Protein Phosphatase Sit4 Affects Lipid Droplet Synthesis and Soraphen A Resistance Independent of Its Role in Regulating Elongator Dependent tRNA Modification

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Strains
2.2. Reagents
2.3. Growth Conditions and Lipid Droplets Quantification
2.4. Drug Assays
3. Results
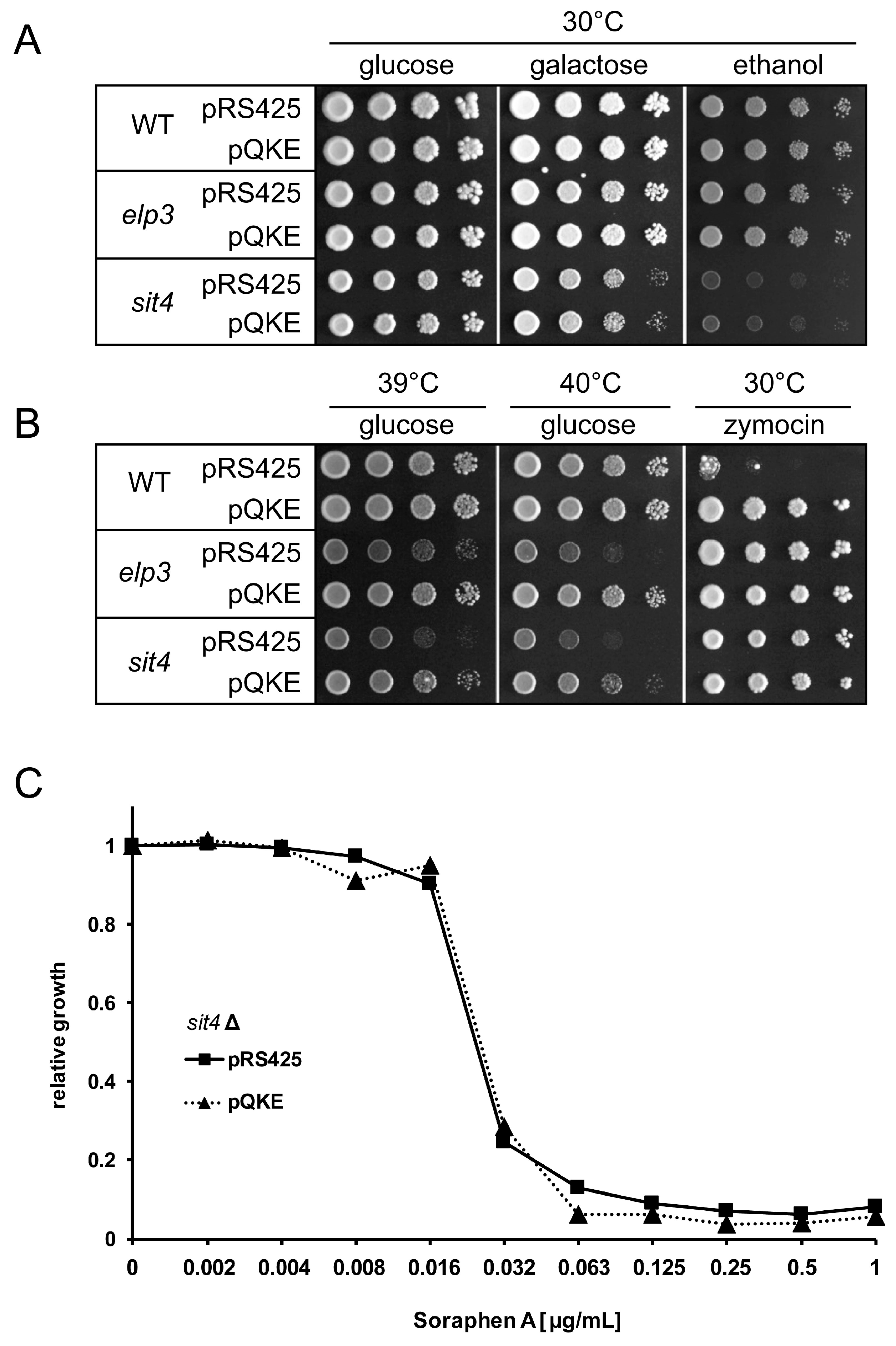
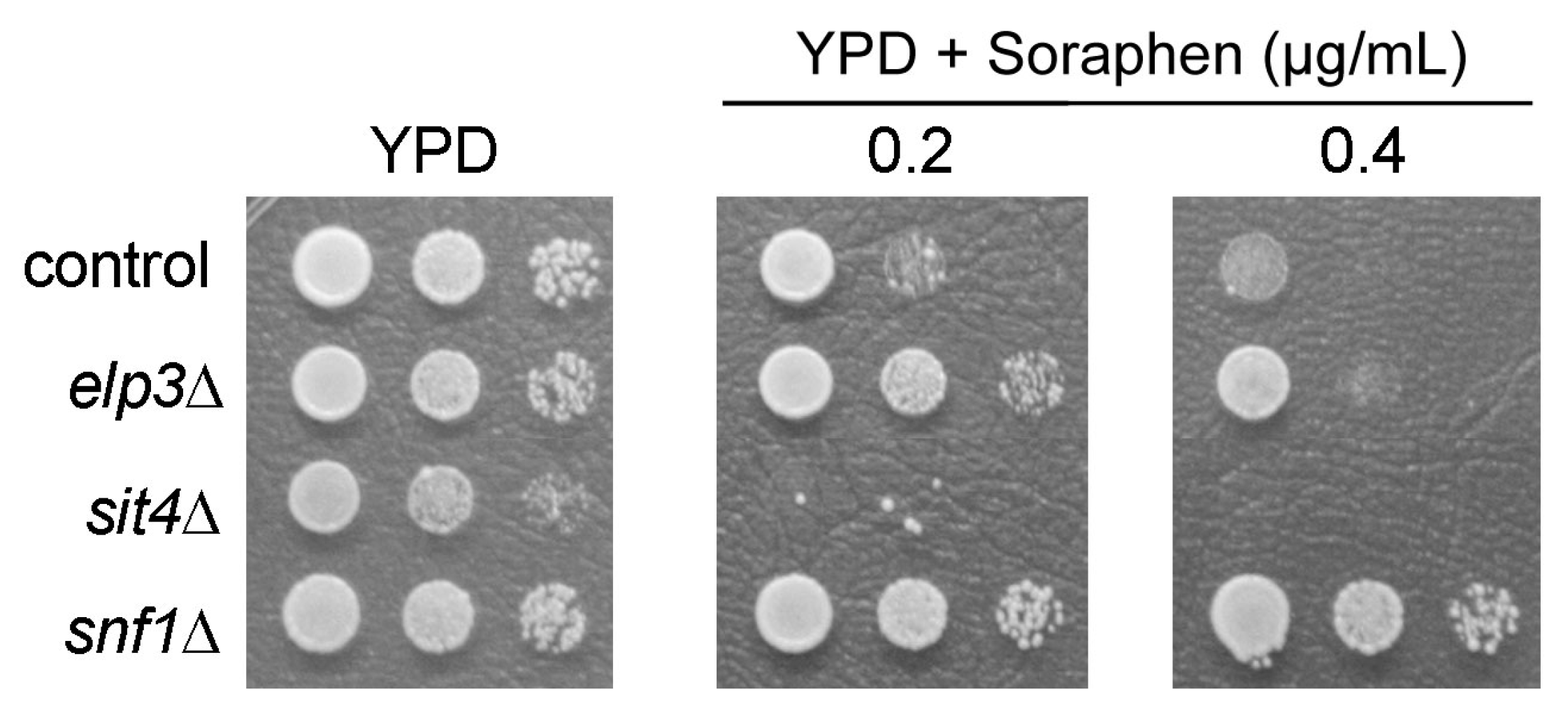
3.1. Analysis of Shared and Non-Shared Phenotypes of sit4 and elp3 Mutants
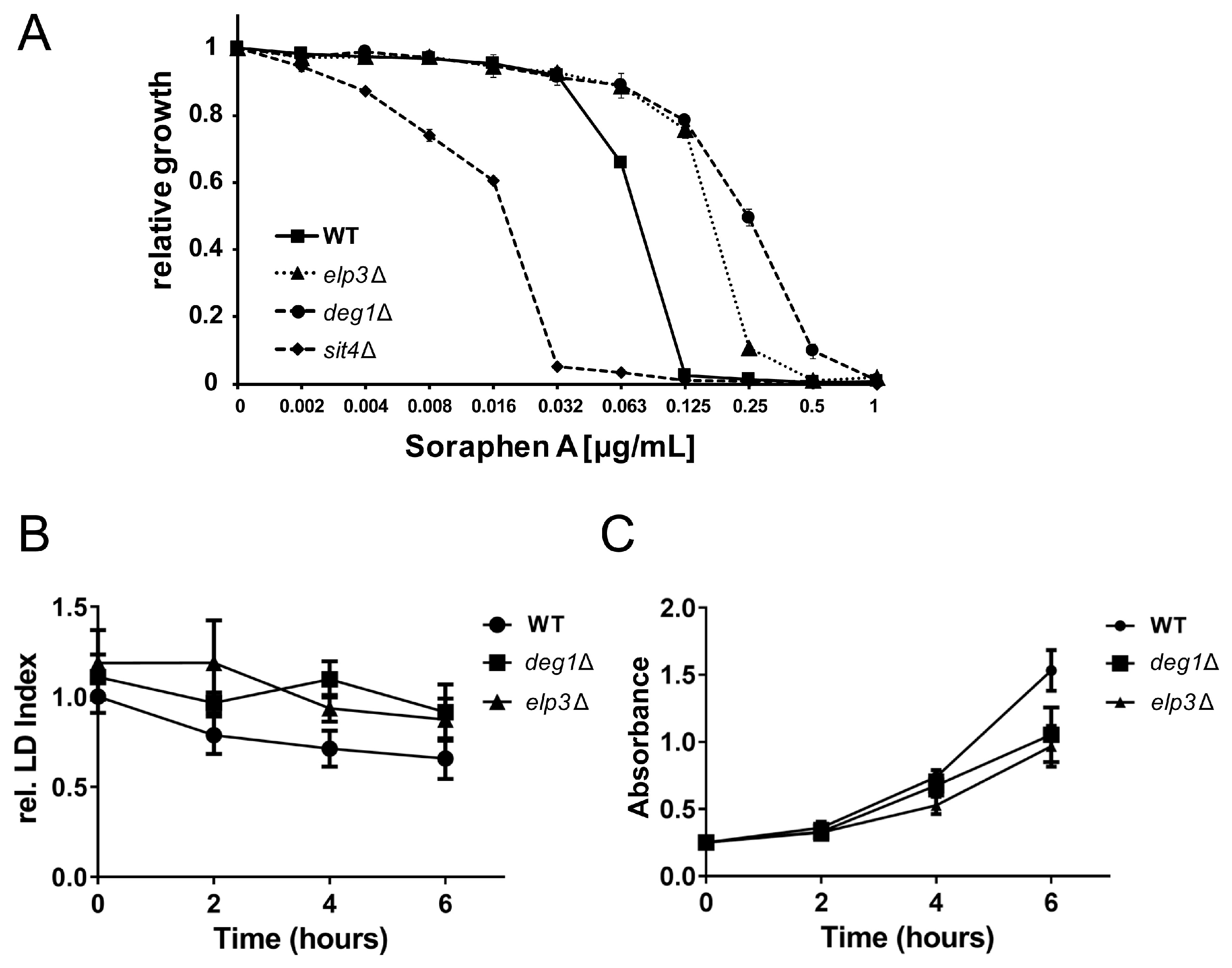
3.2. Role of 5-Methoxycarbonylmethyluridine Modification in Soraphen A Resistance and Lipid Droplet Dynamics
3.3. Screening Other tRNA Modification Mutants for Soraphen A Resistance
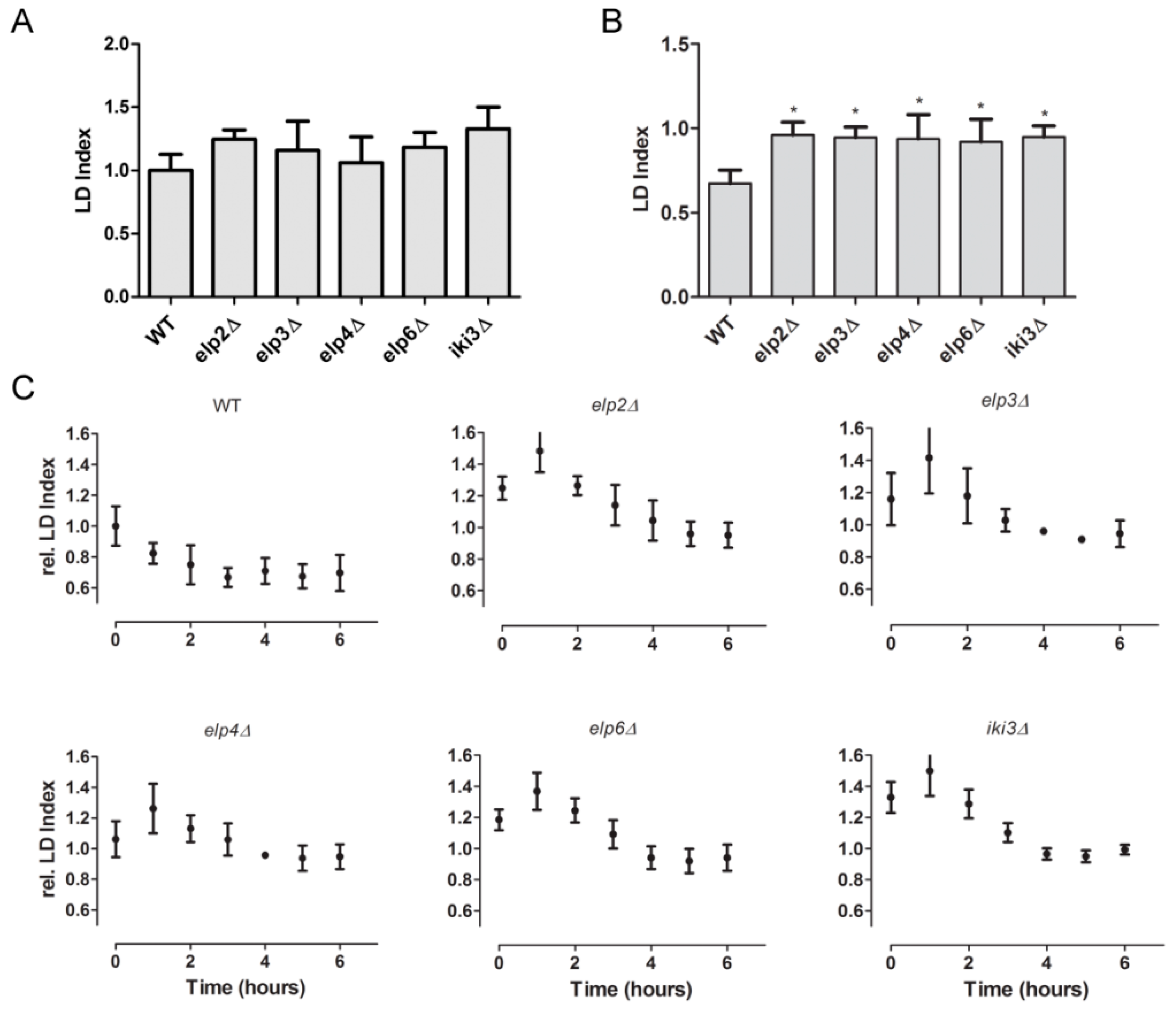
3.4. Changes in Lipid Droplet Dynamics Upon Loss of Ψ38/39
3.5. Rescue of Lipolysis Defect of deg1 Mutants by tRNA Overexpression
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hasslacher, M.; Ivessa, A.S.; Paltauf, F.; Kohlwein, S.D. Acetyl-CoA carboxylase from yeast is an essential enzyme and is regulated by factors that control phospholipid metabolism. J. Biol. Chem. 1993, 268, 10946–10952. [Google Scholar] [PubMed]
- Bozaquel-Morais, B.L.; Madeira, J.B.; Maya-Monteiro, C.M.; Masuda, C.A.; Montero-Lomeli, M. A new fluorescence-based method identifies protein phosphatases regulating lipid droplet metabolism. PLoS ONE 2010, 5, e13692. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Munday, M.R.; Scott, J.; Yang, X.; Carlson, M.; Carling, D. Yeast SNF1 is functionally related to mammalian AMP-activated protein kinase and regulates acetyl-CoA carboxylase in vivo. J. Biol. Chem. 1994, 269, 19509–19515. [Google Scholar] [PubMed]
- Conrad, M.; Schothorst, J.; Kankipati, H.N.; van Zeebroeck, G.; Rubio-Texeira, M.; Thevelein, J.M. Nutrient sensing and signaling in the yeast Saccharomyces cerevisiae. FEMS Microbiol. Rev. 2014, 38, 254–299. [Google Scholar] [CrossRef] [PubMed]
- Madeira, J.B.; Masuda, C.A.; Maya-Monteiro, C.M.; Matos, G.S.; Montero-Lomelí, M.; Bozaquel-Morais, B.L. TORC1 inhibition induces lipid droplet replenishment in yeast. Mol. Cell. Biol. 2015, 35, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Jablonowski, D.; Fichtner, L.; Stark, M.J.R.; Schaffrath, R. The yeast elongator histone acetylase requires Sit4-dependent dephosphorylation for toxin-target capacity. Mol. Biol. Cell 2004, 15, 1459–1469. [Google Scholar] [CrossRef] [PubMed]
- Jablonowski, D.; Täubert, J.-E.; Bär, C.; Stark, M.J.R.; Schaffrath, R. Distinct subsets of Sit4 holophosphatases are required for inhibition of Saccharomyces cerevisiae growth by rapamycin and zymocin. Eukaryot. Cell 2009, 8, 1637–1647. [Google Scholar] [CrossRef] [PubMed]
- Mehlgarten, C.; Jablonowski, D.; Breunig, K.D.; Stark, M.J.R.; Schaffrath, R. Elongator function depends on antagonistic regulation by casein kinase Hrr25 and protein phosphatase Sit4. Mol. Microbiol. 2009, 73, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Schaffrath, R.; Leidel, S.A. Wobble uridine modifications–a reason to live, a reason to die?! RNA Biol. 2017, 14, 1209–1222. [Google Scholar] [CrossRef] [PubMed]
- Dauden, M.I.; Kosinski, J.; Kolaj-Robin, O.; Desfosses, A.; Ori, A.; Faux, C.; Hoffmann, N.A.; Onuma, O.F.; Breunig, K.D.; Beck, M.; et al. Architecture of the yeast Elongator complex. EMBO Rep. 2017, 18, 264–279. [Google Scholar] [CrossRef] [PubMed]
- Setiaputra, D.T.; Cheng, D.T.; Lu, S.; Hansen, J.M.; Dalwadi, U.; Lam, C.H.; To, J.L.; Dong, M.-Q.; Yip, C.K. Molecular architecture of the yeast Elongator complex reveals an unexpected asymmetric subunit arrangement. EMBO Rep. 2017, 18, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Esberg, A.; Huang, B.; Johansson, M.J.O.; Byström, A.S. Elevated levels of two tRNA species bypass the requirement for elongator complex in transcription and exocytosis. Mol. Cell 2006, 24, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Johansson, M.J.O.; Byström, A.S. An early step in wobble uridine tRNA modification requires the Elongator complex. RNA 2005, 11, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Frohloff, F.; Fichtner, L.; Jablonowski, D.; Breunig, K.D.; Schaffrath, R. Saccharomyces cerevisiae Elongator mutations confer resistance to the Kluyveromyces lactis zymocin. EMBO J. 2001, 20, 1993–2003. [Google Scholar] [CrossRef] [PubMed]
- Leidel, S.; Pedrioli, P.G.A.; Bucher, T.; Brost, R.; Costanzo, M.; Schmidt, A.; Aebersold, R.; Boone, C.; Hofmann, K.; Peter, M. Ubiquitin-related modifier Urm1 acts as a sulphur carrier in thiolation of eukaryotic transfer RNA. Nature 2009, 458, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Scheidt, V.; Jüdes, A.; Bär, C.; Klassen, R.; Schaffrath, R. Loss of wobble uridine modification in tRNA anticodons interferes with TOR pathway signaling. Microb. Cell 2014, 1, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Klassen, R.; Grunewald, P.; Thüring, K.L.; Eichler, C.; Helm, M.; Schaffrath, R. Loss of anticodon wobble uridine modifications affects tRNA(Lys) function and protein levels in Saccharomyces cerevisiae. PLoS ONE 2015, 10, e0119261. [Google Scholar] [CrossRef] [PubMed]
- Klassen, R.; Ciftci, A.; Funk, J.; Bruch, A.; Butter, F.; Schaffrath, R. tRNA anticodon loop modifications ensure protein homeostasis and cell morphogenesis in yeast. Nucleic Acids Res. 2016, 44, 10946–10959. [Google Scholar] [CrossRef] [PubMed]
- Zinshteyn, B.; Gilbert, W.V. Loss of a conserved tRNA anticodon modification perturbs cellular signaling. PLoS Genet. 2013, 9, e1003675. [Google Scholar] [CrossRef] [PubMed]
- Nedialkova, D.D.; Leidel, S.A. Optimization of Codon Translation Rates via tRNA Modifications Maintains Proteome Integrity. Cell 2015, 161, 1606–1618. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, N.; Rodnina, M.V. Thio-Modification of tRNA at the Wobble Position as Regulator of the Kinetics of Decoding and Translocation on the Ribosome. J. Am. Chem. Soc. 2017, 139, 5857–5864. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Lu, J.; Byström, A.S. A genome-wide screen identifies genes required for formation of the wobble nucleoside 5-methoxycarbonylmethyl-2-thiouridine in Saccharomyces cerevisiae. RNA 2008, 14, 2183–2194. [Google Scholar] [CrossRef] [PubMed]
- Lecointe, F.; Simos, G.; Sauer, A.; Hurt, E.C.; Motorin, Y.; Grosjean, H. Characterization of yeast protein Deg1 as pseudouridine synthase (Pus3) catalyzing the formation of Ψ38 and Ψ39 in tRNA anticodon loop. J. Biol. Chem. 1998, 273, 1316–1323. [Google Scholar] [CrossRef] [PubMed]
- Gietz, R.D.; Schiestl, R.H. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2007, 2, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Klassen, R.; Schaffrath, R. Role of Pseudouridine Formation by Deg1 for Functionality of Two Glutamine Isoacceptor tRNAs. Biomolecules 2017, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Huang, B.; Esberg, A.; Johansson, M.J.O.; Byström, A.S. The Kluyveromyces lactis γ-toxin targets tRNA anticodons. RNA 2005, 11, 1648–1654. [Google Scholar] [CrossRef] [PubMed]
- Klassen, R.; Krampe, S.; Meinhardt, F. Homologous recombination and the yKu70/80 complex exert opposite roles in resistance against the killer toxin from Pichia acaciae. DNA Repair 2007, 6, 1864–1875. [Google Scholar] [CrossRef] [PubMed]
- Klassen, R.; Wemhoff, S.; Krause, J.; Meinhardt, F. DNA repair defects sensitize cells to anticodon nuclease yeast killer toxins. Mol. Genet. Genom. MGG 2011, 285, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Romero-Aguilar, L.; Montero-Lomeli, M.; Pardo, J.P.; Guerra-Sánchez, G. Lipid Index Determination by Liquid Fluorescence Recovery in the Fungal Pathogen Ustilago Maydis. J. Vis. Exp. JoVE 2018, 134. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.R.; White, J.H.; Folawiyo, Y.; Edlin, A.; Gardiner, D.; Stark, M.J. Two Saccharomyces cerevisiae genes which control sensitivity to G1 arrest induced by Kluyveromyces lactis toxin. Mol. Cell. Biol. 1994, 14, 6306–6316. [Google Scholar] [CrossRef] [PubMed]
- Jablonowski, D.; Zink, S.; Mehlgarten, C.; Daum, G.; Schaffrath, R. tRNAGlu wobble uridine methylation by Trm9 identifies Elongator’s key role for zymocin-induced cell death in yeast. Mol. Microbiol. 2006, 59, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Kurat, C.F.; Natter, K.; Petschnigg, J.; Wolinski, H.; Scheuringer, K.; Scholz, H.; Zimmermann, R.; Leber, R.; Zechner, R.; Kohlwein, S.D. Obese yeast: Triglyceride lipolysis is functionally conserved from mammals to yeast. J. Biol. Chem. 2006, 281, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Bozaquel-Morais, B.L.; Madeira, J.B.; Venâncio, T.M.; Pacheco-Rosa, T.; Masuda, C.A.; Montero-Lomeli, M. A Chemogenomic Screen Reveals Novel Snf1p/AMPK Independent Regulators of Acetyl-CoA Carboxylase. PLoS ONE 2017, 12, e0169682. [Google Scholar] [CrossRef] [PubMed]
- Jüdes, A.; Bruch, A.; Klassen, R.; Helm, M.; Schaffrath, R. Sulfur transfer and activation by ubiquitin-like modifier system Uba4•Urm1 link protein urmylation and tRNA thiolation in yeast. Microb. Cell 2016, 3, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Kon, Y.; Phizicky, E.M. Functional importance of Ψ38 and Ψ39 in distinct tRNAs, amplified for tRNAGln(UUG) by unexpected temperature sensitivity of the s2U modification in yeast. RNA 2015, 21, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Rajakumari, S.; Daum, G. Multiple functions as lipase, steryl ester hydrolase, phospholipase, and acyltransferase of Tgl4p from the yeast Saccharomyces cerevisiae. J. Biol. Chem. 2010, 285, 15769–15776. [Google Scholar] [CrossRef] [PubMed]
- Thiaville, P.C.; Legendre, R.; Rojas-Benítez, D.; Baudin-Baillieu, A.; Hatin, I.; Chalancon, G.; Glavic, A.; Namy, O.; de Crécy-Lagard, V. Global translational impacts of the loss of the tRNA modification t6A in yeast. Microb. Cell 2016, 3, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.-J.; Donnard, E.; Gustafsson, H.T.; Garber, M.; Rando, O.J. Transcriptome-wide Analysis of Roles for tRNA Modifications in Translational Regulation. Mol. Cell 2017, 68, 978–992.e4. [Google Scholar] [CrossRef] [PubMed]
- Sokołowski, M.; Klassen, R.; Bruch, A.; Schaffrath, R.; Glatt, S. Cooperativity between different tRNA modifications and their modification pathways. Biochim. Biophys. Acta 2018, 4, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Karlsborn, T.; Mahmud, A.K.M.F.; Tükenmez, H.; Byström, A.S. Loss of ncm5 and mcm5 wobble uridine side chains results in an altered metabolic profile. Metabol. Off. J. Metabol. Soc. 2016, 12, 177. [Google Scholar] [CrossRef] [PubMed]
- Mülleder, M.; Calvani, E.; Alam, M.T.; Wang, R.K.; Eckerstorfer, F.; Zelezniak, A.; Ralser, M. Functional Metabolomics Describes the Yeast Biosynthetic Regulome. Cell 2016, 167, 553–565.e12. [Google Scholar] [CrossRef] [PubMed]
- Uršič, K.; Ogrizović, M.; Kordiš, D.; Natter, K.; Petrovič, U. Tum1 is involved in the metabolism of sterol esters in Saccharomyces cerevisiae. BMC Microbiol. 2017, 17, 181. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Nomenclature | Description | Sensitivity Score | Response |
|---|---|---|---|---|
| DEG1 | YFL001W | depressed growth rate | +6 | RESISTANT |
| TUM1 | YOR251C | thiouridine modification | +2 | RESISTANT |
| SIT4 | YDL047W | suppressor initiation of transcription | −5 | SENSITIVE |
| SAP185 | YKR028W | Sit4 associated protein | −4 | SENSITIVE |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bozaquel-Morais, B.L.; Vogt, L.; D’Angelo, V.; Schaffrath, R.; Klassen, R.; Montero-Lomelí, M. Protein Phosphatase Sit4 Affects Lipid Droplet Synthesis and Soraphen A Resistance Independent of Its Role in Regulating Elongator Dependent tRNA Modification. Biomolecules 2018, 8, 49. https://doi.org/10.3390/biom8030049
Bozaquel-Morais BL, Vogt L, D’Angelo V, Schaffrath R, Klassen R, Montero-Lomelí M. Protein Phosphatase Sit4 Affects Lipid Droplet Synthesis and Soraphen A Resistance Independent of Its Role in Regulating Elongator Dependent tRNA Modification. Biomolecules. 2018; 8(3):49. https://doi.org/10.3390/biom8030049
Chicago/Turabian StyleBozaquel-Morais, Bruno Leonardo, Leonie Vogt, Valentina D’Angelo, Raffael Schaffrath, Roland Klassen, and Mónica Montero-Lomelí. 2018. "Protein Phosphatase Sit4 Affects Lipid Droplet Synthesis and Soraphen A Resistance Independent of Its Role in Regulating Elongator Dependent tRNA Modification" Biomolecules 8, no. 3: 49. https://doi.org/10.3390/biom8030049
APA StyleBozaquel-Morais, B. L., Vogt, L., D’Angelo, V., Schaffrath, R., Klassen, R., & Montero-Lomelí, M. (2018). Protein Phosphatase Sit4 Affects Lipid Droplet Synthesis and Soraphen A Resistance Independent of Its Role in Regulating Elongator Dependent tRNA Modification. Biomolecules, 8(3), 49. https://doi.org/10.3390/biom8030049