A Recombinant Snake Cathelicidin Derivative Peptide: Antibiofilm Properties and Expression in Escherichia coli


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Clinical Bacterial Strains
2.3. Antimicrobial Activity of the Peptide
2.4. Investigation of Antimicrobial Effects on an Established Biofilm
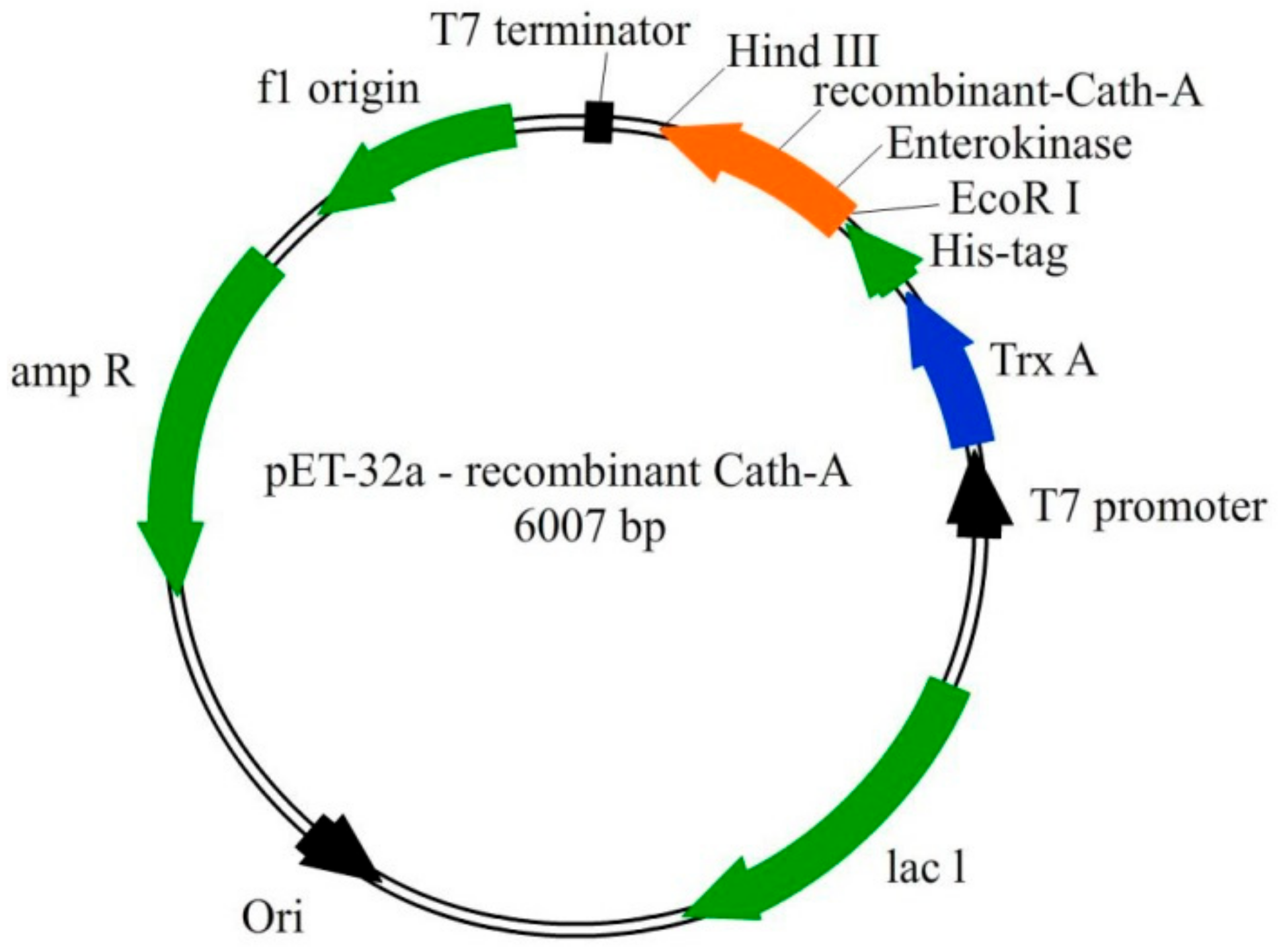
2.5. Recombinant Vector Construct
2.6. Expression of the Fusion Protein
2.7. Recombinant Proteins Isolation and Purification
2.8. Proteolytic Cleavage of Fusion Protein
2.9. Analysis of Protein Concentration
2.10. Antimicrobial Activity Assay of Expressed Peptide
2.11. Statistical Analysis
3. Results
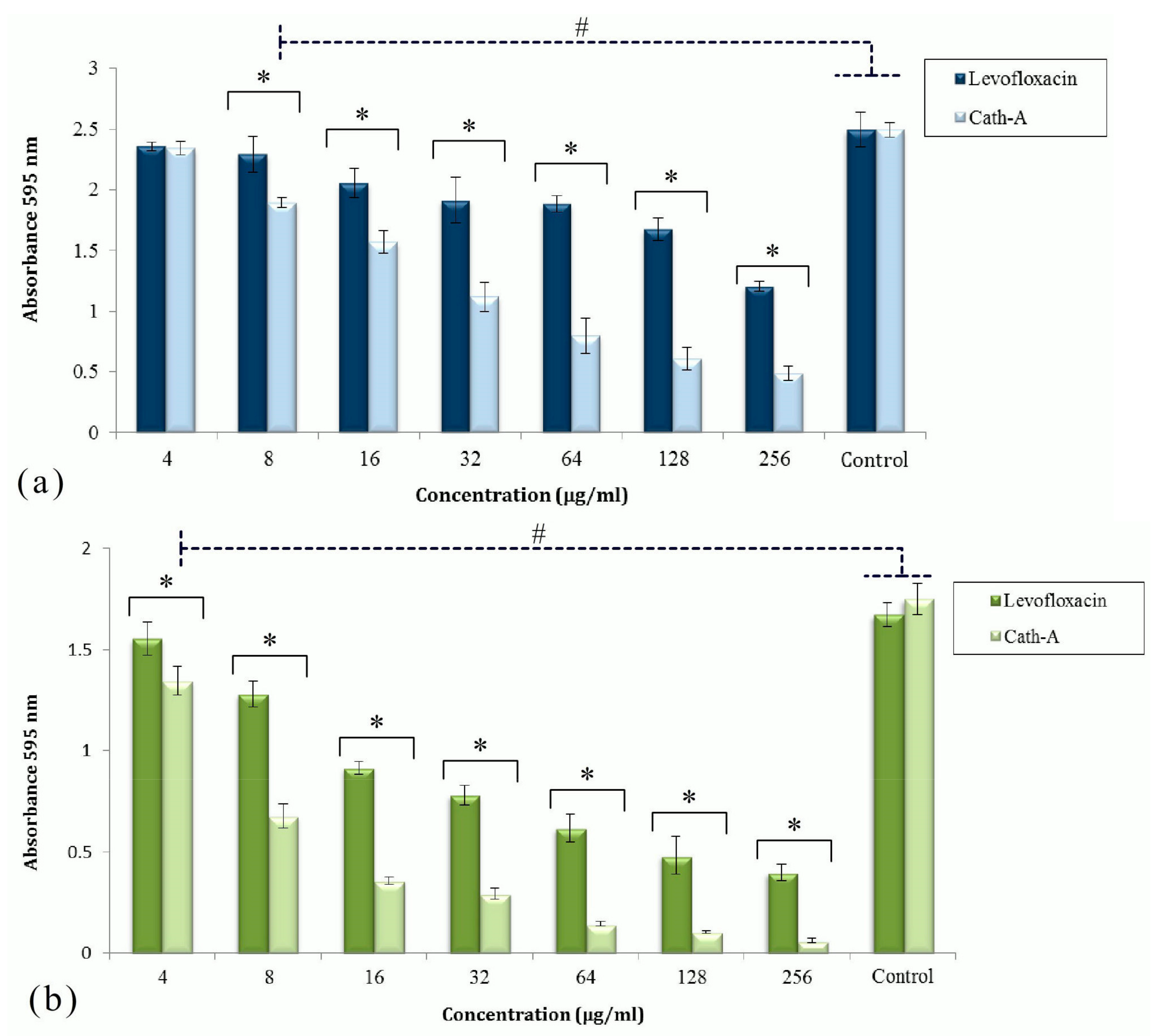
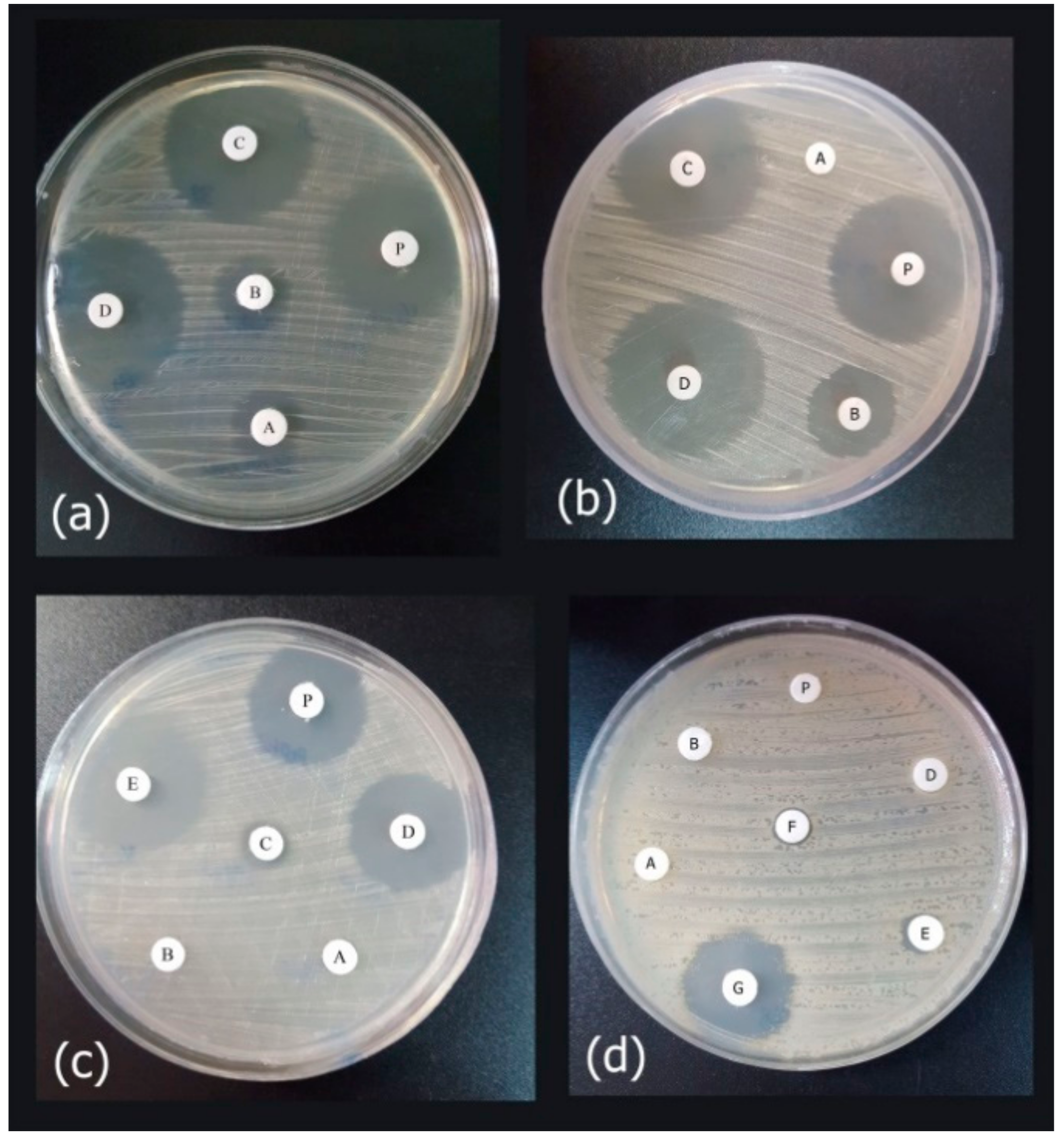
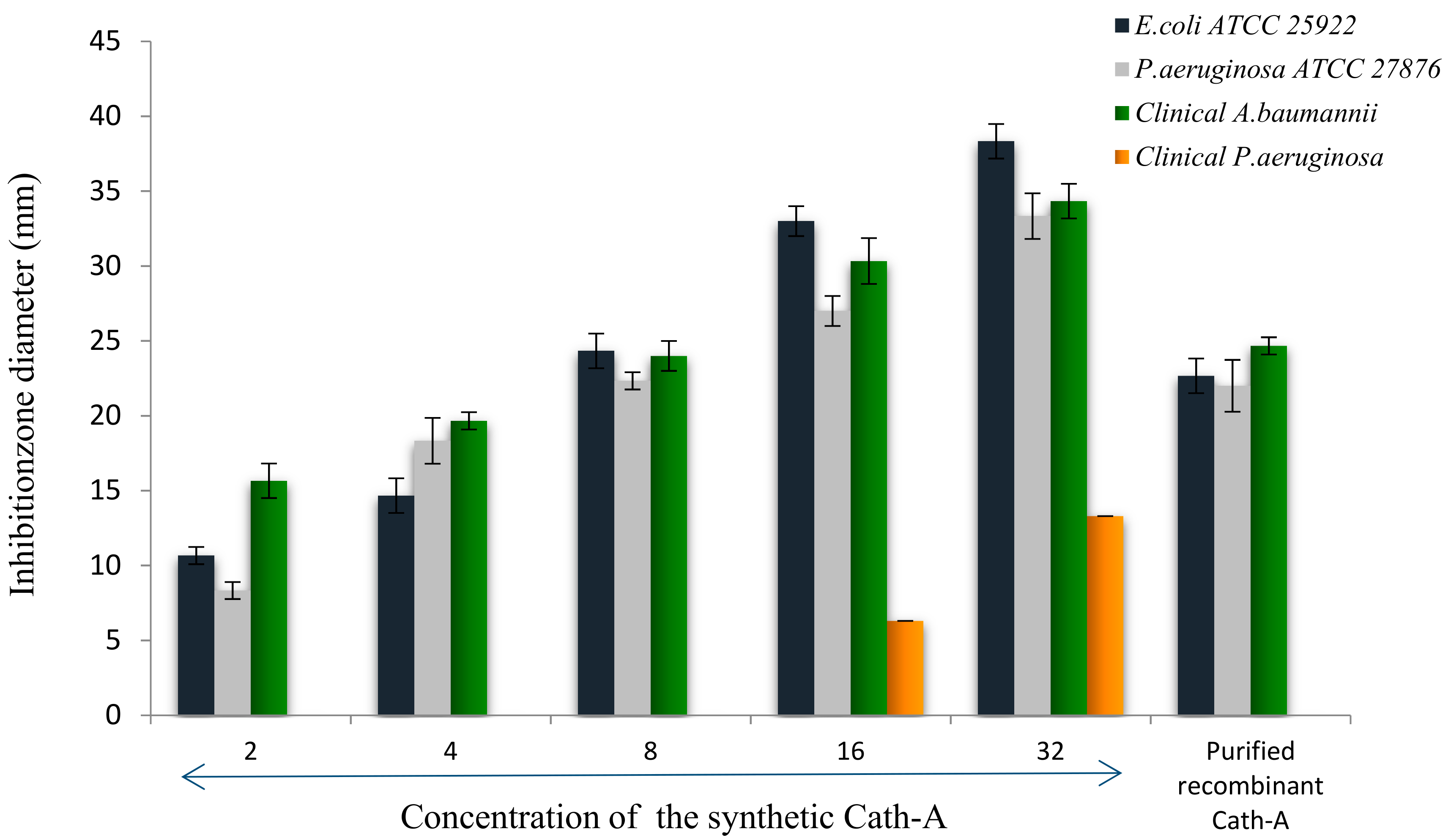
3.1. Antimicrobial Activities of Antibiotics and Peptide
3.2. Disruption of the Established Biofilms
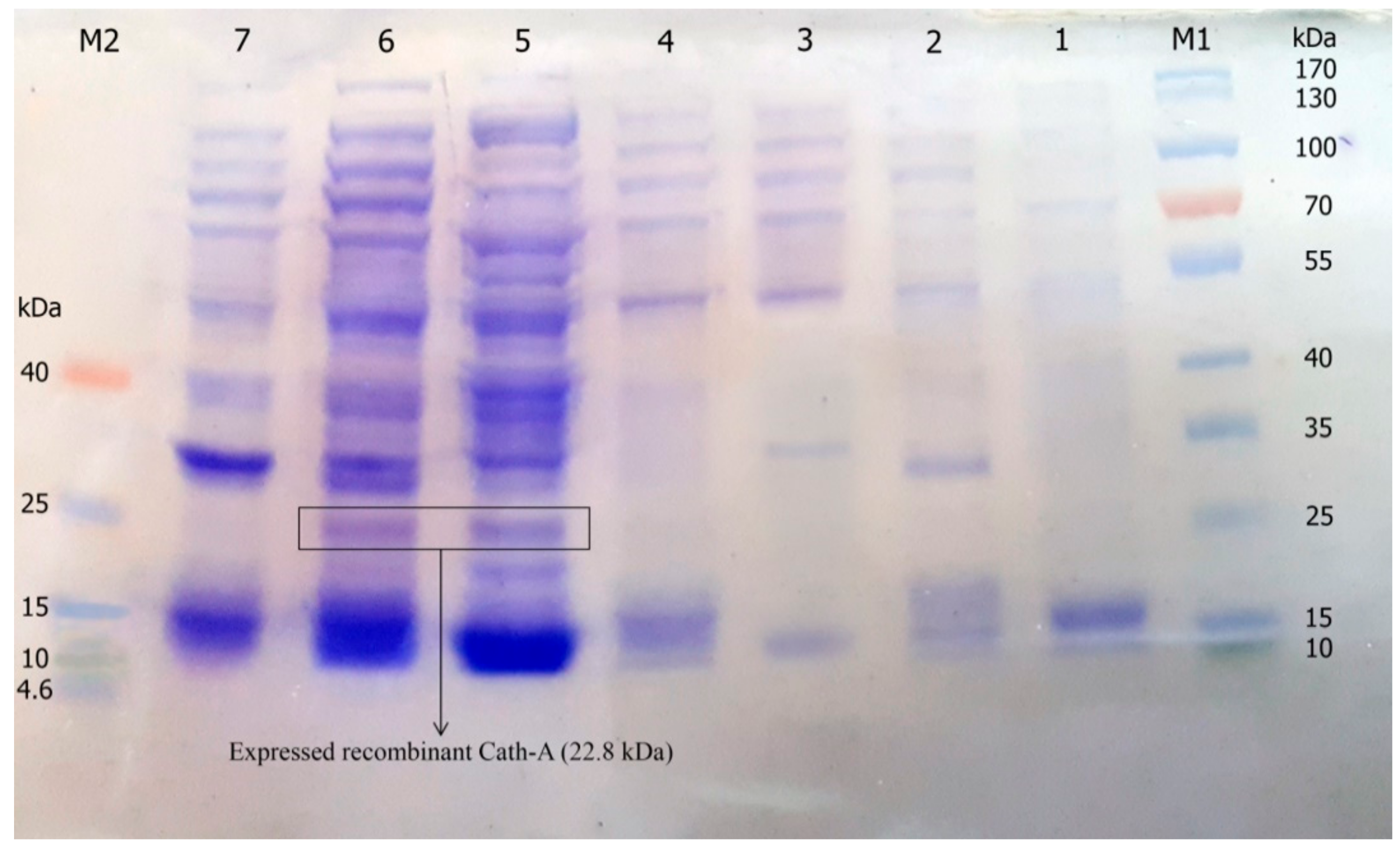
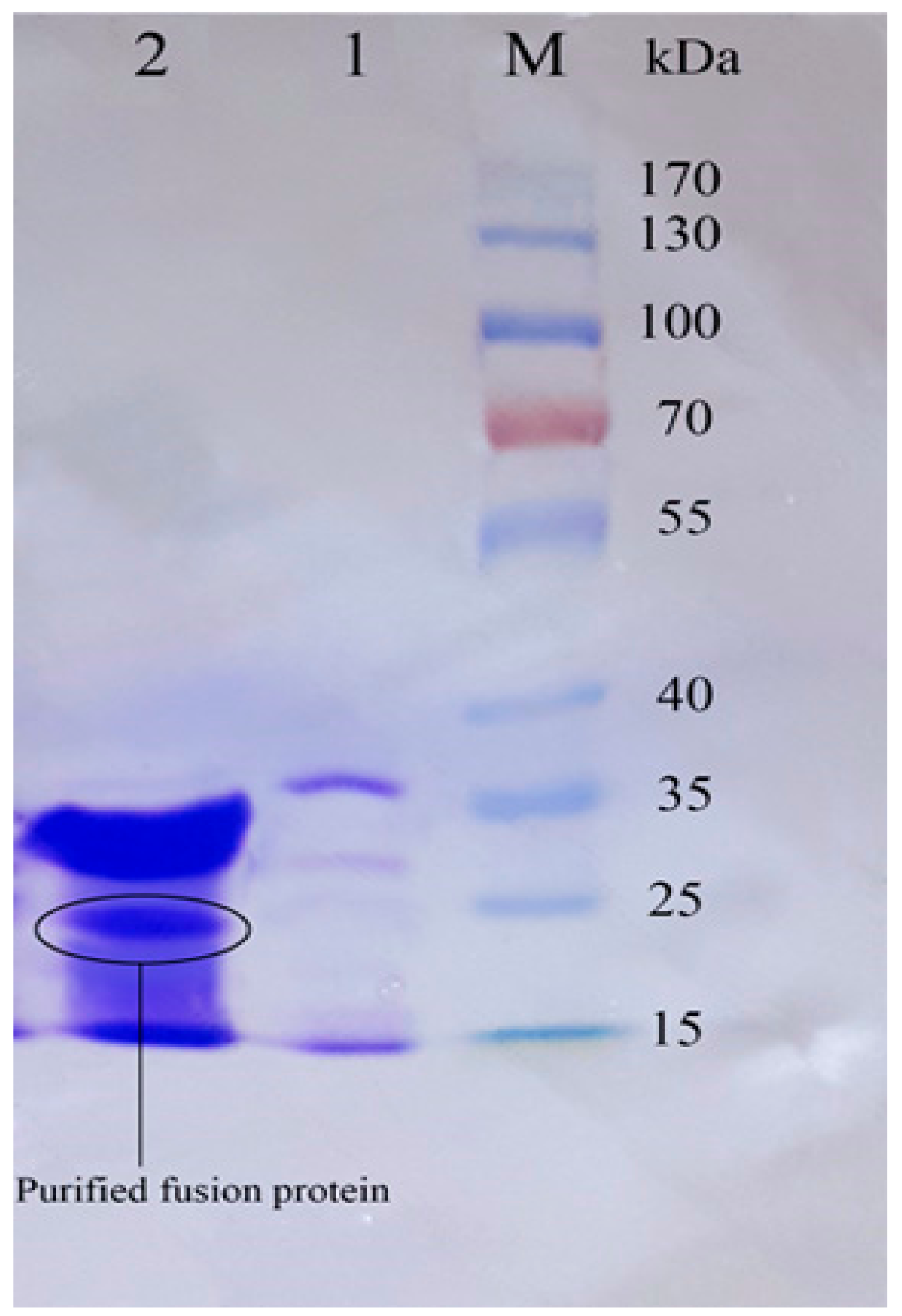
3.3. Expression of Recombinant Peptide in E. coli BL21
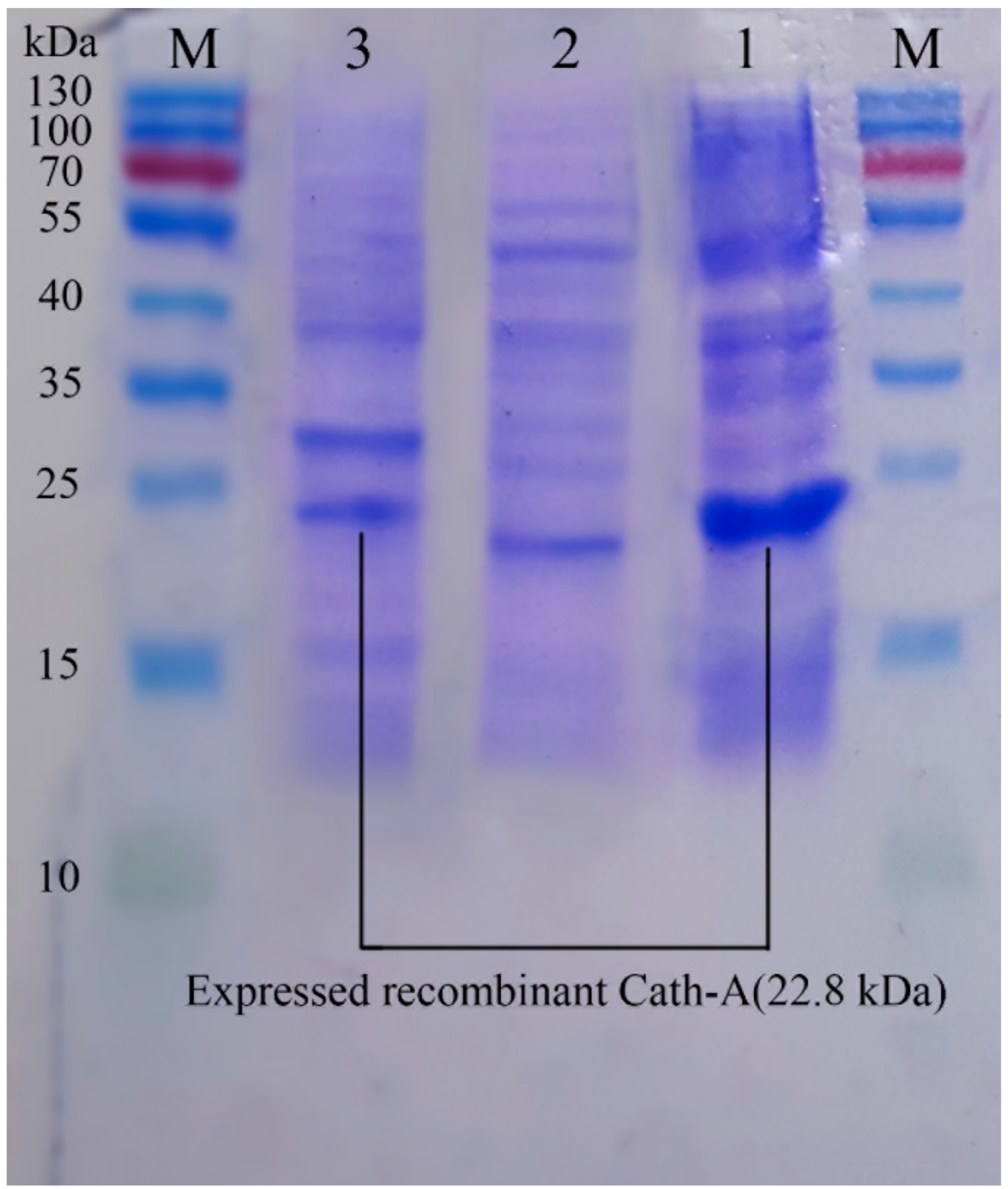
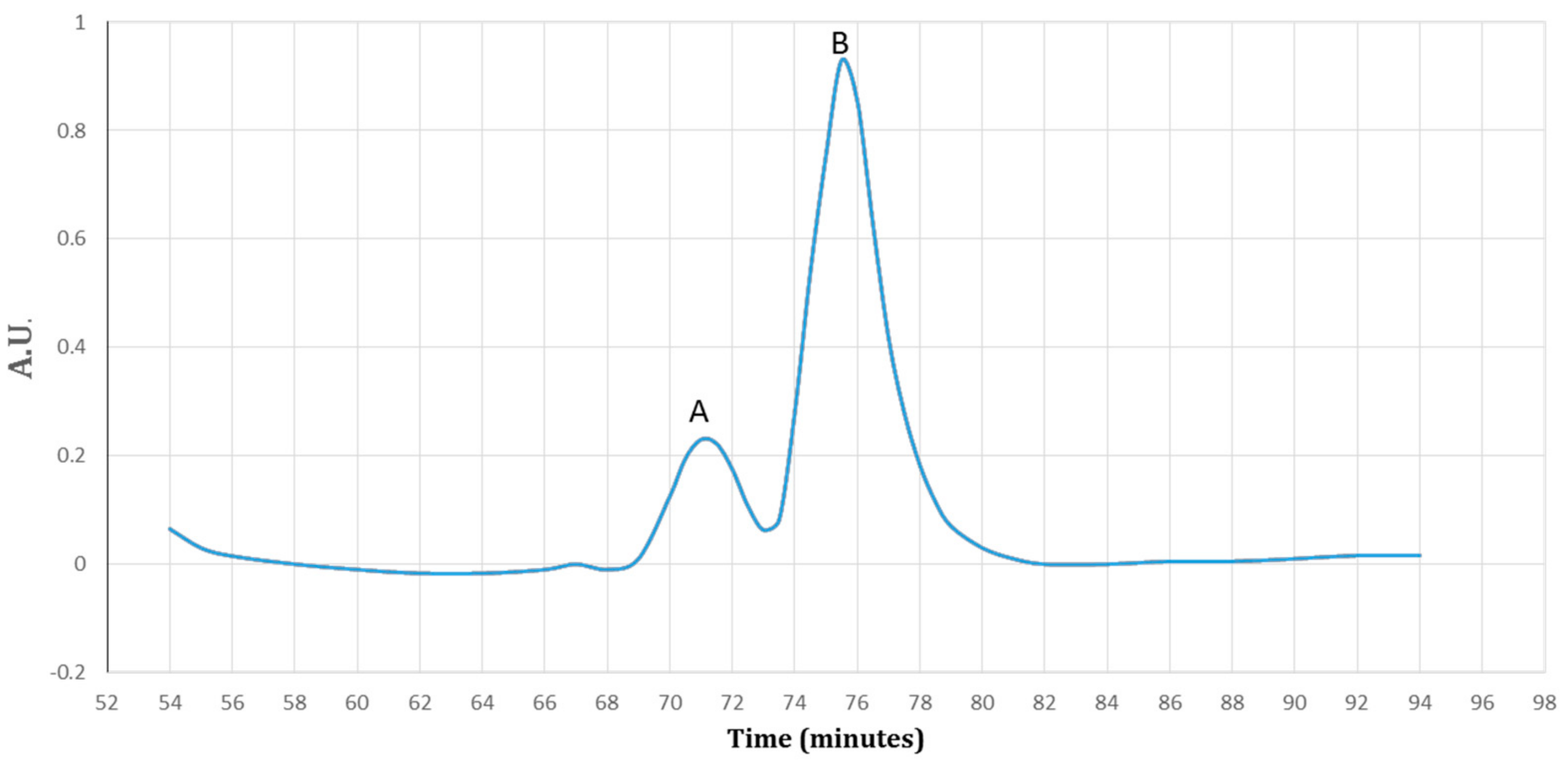
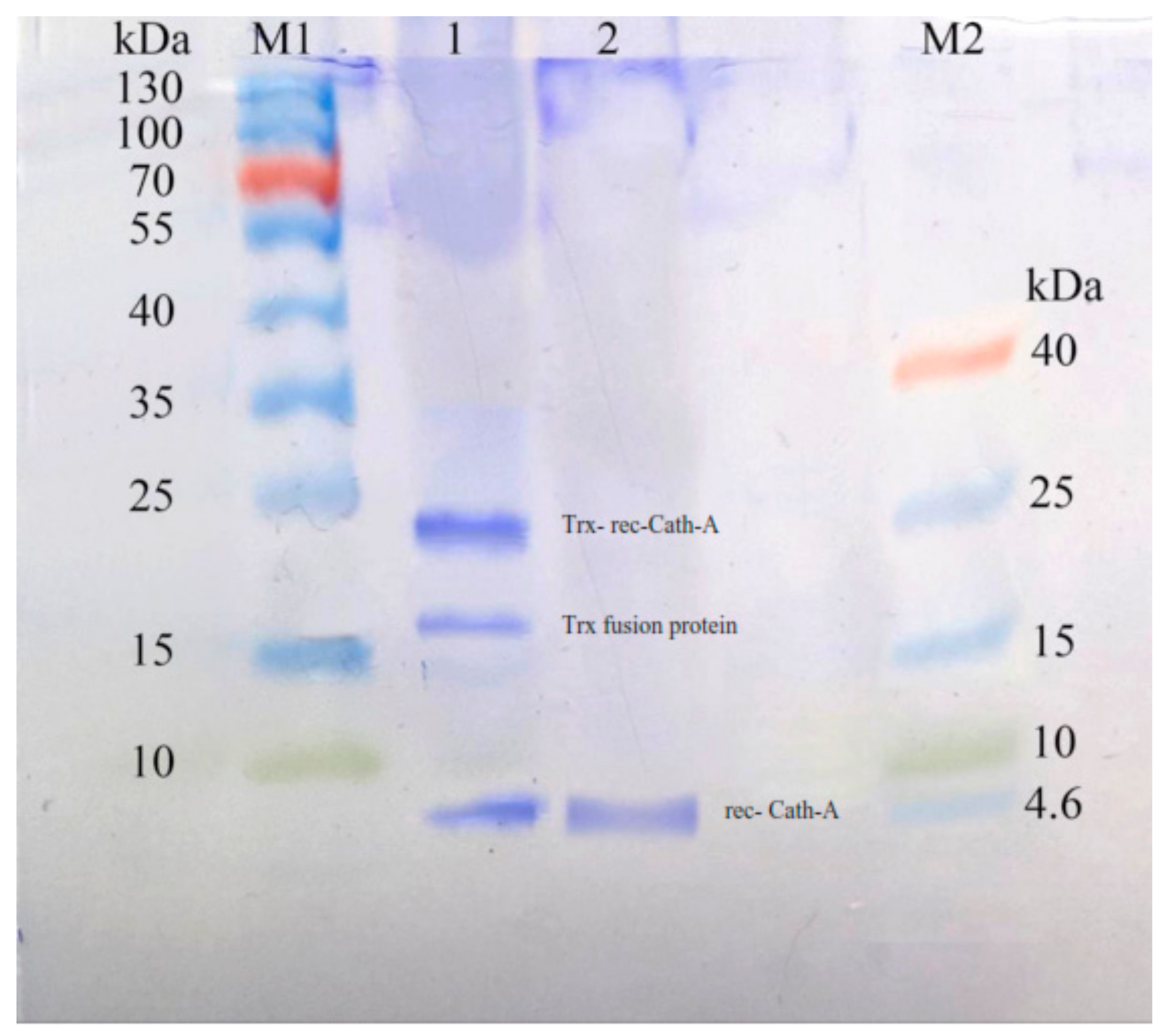
3.4. Purification of Recombinant–Cath-A
3.5. Antimicrobial Activity of the Released Peptide
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rai, V.; Yuet-Meng, C.; Rosenthal, V.D.; Hasan, M.S.; Zaman, M.K.; Mansor, M.; Ponnampala, S.S.L.S.; Abidin, A.S.Z.; Manikavasagam, J.; Phuong, D.M. Device-associated infection and mortality rates, bacterial resistance, and length of stay in hospitals of Malaysia: International nosocomial infection control consortium (INICC)’s findings. Can. J. Infect. Dis. Med. Microbiol. 2016, 31, 107–112. [Google Scholar]
- Cosgrove, S.E. The relationship between antimicrobial resistance and patient outcomes: Mortality, length of hospital stay, and health care costs. Clin. Infect. Dis. 2006, 42, S82–S89. [Google Scholar] [CrossRef] [PubMed]
- Laxminarayan, R.; Matsoso, P.; Pant, S.; Brower, C.; Røttingen, J.-A.; Klugman, K.; Davies, S. Access to effective antimicrobials: A worldwide challenge. Lancet 2016, 387, 168–175. [Google Scholar] [CrossRef]
- WHO. Antimicrobial Resistance: Global Report on Surveillance; WHO: Geneva, Switzerland, 2014; p. 257. [Google Scholar]
- Potron, A.; Poirel, L.; Nordmann, P. Emerging broad-spectrum resistance in Pseudomonas aeruginosa and Acinetobacter baumannii: Mechanisms and epidemiology. Int. J. Antimicrob. Agents 2015, 45, 568–585. [Google Scholar] [CrossRef] [PubMed]
- Rossolini, G.M.; Arena, F.; Pecile, P.; Pollini, S. Update on the antibiotic resistance crisis. Curr. Opin. Pharmacol. 2014, 18, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Dantas, R.C.C.; e Silva, R.T.; Ferreira, M.L.; Gonçalves, I.R.; Araújo, B.F.; de Campos, P.A.; Royer, S.; da Fonseca Batistão, D.W.; Gontijo-Filho, P.P.; Ribas, R.M. Molecular epidemiological survey of bacteremia by multidrug resistant Pseudomonas aeruginosa: The relevance of intrinsic resistance mechanisms. PLoS ONE 2017, 12, e0176774. [Google Scholar] [CrossRef] [PubMed]
- Ruppé, É.; Woerther, P.-L.; Barbier, F. Mechanisms of antimicrobial resistance in Gram-negative bacilli. Ann. Intensive Care 2015, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.; Carmeli, Y.; Falagas, M.; Giske, C.; Harbarth, S.; Hindler, J.; Kahlmeter, G.; Olsson-Liljequist, B. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti-Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Gurung, J.; Khyriem, A.B.; Banik, A.; Lyngdoh, W.V.; Choudhury, B.; Bhattacharyya, P. Association of biofilm production with multidrug resistance among clinical isolates of Acinetobacter baumannii and Pseudomonas aeruginosa from intensive care unit. Indian J. Crit. Care Med. 2013, 17, 214–218. [Google Scholar] [PubMed]
- De Francesco, M.; Ravizzola, G.; Peroni, L.; Bonfanti, C.; Manca, N. Prevalence of multidrug-resistant Acinetobacter baumannii and Pseudomonas aeruginosa in an italian hospital. J. Infect. Public Health 2013, 6, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Royer, S.; Faria, A.L.S.; Seki, L.M.; Chagas, T.P.G.; de Campos, P.A.; da Fonseca Batistão, D.W.; Asensi, M.D.; Gontijo Filho, P.P.; Ribas, R.M. Spread of multidrug-resistant Acinetobacter baumannii and Pseudomonas aeruginosa clones in patients with ventilator-associated pneumonia in an adult intensive care unit at a university hospital. Braz. J. Infect. Dis. 2015, 19, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.E.; Slayton, R.B.; Stevens, V.W.; Jones, M.M.; Khader, K.; Rubin, M.A.; Jernigan, J.A.; Samore, M.H. Attributable mortality of healthcare-associated infections due to multidrug-resistant gram-negative bacteria and methicillin-resistant Staphylococcus Aureus. Infect. Control Hosp. Epidemiol. 2017, 38, 848–856. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.J.; Bae, I.K.; Jang, I.-H.; Jeong, S.H.; Kang, H.-K.; Lee, K. Epidemiology and characteristics of metallo-β-lactamase-producing Pseudomonas aeruginosa. J. Infect. Chemother. 2015, 47, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Erfani, Y.; Yaghuobi, S.; Fallah, F.; Rahbar, M.; Rasti, A.; Ghanati, K. Detection of bla NDM-1, bla VIM, and bla IMP genes in multidrug-resistant Acinetobacter baumannii and Pseudomonas aeruginosa from clinical isolates in Tehran hospitals. Int. J. Adv. Biotechnol. Res. 2017, 8, 500–506. [Google Scholar]
- Mohammadi, F.; Goudarzi, H.; Hashemi, A.; Nojookambari, N.Y.; Khoshnood, S.; Sabzehali, F. Detection of isaba1 in Acinetobacter baumannii strains carrying OXA genes isolated from Iranian burns patients. Arch. Pediatr. Infect. Dis. 2017, 5, e39307. [Google Scholar] [CrossRef]
- Bavasheh, N.; Karmostaji, A. Antibiotic resistance pattern and evaluation of blaOXA-10, blaPER-1, blaVEB, blaSHV genes in clinical isolates of Pseudomonas aeruginosa isolated from hospital in south of Iran in 2014–2015. Infect. Epidemiol. Microbiol. 2017, 3, 1–5. [Google Scholar] [CrossRef]
- Armin, S.; Karimi, A.; Fallah, F.; Tabatabaii, S.R.; Alfatemi, S.M.H.; Khiabanirad, P.; Shiva, F.; Fahimzad, A.; Rahbar, M.; Mansoorghanaii, R. Antimicrobial resistance patterns of Acinetobacter baumannii, Pseudomonas aeruginosa and staphylococcus aureus isolated from patients with nosocomial infections admitted to tehran hospitals. Arch. Pediatr. Infect. Dis. 2015, 3, e32554. [Google Scholar] [CrossRef]
- WHO. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics Geneva; WHO: Geneva, Switzerland, 2017; pp. 1–7. [Google Scholar]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y. Discovery, research, and development of new antibiotics: The who priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Di Domenico, E.G.; Farulla, I.; Prignano, G.; Gallo, M.T.; Vespaziani, M.; Cavallo, I.; Sperduti, I.; Pontone, M.; Bordignon, V.; Cilli, L.; et al. Biofilm is a major virulence determinant in bacterial colonization of chronic skin ulcers independently from the multidrug resistant phenotype. Int. J. Mol. Sci. 2017, 18, 1077. [Google Scholar] [CrossRef] [PubMed]
- Johani, K.; Abualsaud, D.; Costa, D.M.; Hu, H.; Whiteley, G.; Deva, A.; Vickery, K. Characterization of microbial community composition, antimicrobial resistance and biofilm on intensive care surfaces. J. Infect. Public Health 2018, 11, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Wieland, K.; Chhatwal, P.; Vonberg, R.-P. Nosocomial outbreaks caused by Acinetobacter baumannii and Pseudomonas aeruginosa: Results of a systematic review. Am. J. Infect. Control 2018, 46, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Andrea, A.; Molchanova, N.; Jenssen, H. Antibiofilm peptides and peptidomimetics with focus on surface immobilization. Biomolecule 2018, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente-Núñez, C.; Cardoso, M.H.; de Souza Cândido, E.; Franco, O.L.; Hancock, R.E.W. Synthetic antibiofilm peptides. Biochim. Biophys. Acta Biomembr. 2016, 1858, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Haney, E.F.; Gill, E.E. The immunology of host defence peptides: Beyond antimicrobial activity. Nat. Rev. Immunol. 2016, 16, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kizhakkedathu, J.; Straus, S. Antimicrobial peptides: Diversity, mechanism of action and strategies to improve the activity and biocompatibility in vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Pletzer, D.; Hancock, R.E. Anti-biofilm peptides: Potential as broad-spectrum agents. J. Bacteriol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Mojsoska, B.; Jenssen, H. Peptides and peptidomimetics for antimicrobial drug design. Pharmaceuticals 2015, 8, 366–415. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Mishra, B.; Lau, K.; Lushnikova, T.; Golla, R.; Wang, X. Antimicrobial peptides in 2014. Pharmaceuticals 2015, 8, 123–150. [Google Scholar] [CrossRef] [PubMed]
- Haney, E.F.; Mansour, S.C.; Hancock, R.E. Antimicrobial peptides: An introduction. In Antimicrobial Peptides: Methods and Protocols; Humana Press: New York, NY, USA, 2017; pp. 3–22. [Google Scholar]
- Kang, H.-K.; Kim, C.; Seo, C.H.; Park, Y. The therapeutic applications of antimicrobial peptides (AMPs): A patent review. J. Microbiol. 2017, 55, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Batoni, G.; Maisetta, G.; Esin, S. Antimicrobial peptides and their interaction with biofilms of medically relevant bacteria. Biochim. Biophys. Acta Biomembr. 2016, 1858, 1044–1060. [Google Scholar] [CrossRef] [PubMed]
- Di Luca, M.; Maccari, G.; Nifosì, R. Treatment of microbial biofilms in the post-antibiotic era: Prophylactic and therapeutic use of antimicrobial peptides and their design by bioinformatics tools. Pathog. Dis. 2014, 70, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Mansour, S.C.; de la Fuente-Núñez, C.; Hancock, R.E. Peptide IDR-1018: Modulating the immune system and targeting bacterial biofilms to treat antibiotic-resistant bacterial infections. J. Pept. Sci. 2015, 21, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-J.; Park, S.J.; Mishig-Ochir, T.; Lee, B.-J. Antimicrobial peptides: Therapeutic potentials. Expert Rev. Anti-Infect. Ther. 2014, 12, 1477–1486. [Google Scholar] [CrossRef] [PubMed]
- Li, Y. Recombinant production of antimicrobial peptides in Escherichia coli: A review. Protein Expr. Purif. 2011, 80, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Parachin, N.S.; Mulder, K.C.; Viana, A.A.B.; Dias, S.C.; Franco, O.L. Expression systems for heterologous production of antimicrobial peptides. Peptides 2012, 38, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Kościuczuk, E.M.; Lisowski, P.; Jarczak, J.; Strzałkowska, N.; Jóźwik, A.; Horbańczuk, J.; Krzyżewski, J.; Zwierzchowski, L.; Bagnicka, E. Cathelicidins: Family of antimicrobial peptides. A review. Mol. Biol. Rep. 2012, 39, 10957–10970. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yang, B.; Zhou, H.; Sun, L.; Dou, J.; Qian, H.; Huang, W.; Mei, Y.; Han, J. Structure–activity relationships of a snake cathelicidin-related peptide, bf-15. Peptides 2011, 32, 2497–2503. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hong, J.; Liu, X.; Yang, H.; Liu, R.; Wu, J.; Wang, A.; Lin, D.; Lai, R. Snake cathelicidin from Bungarus fasciatus is a potent peptide antibiotics. PLoS ONE 2008, 3, e3217. [Google Scholar] [CrossRef] [PubMed]
- Andreev, K.; Bianchi, C.; Laursen, J.S.; Citterio, L.; Hein-Kristensen, L.; Gram, L.; Kuzmenko, I.; Olsen, C.A.; Gidalevitz, D. Guanidino groups greatly enhance the action of antimicrobial peptidomimetics against bacterial cytoplasmic membranes. Biochim. Biophys. Acta Biomembr. 2014, 1838, 2492–2502. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.M.; Edwards, M.A.; Li, J.; Yip, C.M.; Deber, C.M. Roles of hydrophobicity and charge distribution of cationic antimicrobial peptides in peptide-membrane interactions. J. Biol. Chem. 2012. [Google Scholar] [CrossRef] [PubMed]
- Tajbakhsh, M.; Karimi, A.; Tohidpour, A.; Abbasi, N.; Fallah, F.; Akhavan, M.M. The antimicrobial potential of a new derivative of cathelicidin from Bungarus fasciatus against methicillin-resistant Staphylococcus aureus. J. Microbiol. 2018, 56, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Chung, P.Y.; Khanum, R. Antimicrobial peptides as potential anti-biofilm agents against multidrug-resistant bacteria. J. Microbiol. Immunol. Infect. 2017, 50, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Zhao, D.-M.; Wen, Y. Expression, purification and antibacterial activity of the channel catfish hepcidin mature peptide. Protein Expr. Purif. 2014, 94, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Herbel, V.; Schäfer, H.; Wink, M. Recombinant production of snakin-2 (an antimicrobial peptide from tomato) in E. coli and analysis of its bioactivity. Molecules 2015, 20, 14889–14901. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.-T.; Wu, C.-L.; Yip, B.-S.; Yu, H.-Y.; Cheng, H.-T.; Chih, Y.-H.; Cheng, J.-W. High level expression and purification of the clinically active antimicrobial peptide P-113 in Escherichia coli. Molecules 2018, 23, 800. [Google Scholar] [CrossRef] [PubMed]
- Ishida, H.; Nguyen, L.T.; Gopal, R.; Aizawa, T.; Vogel, H.J. Overexpression of antimicrobial, anticancer, and transmembrane peptides in Escherichia coli through a calmodulin-peptide fusion system. J. Am. Chem. Soc. 2016, 138, 11318–11326. [Google Scholar] [CrossRef] [PubMed]
- LaVallie, E.R.; Lu, Z.; Diblasio-Smith, E.A.; Collins-Racie, L.A.; McCoy, J.M. Thioredoxin as a fusion partner for production of soluble recombinant proteins in Escherichia coli. Meth. Enzymol. 2000, 326, 322–340. [Google Scholar] [PubMed]
- Clinical and Laboratory Standards Institute (CLSI). Performance Standards for Antimicrobial Susceptibility Testing; Twenty-Fifth Informational Supplement m100-s26; CLSI: Wayne, PA, USA, 2016. [Google Scholar]
- Jiang, Z.; Vasil, A.; Vasil, M.; Hodges, R. “Specificity determinants” improve therapeutic indices of two antimicrobial peptides piscidin 1 and dermaseptin S4 against the gram-negative pathogens Acinetobacter baumannii and Pseudomonas aeruginosa. Pharmaceuticals 2014, 7, 366–391. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, G.A. Microtiter dish biofilm formation assay. J. Vis. Exp. 2011, 47, e2437. [Google Scholar] [CrossRef] [PubMed]
- Merritt, J.H.; Kadouri, D.E.; O’Toole, G.A. Growing and analyzing static biofilms. Curr. Protoc. Microbiol. 2011, 22, 1–17. [Google Scholar] [CrossRef]
- Kruger, N.J. The bradford method for protein quantitation. In The Protein Protocols Handbook; Humana Press: New York, NY, USA, 2009; pp. 17–24. [Google Scholar]
- De la Fuente-Núñez, C.; Reffuveille, F.; Mansour, S.C.; Reckseidler-Zenteno, S.L.; Hernández, D.; Brackman, G.; Coenye, T.; Hancock, R.E. D-enantiomeric peptides that eradicate wild-type and multidrug-resistant biofilms and protect against lethal Pseudomonas aeruginosa infections. Chem. Biol. 2015, 22, 196–205. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente-Núñez, C.; Reffuveille, F.; Haney, E.F.; Straus, S.K.; Hancock, R.E. Broad-spectrum anti-biofilm peptide that targets a cellular stress response. PLoS Pathog. 2014, 10, e1004152. [Google Scholar] [CrossRef] [PubMed]
- Pletzer, D.; Coleman, S.R.; Hancock, R.E.W. Anti-biofilm peptides as a new weapon in antimicrobial warfare. Curr. Opin. Microbiol. 2016, 33, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Farshadzadeh, Z.; Hashemi, F.B.; Rahimi, S.; Pourakbari, B.; Esmaeili, D.; Haghighi, M.A.; Majidpour, A.; Shojaa, S.; Rahmani, M.; Gharesi, S. Wide distribution of carbapenem resistant Acinetobacter baumannii in burns patients in iran. Front. Microbiol. 2015, 6, 1146. [Google Scholar] [CrossRef] [PubMed]
- Olejnickova, K.; Hola, V.; Ruzicka, F. Catheter-related infections caused by Pseudomonas aeruginosa: Virulence factors involved and their relationships. Pathog. Dis. 2014, 72, 87–94. [Google Scholar] [PubMed]
- Mishra, B.; Wang, G. Individual and combined effects of engineered peptides and antibiotics on Pseudomonas aeruginosa biofilms. Pharmaceuticals 2017, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Sambanthamoorthy, K.; Palys, T.; Paranavitana, C. The human antimicrobial peptide LL-37 and its fragments possess both antimicrobial and antibiofilm activities against multidrug-resistant Acinetobacter baumannii. Peptides 2013, 49, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Reffuveille, F.; de la Fuente-Núñez, C.; Mansour, S.; Hancock, R.E. A broad-spectrum anti-biofilm peptide enhances antibiotic action against bacterial biofilms. Antimicrob. Agents Chemother. 2014. [Google Scholar] [CrossRef] [PubMed]
- Azizi, O.; Shakibaie, M.R.; Modarresi, F.; Shahcheraghi, F. Molecular detection of class-D OXA carbapenemase genes in biofilm and non-biofilm forming clinical isolates of Acinetobacter baumannii. Jundishapur J. Microbiol. 2015, 8, e21042. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Li, H.; Zhang, C.; Liang, B.; Li, J.; Wang, L.; Du, X.; Liu, X.; Qiu, S.; Song, H. Relationship between antibiotic resistance, biofilm formation, and biofilm-specific resistance in Acinetobacter baumannii. Front. Microbiol. 2016, 7, 483. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.H.; Kwon, K.C.; Kim, S.; Park, Y.; Koo, S.H. Association between biofilm formation and antimicrobial resistance in carbapenem-resistant Pseudomonas aeruginosa. Ann. Clin. Lab. Sci. 2018, 48, 363–368. [Google Scholar] [PubMed]
- Joo, H.-S.; Fu, C.-I.; Otto, M. Bacterial strategies of resistance to antimicrobial peptides. Philos. Trans. R. Soc. B 2016, 371, 20150292. [Google Scholar] [CrossRef] [PubMed]
- Fahimirad, S.; Abtahi, H.; Razavi, S.H.; Alizadeh, H.; Ghorbanpour, M. Production of recombinant antimicrobial polymeric protein beta casein-E 50-52 and its antimicrobial synergistic effects assessment with thymol. Molecules 2017, 22, 822. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Xu, Z.; Fang, X.; Wang, F.; Cen, P. High-level expression of soluble human β-defensin-2 in Escherichia coli. Process Biochem. 2004, 39, 2199–2205. [Google Scholar] [CrossRef]
- Moon, J.-Y.; Henzler-Wildman, K.A.; Ramamoorthy, A. Expression and purification of a recombinant LL-37 from Escherichia coli. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Luan, C.; Zhang, H.W.; Song, D.G.; Xie, Y.G.; Feng, J.; Wang, Y.Z. Expressing antimicrobial peptide cathelicidin-BF in Bacillus subtilis using SUMO technology. Appl. Microbiol. Biotechnol. 2014, 98, 3651–3658. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Wu, R.; Zhang, L.; Ahmad, B.; Si, D.; Zhang, R. Expression, purification, and characterization of a novel hybrid peptide with potent antibacterial activity. Molecules 2018, 23, 1491. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microorganisms | MIC (µg/mL) | |||||||
|---|---|---|---|---|---|---|---|---|
| Ap | PIP | CAZ | CP | T | LEV | IMI | Cath-BF | |
| P. aeruginosa 1 | >256 | >256 | 256 | 128 | 128 | 64 | 32 | 32 |
| P. aeruginosa 2 | >256 | >256 | 64 | 256 | 128 | 8 | 8 | 64 |
| P. aeruginosa 3 | >256 | >256 | >256 | >256 | >256 | 16 | >256 | >256 |
| P. aeruginosa 4 | >256 | >256 | 256 | 128 | 128 | 32 | 32 | 128 |
| P. aeruginosa 5 | >256 | >256 | 16 | 32 | >256 | 256 | 32 | 16 |
| P. aeruginosa 6 | >256 | 64 | 64 | 64 | 32 | 16 | 8 | 64 |
| P. aeruginosa 7 | >256 | >256 | >256 | 128 | >256 | 64 | 256 | >256 |
| P. aeruginosa 8 | >256 | >256 | >256 | 64 | 32 | 64 | 32 | 256 |
| A. baumannii 1 | >256 | >256 | 256 | 256 | 256 | 16 | >256 | 16 |
| A. baumannii 2 | >256 | >256 | 128 | 64 | 32 | 16 | >256 | 8 |
| A. baumannii 3 | >256 | >256 | >256 | 128 | >256 | 16 | 256 | 8 |
| A. baumannii 4 | >256 | >256 | 64 | 32 | >256 | 8 | 256 | 8 |
| A. baumannii 5 | >256 | >256 | 128 | 64 | >256 | 16 | 256 | 8 |
| A. baumannii 6 | >256 | >256 | 128 | 128 | >256 | >256 | 256 | 8 |
| A. baumannii 7 | >256 | >256 | 64 | 64 | 64 | 16 | 128 | 16 |
| A. baumannii 8 | >256 | >256 | 128 | 32 | 256 | 32 | 256 | 16 |
| A. baumannii 9 | >256 | >256 | >256 | 128 | 128 | 64 | >256 | 32 |
| A. baumannii 10 | 256 | >256 | 128 | 32 | 64 | 32 | 128 | 8 |
| P. aeruginosa ATCCC 27863 | 128 | 4 | 2 | 1 | 32 | 0.5 | 4 | 8 |
| E. coli ATCC 25922 | 2 | 0.5 | 0.5 | <0.25 | 0.5 | 0.25 | 1 | 8 |
| Fraction | Volume (mL) | Concentration (mg/mL) | Total Protein (mg) | Yield (%) |
|---|---|---|---|---|
| Crude supernatant | 60 | 2.57 ± 0.07 | 154.6 ± 4.2 | 100 |
| Ni-NTA Histrap | 45 | 1.42 ± 0.09 | 64.2 ± 4.2 | 41.5 |
| Pick-II | 20 | 0.576 ± 0.015 | 11.52 ± 0.30 | 7.45 |
| Ultrafilter | 10 | 0.394 ± 0.019 | 3.94 ± 0.19 | 3.2 |
| rec-Cath-BF (Dialysis) | 5 | 0.0176 ± 0.008 | 0.088 ± 0.04 | 0.34 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tajbakhsh, M.; Akhavan, M.M.; Fallah, F.; Karimi, A. A Recombinant Snake Cathelicidin Derivative Peptide: Antibiofilm Properties and Expression in Escherichia coli. Biomolecules 2018, 8, 118. https://doi.org/10.3390/biom8040118
Tajbakhsh M, Akhavan MM, Fallah F, Karimi A. A Recombinant Snake Cathelicidin Derivative Peptide: Antibiofilm Properties and Expression in Escherichia coli. Biomolecules. 2018; 8(4):118. https://doi.org/10.3390/biom8040118
Chicago/Turabian StyleTajbakhsh, Mercedeh, Maziar Mohammad Akhavan, Fatemeh Fallah, and Abdollah Karimi. 2018. "A Recombinant Snake Cathelicidin Derivative Peptide: Antibiofilm Properties and Expression in Escherichia coli" Biomolecules 8, no. 4: 118. https://doi.org/10.3390/biom8040118
APA StyleTajbakhsh, M., Akhavan, M. M., Fallah, F., & Karimi, A. (2018). A Recombinant Snake Cathelicidin Derivative Peptide: Antibiofilm Properties and Expression in Escherichia coli. Biomolecules, 8(4), 118. https://doi.org/10.3390/biom8040118