 116, 2224–2233. [Google Scholar] [CrossRef] [PubMed]
- Shah, W.; Yan, X.; Jing, L.; Zhou, Y.; Chen, H.; Wang, Y. A reversed CD4/CD8 ratio of tumor-infiltrating lymphocytes and a high percentage of CD4(+)FOXP3(+) regulatory T cells are significantly associated with clinical outcome in squamous cell carcinoma of the cervix. Cell Mol. Immunol. 2011, 8, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Gerber, A.L.; Münst, A.; Schlapbach, C.; Shafighi, M.; Kiermeir, D.; Hüsler, R.; Hunger, R.E. High expression of FOXP3 in primary melanoma is associated with tumour progression. Br. J. Dermatol. 2014, 170, 103–109. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nor Effa, S.Z.; Yaacob, N.S.; Mohd Nor, N. Crosstalk between PPARγ Ligands and Inflammatory-Related Pathways in Natural T-Regulatory Cells from Type 1 Diabetes Mouse Model. Biomolecules 2018, 8, 135. https://doi.org/10.3390/biom8040135
Nor Effa SZ, Yaacob NS, Mohd Nor N. Crosstalk between PPARγ Ligands and Inflammatory-Related Pathways in Natural T-Regulatory Cells from Type 1 Diabetes Mouse Model. Biomolecules. 2018; 8(4):135. https://doi.org/10.3390/biom8040135
Chicago/Turabian StyleNor Effa, S. Zulkafli, Nik Soriani Yaacob, and Norazmi Mohd Nor. 2018. "Crosstalk between PPARγ Ligands and Inflammatory-Related Pathways in Natural T-Regulatory Cells from Type 1 Diabetes Mouse Model" Biomolecules 8, no. 4: 135. https://doi.org/10.3390/biom8040135
APA StyleNor Effa, S. Z., Yaacob, N. S., & Mohd Nor, N. (2018). Crosstalk between PPARγ Ligands and Inflammatory-Related Pathways in Natural T-Regulatory Cells from Type 1 Diabetes Mouse Model. Biomolecules, 8(4), 135. https://doi.org/10.3390/biom8040135