In Silico HCT116 Human Colon Cancer Cell-Based Models En Route to the Discovery of Lead-Like Anticancer Drugs





Abstract
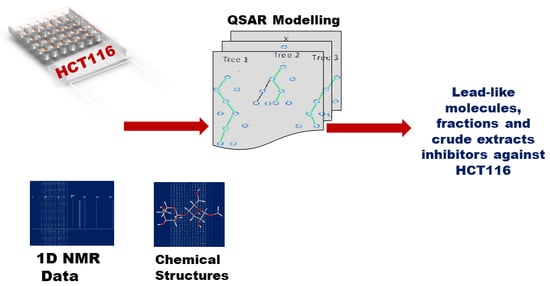
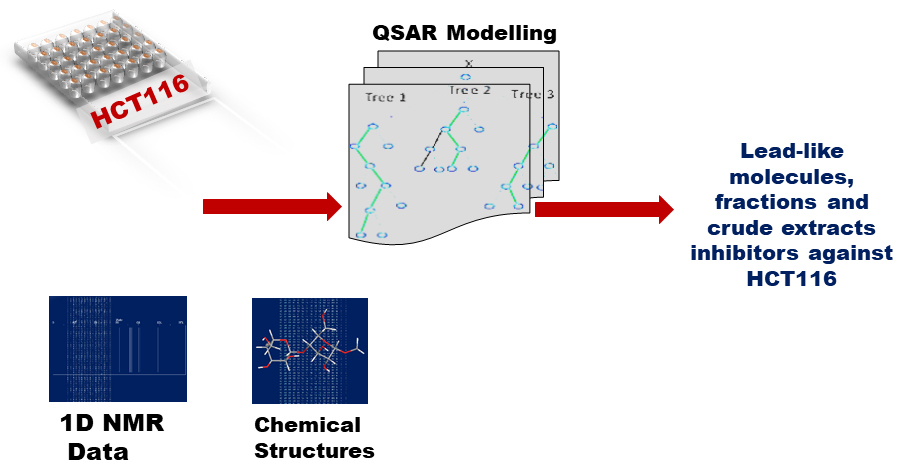
1. Introduction
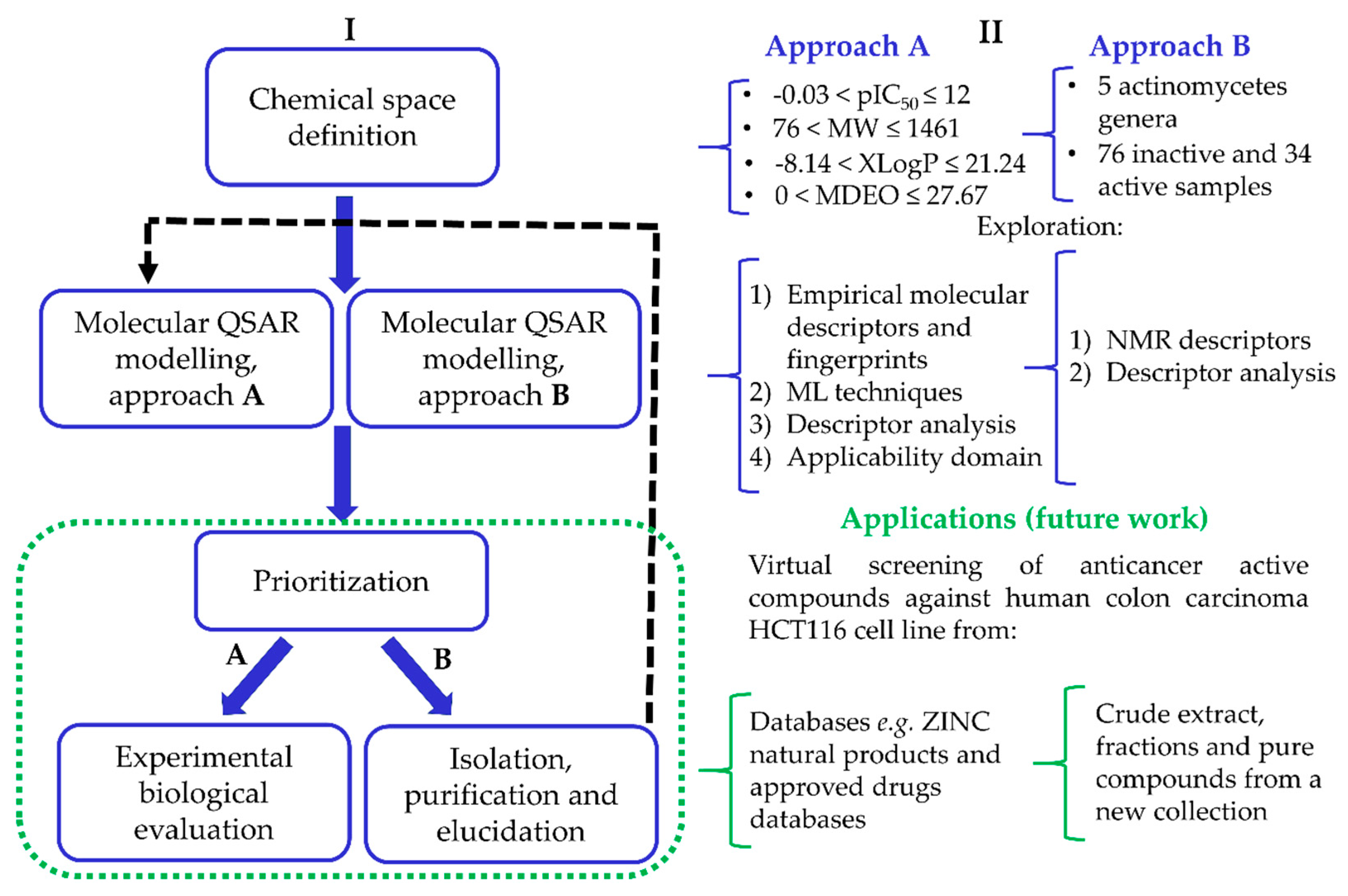
2. Materials and Methods
2.1. Data Sets
2.1.1. Approach A
2.1.2. Approach B
2.2. Descriptors
2.2.1. Approach A
2.2.2. Approach B
2.3. Selection of Training and Test Sets
2.3.1. Approach A
2.3.2. Approach B
2.4. Selection of Descriptors and Optimization of QSAR Models
Approach A
2.5. Machine Learning Techniques
2.5.1. k-Nearest Neighbors
2.5.2. Random Forests
2.5.3. Support Vector Machines
2.6. Anticancer Screening in HCT116 Human Colon Carcinoma Cells
2.6.1. Cell Culture
2.6.2. Crude Extract and 5-FU Exposure
2.6.3. Evaluation of Cytotoxicity
3. Results and Discussion
3.1. Chemical Space of the HCT116 Models
3.1.1. Approach A
3.1.2. Approach B
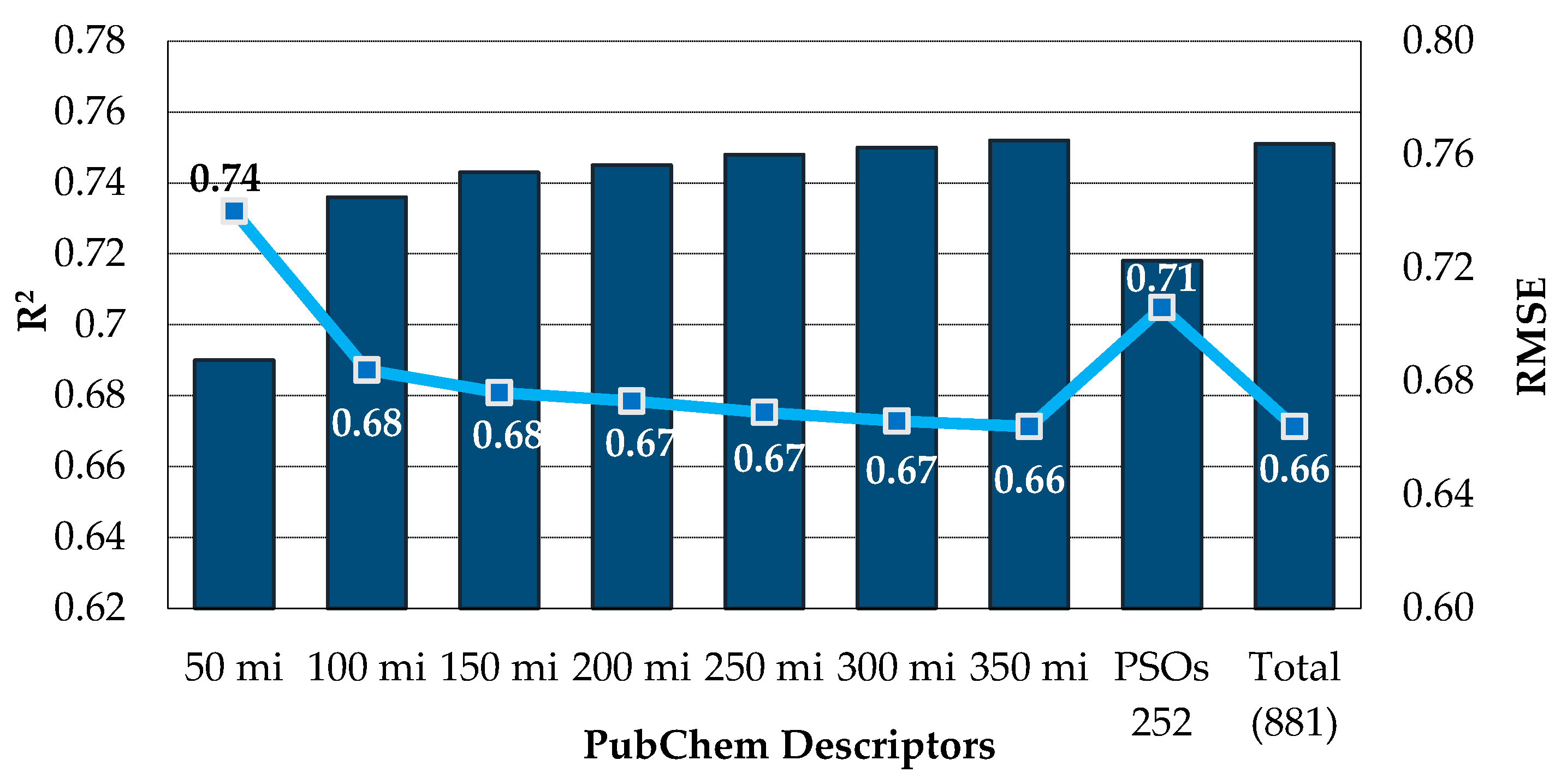
3.2. Exploration of Empirical Molecular Descriptors and Fingerprints for QSAR Approach A
3.2.1. Exploration of Other State-of-the-Art Machine Learning Techniques
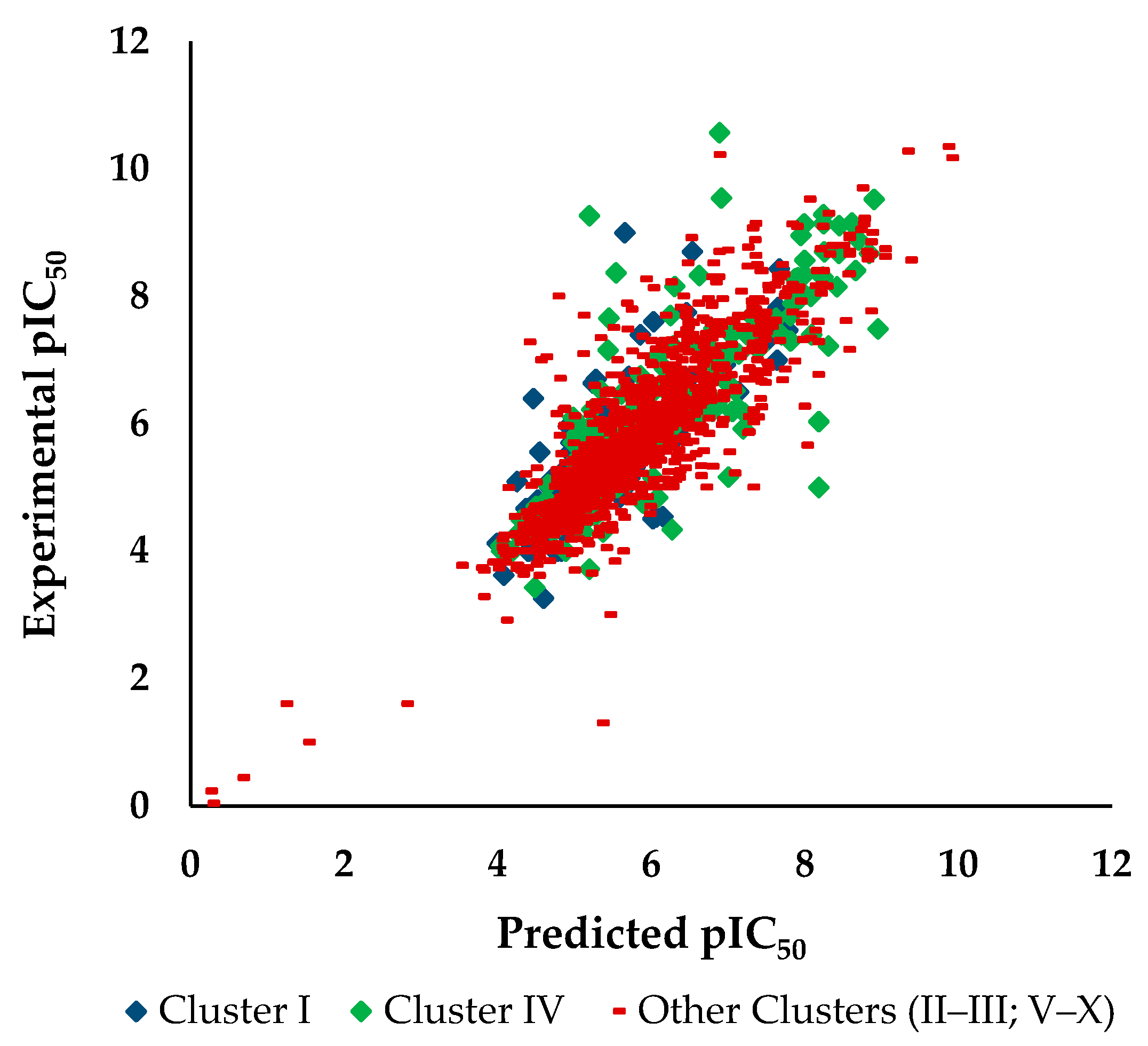
3.2.2. Analysis of PubChem Fingerprints Identified as Relevant for Modeling the pIC50 Against HCT116
3.2.3. Applicability Domain of the pIC50 Against HCT116 Model
3.3. Exploration of NMR Descriptors for QSAR Approach B
Analysis of NMR Descriptors Identified as Relevant for Modeling HCT116 Activity in the Best RF Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer. Cancer Today. Available online: http://gco.iarc.fr/today (accessed on 23 March 2017).
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [PubMed]
- Nantasenamat, C.; Prachayasittikul, V. Maximizing computational tools for successful drug discovery. Expert Opin. Drug Discov. 2015, 10, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, J. Chemoinformatics: Achievements and challenges, a personal view. Molecules 2016, 21, 151. [Google Scholar] [CrossRef] [PubMed]
- Mueller, R.; Dawson, E.S.; Meiler, J.; Rodriguez, A.L.; Chauder, B.A.; Bates, B.S.; Felts, A.S.; Lamb, J.P.; Menon, U.N.; Jadhav, S.B.; et al. Discovery of 2-(2-benzoxazoyl amino)-4-aryl-5-cyanopyrimidine as negative allosteric modulators (NAMs) of metabotropic glutamate receptor 5 (mGlu5): From an artificial neural network virtual screen to an in vivo tool compound. ChemMedChem 2012, 7, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed]
- Katsila, T.; Spyroulias, G.A.; Patrinos, G.P.; Matsoukas, M.-T. Computational approaches in target identification and drug discovery. Comput. Struct. Biotechnol. J. 2016, 14, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.E. What has computer-aided molecular design ever done for drug discovery? Expert Opin. Drug Discov. 2006, 1, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.Y.; Woo, Y.; Hyun, J.; Yong, Y.; Koh, D.; Lee, Y.H.; Lim, Y. Relationship between the structures of flavonoids and their NF-kappa B-dependent transcriptional activities. Bioorg. Med. Chem. Lett. 2011, 21, 6036–6041. [Google Scholar] [CrossRef] [PubMed]
- Hyun, J.; Shin, S.Y.; So, K.M.; Lee, Y.H.; Lim, Y. Isoflavones inhibit the clonogenicity of human colon cancer cells. Bioorg. Med. Chem. Lett. 2012, 22, 2664–2669. [Google Scholar] [CrossRef] [PubMed]
- Simon, L.; Imane, A.; Srinivasan, K.K.; Pathak, L.; Daoud, I. In silico drug-designing studies on flavanoids as anticolon cancer agents: Pharmacophore mapping, molecular docking, and Monte Carlo method-based QSAR modeling. Interdiscip. Sci. 2017, 9, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.-Y.; Shiao, H.-Y.; Hsu, Y.C.; Chu, C.-Y.; Wang, W.-C.; Lee, Y.-C.; Lin, W.-H.; Chen, C.-H.; Hsu, J.T.A.; Chang, C.-W.; et al. 3D-QSAR-assisted drug design: Identification of a potent quinazoline-based Aurora kinase inhibitor. ChemMedChem 2013, 8, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Girgis, A.S.; Stawinski, J.; Ismail, N.S.M.; Farag, H. Synthesis and QSAR study of novel cytotoxic spiro[3H-indole-3,2′(1′H)-pyrrolo[3,4-c]pyrrole]-2,3′,5′(1H,2′aH,4′H)-triones. Eur. J. Med. Chem. 2012, 47, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Girgis, A.S.; Panda, S.S.; Farag, I.S.A.; El-Shabiny, A.M.; Moustafa, A.M.; Ismail, N.S.M.; Pillai, G.G.; Panda, C.S.; Hall, C.D.; Katritzky, A.R. Synthesis, and QSAR analysis of anti-oncological active spiro-alkaloids. Org. Biomol. Chem. 2015, 13, 1741–1753. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S.; Shin, S.Y.; Ahn, S.; Koh, D.; Lee, Y.H.; Lim, Y. Biological evaluation of 2-pyrazolinyl-1-carbothioamide derivatives against HCT116 human colorectal cancer cell lines and elucidation on QSAR and molecular binding modes. Bioorg. Med. Chem. 2017, 25, 5423–5432. [Google Scholar] [CrossRef] [PubMed]
- Zolnowska, B.; Slawinski, J.; Belka, M.; Baczek, T.; Kawiak, A.; Chojnacki, J.; Pogorzelska, A.; Szafranski, K. Synthesis, molecular structure, metabolic stability and QSAR studies of a novel series of anticancer N-acylbenzenesulfonamides. Molecules 2015, 20, 19101–19129. [Google Scholar] [CrossRef] [PubMed]
- Slawinski, J.; Zolnowska, B.; Brzozowski, Z.; Kawiak, A.; Belka, M.; Baczek, T. Synthesis and QSAR study of novel 6-chloro-3-(2-arylmethylene-1-methylhydrazino)-1,4,2-benzodithiazine 1,1-dioxide derivatives with anticancer activity. Molecules 2015, 20, 5754–5770. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-J.; Zhang, T.; Yu, S.-L.; Dai, X.-J.; Wu, Y.; Tao, J.-C. Synthesis, cytotoxic activity, and 2D and 3D QSAR studies of 19-carboxyl-modified novel isosteviol derivatives as potential anticancer agents. Chem. Biol. Drug Des. 2017, 89, 870–887. [Google Scholar] [CrossRef] [PubMed]
- Gabr, M.T.; El-Gohary, N.S.; El-Bendary, E.R.; El-Kerdawy, M.M. EGFR tyrosine kinase targeted compounds: In vitro antitumor activity and molecular modeling studies of new benzothiazole and pyrimido[2,1-b]benzothiazole derivatives. EXCLI J. 2014, 13, 573–585. [Google Scholar] [PubMed]
- Banfi, S.; Caruso, E.; Buccafurni, L.; Murano, R.; Monti, E.; Gariboldi, M.; Papa, E.; Gramatica, P. Comparison between 5,10,15,20-tetraaryl- and 5,15-diarylporphyrins as photosensitizers: Synthesis, photodynamic activity, and quantitative structure-activity relationship modeling. J. Med. Chem. 2006, 49, 3293–3304. [Google Scholar] [CrossRef] [PubMed]
- Gramatica, P.; Papa, E.; Luini, M.; Monti, E.; Gariboldi, M.B.; Ravera, M.; Gabano, E.; Gaviglio, L.; Osella, D. Antiproliferative Pt(IV) complexes: Synthesis, biological activity, and quantitative structure–activity relationship modeling. J. Biol. Inorg. Chem. 2010, 15, 1157–1169. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Drugs and drug candidates from marine sources: An assessment of the current “State of play”. Planta Med. 2016, 82, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H.; Moore, B.S. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 19, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Gaudencio, S.P.; Pereira, F. Dereplication: Racing to speed up the natural products discovery process. Nat. Prod. Rep. 2015, 32, 779–810. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Don Duy, N.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with global natural products social molecular networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Lang, G.; Mayhudin, N.A.; Mitova, M.I.; Sun, L.; van der Sar, S.; Blunt, J.W.; Cole, A.L.J.; Ellis, G.; Laatsch, H.; Munro, M.H.G. Evolving trends in the dereplication of natural product extracts: New methodology for rapid, small-scale investigation of natural product extracts. J. Nat. Prod. 2008, 71, 1595–1599. [Google Scholar] [CrossRef] [PubMed]
- Camp, D.; Davis, R.A.; Campitelli, M.; Ebdon, J.; Quinn, R.J. Drug-like properties: Guiding principles for the design of natural product libraries. J. Nat. Prod. 2012, 75, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Davo, A.; Dias, T.; Gomes, S.E.; Rodrigues, S.; Parera-Valadezl, Y.; Borralho, P.M.; Pereira, F.; Rodrigues, C.M.P.; Santos-Sanches, I.; Gaudencio, S.P. The Madeira archipelago as a significant source of marine-derived actinomycete diversity with anticancer and antimicrobial potential. Front. Microbiol. 2016, 7, 1594. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrian-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15-ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- White, R.E.; Manitpisitkul, P. Pharmacokinetic theory of cassette dosing in drug discovery screening. Drug Metab. Dispos. 2001, 29, 957–966. [Google Scholar] [PubMed]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Aires-de-Sousa, J. JATOON: Java tools for neural networks. Chemom. Intell. Lab. Syst. 2002, 61, 167–173. [Google Scholar] [CrossRef]
- Akaike, H. New look at statistical-model identification. IEEE Trans. Autom. Control 1974, 19, 716–723. [Google Scholar] [CrossRef]
- Hall, M.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The WEKA data mining software: An update. SIGKDD Explor. Newsl. 2009, 11, 10–18. [Google Scholar] [CrossRef]
- Hall, M.A.; Smith, L.A. Correlation-based feature selection for machine learning. In Proceedings of the Twelfth International FLAIRS Conference, Orlando, FL, USA, 1–5 May 1999; AAAI Press: Menlo Park, CA, USA, 1999; pp. 235–239. [Google Scholar]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random forest: A classification and regression tool for compound classification and QSAR modeling. J. Chem. Inf. Comput. Sci. 2003, 43, 1947–1958. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria. Available online: http://www.R-project.org (accessed on 12 September 2016).
- Liaw, A.; Wiener, M. Classification and regression by randomforest. R News 2002, 2, 18–22. [Google Scholar]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Chang, C.-C.; Lin, C.J. Libsvm: A library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011, 2, 27. [Google Scholar] [CrossRef]
- Gleeson, M.P. Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 2008, 51, 817–834. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.; Latino, D.A.R.S.; Gaudencio, S.P. A chemoinformatics approach to the discovery of lead-like molecules from marine and microbial sources en route to antitumor and antibiotic drugs. Mar. Drugs 2014, 12, 757–778. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.; Latino, D.A.R.S.; Gaudencio, S.P. QSAR-assisted virtual screening of lead-like molecules from marine and microbial natural sources for antitumor and antibiotic drug discovery. Molecules 2015, 20, 4848–4873. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Guha, R.; Wild, D.; Chen, T. Ensemble feature selection: Consistent descriptor subsets for multiple QSAR models. J. Chem. Inf. Model. 2007, 47, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Klementz, D.; Doering, K.; Lucas, X.; Telukunta, K.K.; Erxleben, A.; Deubel, D.; Erber, A.; Santillana, I.; Thomas, O.S.; Bechthold, A.; et al. StreptomeDB 2.0-an extended resource of natural products produced by streptomycetes. Nucleic Acids Res. 2016, 44, D509–D514. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, X.; Xiang, W.; He, L.; Tang, M.; Wang, F.; Wang, T.; Yang, Z.; Yi, Y.; Wang, H.; et al. Development of purine-based hydroxamic acid derivatives: Potent histone deacetylase inhibitors with marked in vitro and in vivo antitumor activities. J. Med. Chem. 2016, 59, 5488–5504. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.R.; Wei, J.L.; Mo, X.F.; Yuan, Z.W.; Wang, J.L.; Zhang, C.; Xie, Y.Y.; You, Q.D.; Sun, H.P. Discovery and optimization of new benzofuran derivatives against p53-independent malignant cancer cells through inhibition of HIF-1 pathway. Bioorg. Med. Chem. Lett. 2016, 26, 2713–2718. [Google Scholar] [CrossRef] [PubMed]
- Vymetalova, L.; Havlicek, L.; Sturc, A.; Skraskova, Z.; Jorda, R.; Pospisil, T.; Strnad, M.; Krystof, V. 5-substituted 3-isopropyl-7-[4-(2-pyridyl)benzyl]amino-1(2)H-pyrazolo[4,3-d]pyrimidines with anti-proliferative activity as potent and selective inhibitors of cyclin-dependent kinases. Eur. J. Med. Chem. 2016, 110, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Q.; Fedorowicz, A.; Singh, H.; Soderholm, S.C. Application of the random forest method in studies of local lymph node assay based skin sensitization data. J. Chem. Inf. Model. 2005, 45, 952–964. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clusters 1 | Training Set 2 | Test Set 2 | Average/Maximum pIC50 3 |
|---|---|---|---|
I—ChEMBL1078221 | 650 | 129 | 5.24/11.00 |
II—ChEMBL1078389 | 569 | 135 | 5.43/9.52 |
III—ChEMBL148968 | 438 | 105 | 5.42/9.60 |
IV—ChEMBL116081 | 713 | 200 | 5.83/11.51 |
V—ChEMBL1083086 | 885 | 208 | 5.32/9.26 |
VI—ChEMBL104408 | 405 | 119 | 5.80/9.31 |
VII—ChEMBL1090871 | 661 | 159 | 5.39/12.00 |
VIII—ChEMBL116614 | 626 | 152 | 5.76/9.24 |
IX—ChEMBL1078573 | 513 | 125 | 5.74/10.35 |
X—ChEMBL1830679 | 415 | 132 | 5.77/9.05 |
| Actinomycetes Genera | Set (Number/Sample Types) | Activity Class/Average IC50 1 |
|---|---|---|
| Actinomadura | Tr 2 set (2, crude extracts) | inactive/≥156 |
| Micromonospora | Tr 2 set (4, 1 crude extract and 3 fractions) | active/33.95 |
| Micromonospora | Tr 2 set (11, 3 crude extracts and 8 fractions) | inactive/≥156 |
| Salinispora | Tr 2 set (1, 1 fraction) | active/9.8 |
| Salinispora | Tr 2 set (20, 9 crude extracts and 11 fractions) | inactive/≥156 |
| Streptomyces | Tr 2 set (20, 11 crude extracts and 9 fractions) | active/16.26 |
| Streptomyces | Tr 2 set (16, 9 crude extracts and 7 fractions) | inactive/≥15 |
| Actinomadura | Te 3 set (1, crude extract) | inactive/≥156 |
| Brevibacterium | Te 3 set (1, crude extract) | inactive/≥156 |
| Micromonospora | Te 3 set (1, crude extract) | active/7.9 |
| Micromonospora | Te 3 set (11, 1 crude extract, 5 fractions, and 5 pure compounds) | inactive/≥156 |
| Salinispora | Te 3 set (1, crude fraction) | active/4.94 |
| Salinispora | Te 3 set (7, 5 crude extracts and 2 fractions) | inactive/≥156 |
| Streptomyces | Te 3 set (7, 2 crude extracts and 5 fractions) | active/26.31 |
| Streptomyces | Te 3 set (7, 3 crude extracts and 4 fractions) | inactive/≥156 |
| Descriptors (#) | CFS Search Type | NO. of Selected Descriptors | R2 | RMSE | MAE | % error ≥ 1/% error < 1 1 |
|---|---|---|---|---|---|---|
| E-State (79) 2 | GSW 4 | 13 | 0.174 | 1.208 | 0.927 | 38/62 |
| MACCS (166) 2 | PSOs 5 | 34 | 0.512 | 0.937 | 0.665 | 22/78 |
| Sub (307) 2 | PSOs 5 | 63 | 0.372 | 1.055 | 0.797 | 30/70 |
| SubC (307) 2 | BF 6 | 63 | 0.509 | 0.942 | 0.671 | 23/77 |
| AP2D (780) 2 | PSOs 5 | 120 | 0.442 | 1.007 | 0.702 | 23/77 |
| APC2D (780) 2 | PSOs 5 | 174 | 0.589 | 0.866 | 0.589 | 18/82 |
| PubChem (881) 2 | PSOs 5 | 252 | 0.696 | 0.742 | 0.500 | 14/86 |
| CDK (1024) 2 | PSOs 5 | 283 | 0.725 | 0.707 | 0.474 | 12/88 |
| CDK Ext (1024) 2 | PSOs 5 | 257 | 0.718 | 0.717 | 0.476 | 13/87 |
| CDK graph (1024) 2 | PSOs 5 | 179 | 0.644 | 0.807 | 0.546 | 16/84 |
| KR (4860) 2 | PSOs 5 | 192 | 0.604 | 0.847 | 0.591 | 19/81 |
| KRC (4860) 2 | PSOs 5 | 160 | 0.618 | 0.832 | 0.579 | 18/82 |
| 1D2D (1438) 3 | PSOs 5 | 416 | 0.703 | 0.737 | 0.493 | 13/87 |
| 1D2D3D (1869) 3 | PSOs 5 | 489 | 0.705 | 0.733 | 0.493 | 13/87 |
| Models | ML | |||
|---|---|---|---|---|
| RF 1 | SVM 2 | K-NN 2 | ||
| 1D2D 3 | R2 | 0.730 | 0.647 | 0.703 |
| RMSE | 0.708 | 0.800 | 0.737 | |
| MAE | 0.523 | 0.566 | 0.493 | |
| % error ≥ 1/% error < 1 7 | 13/87 | 16/84 | 13/87 | |
| 1D2D3D 4 | R2 | 0.729 | 0.615 | 0.705 |
| RMSE | 0.713 | 0.842 | 0.733 | |
| MAE | 0.525 | 0.572 | 0.493 | |
| % error ≥ 1/% error < 1 7 | 13/87 | 17/83 | 13/87 | |
| PubChem 5 | R2 | 0.751 | 0.677 | 0.696 |
| RMSE | 0.664 | 0.762 | 0.742 | |
| MAE | 0.466 | 0.535 | 0.500 | |
| % error ≥ 1/% error < 1 7 | 12/88 | 15/85 | 14/86 | |
| CDK 6 | R2 | 0.753 | 0.744 | 0.725 |
| RMSE | 0.665 | 0.674 | 0.707 | |
| MAE | 0.471 | 0.469 | 0.474 | |
| % error ≥ 1/% error < 1 7 | 11/89 | 12/88 | 12/88 | |
| Clusters | Training Set | Test Set | ||||||
|---|---|---|---|---|---|---|---|---|
| # | R2 | RMSE | MAE | # | R2 | RMSE | MAE | |
| I | 650 | 0.702 | 0.721 | 0.479 | 129 | 0.613 | 0.673 | 0.455 |
| II | 569 | 0.766 | 0.627 | 0.446 | 135 | 0.781 | 0.619 | 0.461 |
| III | 438 | 0.792 | 0.648 | 0.459 | 105 | 0.697 | 0.737 | 0.516 |
| IV | 713 | 0.759 | 0.703 | 0.459 | 200 | 0.682 | 0.821 | 0.533 |
| V | 885 | 0.685 | 0.658 | 0.481 | 208 | 0.658 | 0.734 | 0.489 |
| VI | 405 | 0.649 | 0.646 | 0.460 | 119 | 0.637 | 0.616 | 0.462 |
| VII | 661 | 0.790 | 0.652 | 0.445 | 159 | 0.776 | 0.625 | 0.430 |
| VIII | 626 | 0.636 | 0.708 | 0.512 | 152 | 0.706 | 0.585 | 0.432 |
| IX | 513 | 0.846 | 0.599 | 0.412 | 125 | 0.794 | 0.720 | 0.487 |
| X | 415 | 0.746 | 0.628 | 0.440 | 132 | 0.767 | 0.659 | 0.448 |
| Code | DI 1 | Chemical Pattern |
|---|---|---|
| HEC_2 | 17th | ≥16C |
| HEC_19 | 16th | ≥2O |
| ESSSR_157 | 10th | ≥3 any ring size 5 |
| ESSSR_261 | 5th | ≥4 aromatic rings |
| SAP_301 | 18th | N-O |
| SAP_305 | 19th | N-S |
| SANN_335 | 7th |  ~Any bond order but no aromatic bond ~Any bond order but no aromatic bond |
| SANN_338 | 4th |  ~Any bond order but no aromatic bond ~Any bond order but no aromatic bond |
| SANN_339 | 11th |  ~Any bond order but no aromatic bond ~Any bond order but no aromatic bond |
| SANN_346 | 15th |  ~Any bond order but no aromatic bond ~Any bond order but no aromatic bond |
| DANh_432 | 8th |  |
| SSP_514 | 12th |  |
| SSP_518 | 6th |  |
| SSP_615 | 13th |  |
| SSP_631 | 20th |  |
| SSP_643 | 3rd |  |
| SSP_672 | 14th |  |
| CSP_713 | 1st |  |
| CSP_755 | 9th |  |
| CSP_819 | 2nd |  |
| Model | # 2 | Training 1/Test Sets | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| TP 3 | TN 4 | FP 5 | FN 6 | SE 7 | SP 8 | Q 9 | G-Mean 10 | |||
| 13C | 0.5 | 400 | 12/3 | 38/20 | 11/7 | 13/6 | 0.48/0.33 | 0.78/0.74 | 0.68/0.64 | 0.61/0.50 |
| 1 | 200 | 13/3 | 35/16 | 8/7 | 12/6 | 0.52/0.33 | 0.81/0.70 | 0.71/0.59 | 0.65/0.48 | |
| 1.5 | 133 | 12/2 | 42/20 | 7/7 | 13/7 | 0.48/0.22 | 0.86/0.74 | 0.73/0.61 | 0.64/0.41 | |
| 1H | 0.05 | 240 | 13/2 | 41/20 | 8/7 | 12/7 | 0.52/0.22 | 0.84/0.74 | 0.73/0.61 | 0.66/0.41 |
| 0.1 | 120 | 15/2 | 40/19 | 9/8 | 10/7 | 0.60/0.22 | 0.82/0.70 | 0.74/0.58 | 0.70/0.40 | |
| 0.2 | 61 | 14/2 | 39/18 | 10/9 | 11/7 | 0.56/0.22 | 0.80/0.67 | 0.72/0.56 | 0.67/0.39 | |
| 1H and 13C | 0.05; 0.5 | 640 | 13/4 | 44/21 | 5/6 | 12/5 | 0.52/0.44 | 0.90/0.78 | 0.77/0.69 | 0.68/0.59 |
| 0.1; 0.5 | 520 | 14/5 | 44/19 | 5/8 | 11/4 | 0.56/0.56 | 0.90/0.70 | 0.78/0.67 | 0.71/0.63 | |
| 0.1; 1 | 320 | 13/3 | 44/19 | 5/8 | 12/6 | 0.52/0.33 | 0.90/0.70 | 0.77/0.61 | 0.68/0.48 | |
| Sets | TP 1 | TN 2 | FP 3 | FN 4 | SE 5 | SP 6 | Q 7 | G-Mean 8 |
|---|---|---|---|---|---|---|---|---|
| Training | 18 | 36 | 13 | 7 | 0.72 | 0.74 | 0.73 | 0.73 |
| Test | 6 | 17 | 10 | 3 | 0.67 | 0.63 | 0.64 | 0.65 |
| Code | Activity Class | Probability of Being Moderate-Active-to-Active |
|---|---|---|
| PTM-99_F2_F27 | Inactive | 0.26 |
| PTM-99_F2_F31 | Inactive | 0.42 |
| PTM-420_F4_F15 | Moderate-active-to-active | 0.64 |
| PTM-420_F5_F42 | Moderate-active-to-active | 0.53 |
| PTM-420_F5_F43 | Moderate-active-to-active | 0.55 |
| H or C (# 1) | NMR Range (ppm) | DI 2 | Importance for Classes | Pattern Identification | |
|---|---|---|---|---|---|
| MAct-Act 3 | InAct 4 | ||||
| H (14) | 1.3019–1.4019 | 1st | 5.43 | 5.97 | Saturated  |
| H (44) | 4.3019–4.4019 | 2nd | 5.90 | 4.46 | Z = O, N, X 5  |
| H (2) | 0.1019–0.2019 | 3rd | 6.43 | 4.01 | Saturated  |
| H (3) | 0.2019–0.3019 | 4th | 4.79 | 4.20 | Saturated |
| H (4) | 0.3019–0.4019 | 5th | 3.94 | 4.60 | Saturated |
| H (45) | 4.4019–4.5019 | 6th | 4.49 | 4.13 | Z = O, N, X 5 |
| H (5) | 0.4019–0.5019 | 7th | 3.27 | 4.04 | Saturated |
| C (271) | 74.9927–75.4927 | 8th | 2.00 | 2.98 | Alcohol and ethers  Alkynes  |
| H (6) | 0.5019–0.6019 | 9th | 1.77 | 3.25 | Saturated |
| H (52) | 5.1019–5.2019 | 10th | 1.87 | 2.67 | Vinylic  |
| H (32) | 3.1019–3.2019 | 12th | 0.881 | 2.87 | Z = O, N, X 5 |
| H (51) | 5.0019–5.1019 | 15th | 0.0887 | 2.73 | Vinylic |
| C (170) | 24.4927–24.9927 | 20th | 0.712 | 2.14 | Allylic  N-Alkyl amines  Saturated  |
| C (352) | 115.4927–115.9927 | 21th | 2.12 | 0.833 | Aromatic  Olefinic  Nitrile  |
| C (280) | 79.4927–79.9927 | 26th | 0.0743 | 1.88 | Alcohol and ethers  Alkynes  |
| H (73) | 7.2019–7.3019 | 32th | 0.083 | 1.91 | Aromatic Conjugated olefinic  |
| H (13) | 1.2019–1.3019 | 49th | 1.42 | 0.0695 | Saturated |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruz, S.; Gomes, S.E.; Borralho, P.M.; Rodrigues, C.M.P.; Gaudêncio, S.P.; Pereira, F. In Silico HCT116 Human Colon Cancer Cell-Based Models En Route to the Discovery of Lead-Like Anticancer Drugs. Biomolecules 2018, 8, 56. https://doi.org/10.3390/biom8030056
Cruz S, Gomes SE, Borralho PM, Rodrigues CMP, Gaudêncio SP, Pereira F. In Silico HCT116 Human Colon Cancer Cell-Based Models En Route to the Discovery of Lead-Like Anticancer Drugs. Biomolecules. 2018; 8(3):56. https://doi.org/10.3390/biom8030056
Chicago/Turabian StyleCruz, Sara, Sofia E. Gomes, Pedro M. Borralho, Cecília M. P. Rodrigues, Susana P. Gaudêncio, and Florbela Pereira. 2018. "In Silico HCT116 Human Colon Cancer Cell-Based Models En Route to the Discovery of Lead-Like Anticancer Drugs" Biomolecules 8, no. 3: 56. https://doi.org/10.3390/biom8030056
APA StyleCruz, S., Gomes, S. E., Borralho, P. M., Rodrigues, C. M. P., Gaudêncio, S. P., & Pereira, F. (2018). In Silico HCT116 Human Colon Cancer Cell-Based Models En Route to the Discovery of Lead-Like Anticancer Drugs. Biomolecules, 8(3), 56. https://doi.org/10.3390/biom8030056