Biophysical Investigations Elucidating the Mechanisms of Action of Antimicrobial Peptides and Their Synergism


Abstract
1. Introduction
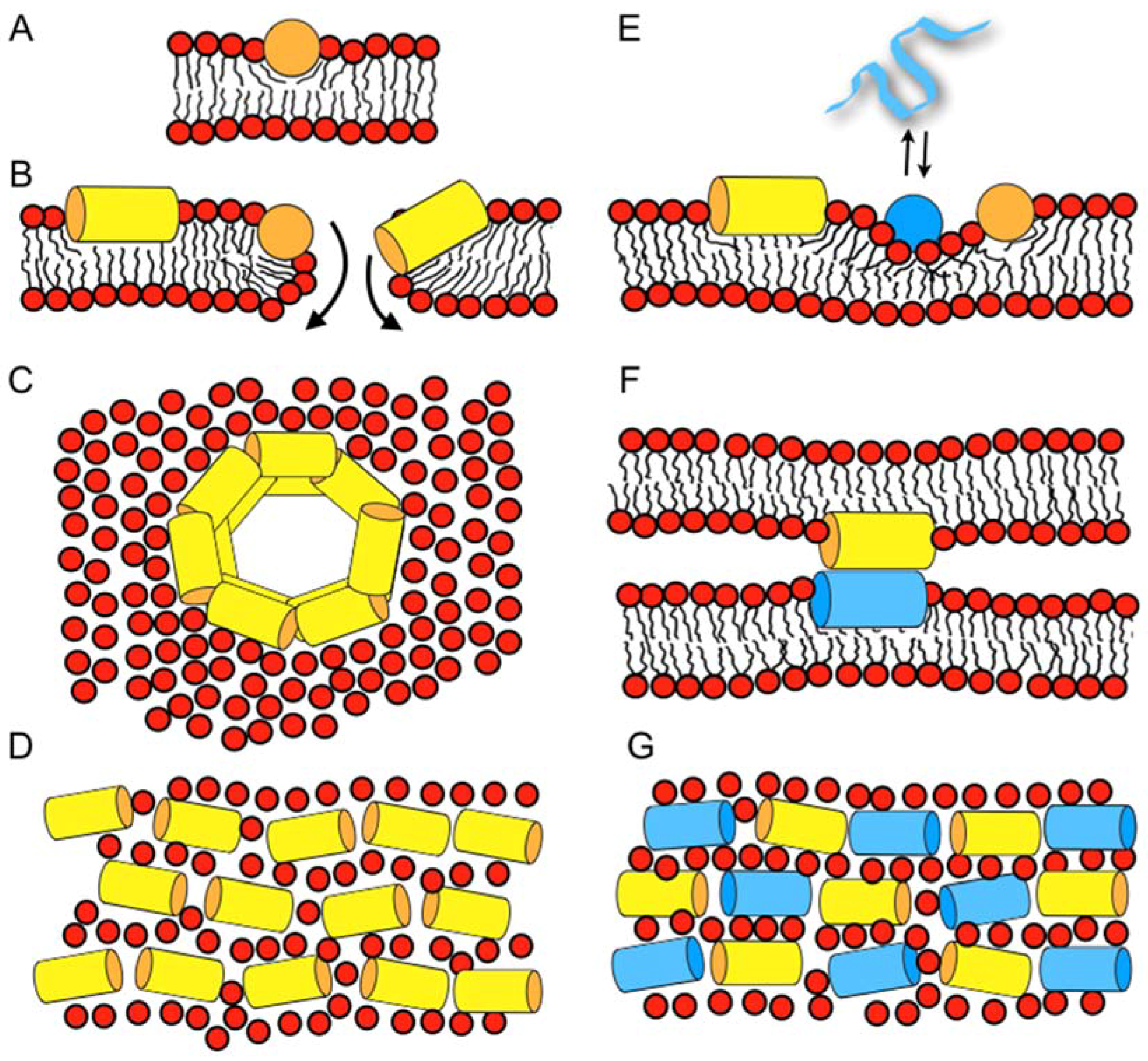
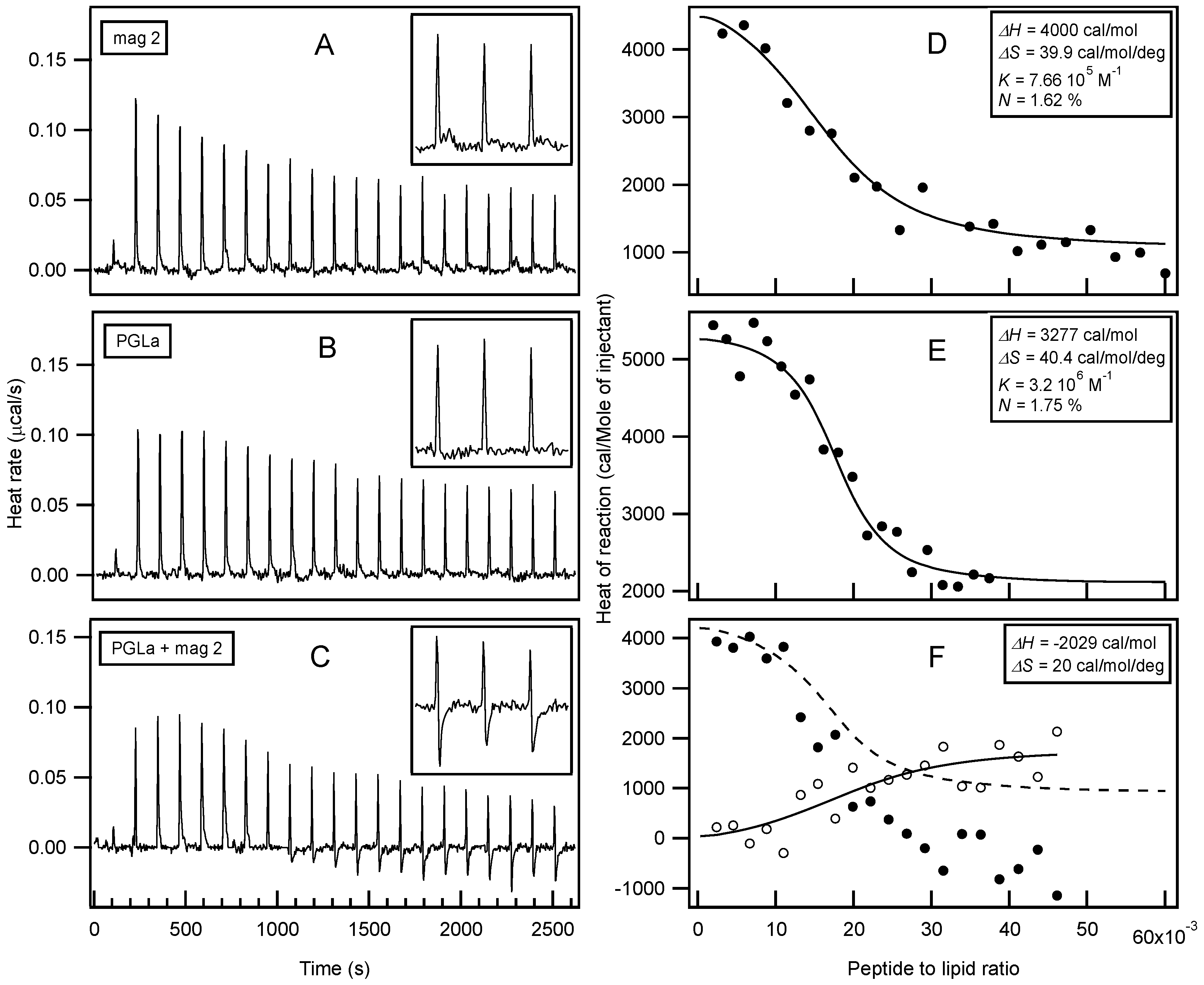
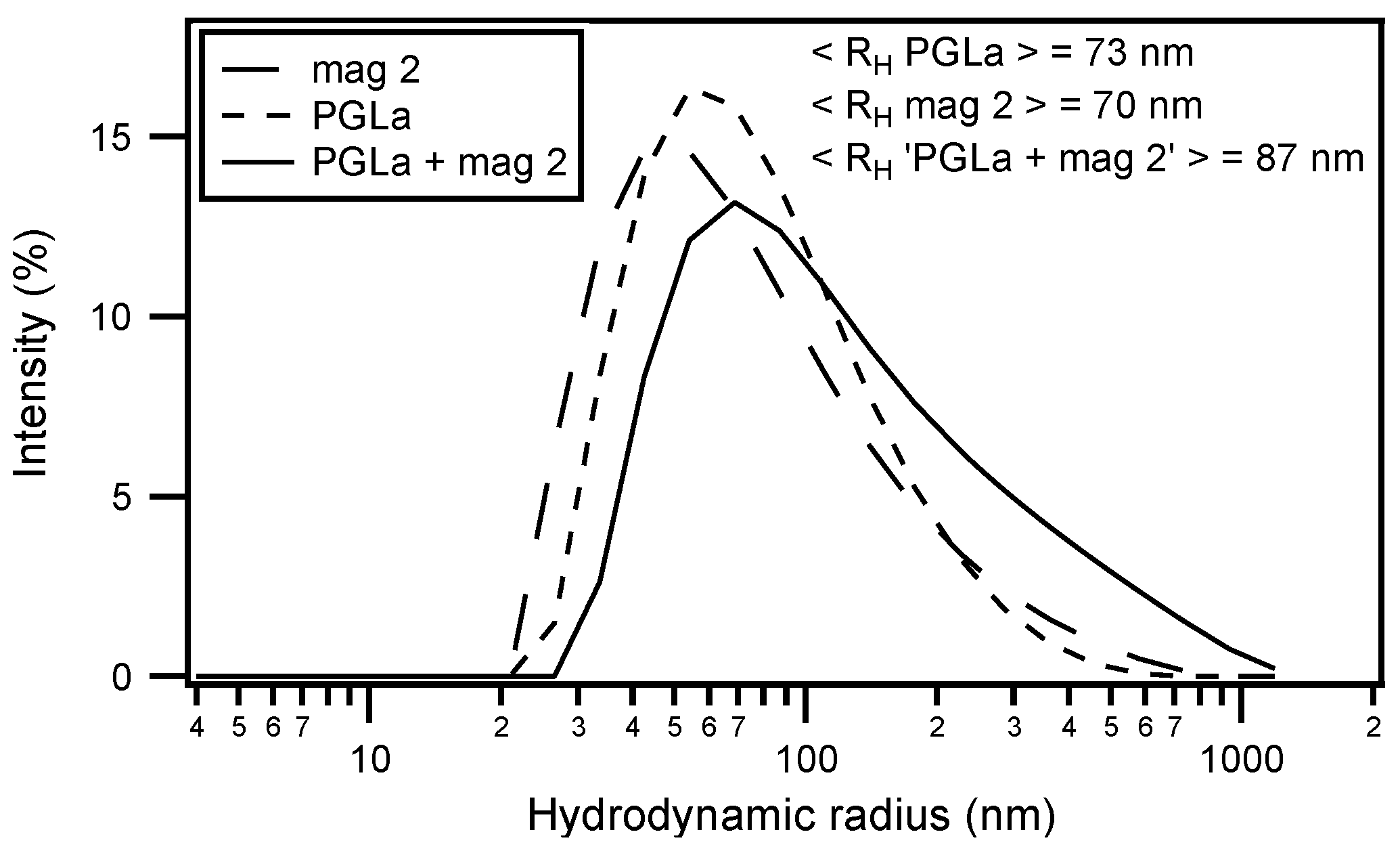
2. Synergistic Enhancement of the Activities of Antimicrobial Peptides
Acknowledgments
Conflicts of Interest
Abbreviations
| Aib | α-aminobutyric acid |
| AMP | antimicrobial peptide |
| CD | circular dichroism |
| DLPC | 1, 2-lauroyl-sn-glycero-3-phosphocholine |
| DLS | dynamic light scattering |
| DMPC | 1, 2-dimyristoyl-sn-glycero-3-phosphocholine |
| DMPG | 1, 2-dimyristoyl-sn-glycero-3-phospho-(1′-rac-glycerol) |
| DOPC | 1, 2-dioleoyl-sn-glycero-3-phosphocholine |
| DOPG | 1, 2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) |
| GUV | giant unilamellar vesicle |
| IP | in-plane |
| ITC | isothermal titration calorimetry |
| LUV | large unilamellar vesicle |
| MD | molecular dynamics |
| NMR | nuclear magnetic resonance |
| PC | phosphatidylcholine |
| PE | phosphatidylethanolamine |
| PG | phosphatidylglycerol |
| POPC | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine |
| POPE | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine |
| POPG | 1-palmitoyl-2-oleoyl -sn-glycero-3- phospho-(1′-rac-glycerol) |
| POPS | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine |
| SMART | Soft Membranes Adapt and Respond, also Transiently |
| TM | transmembrane |
References
- Boman, H.G. Peptide antibiotics and their role in innate immunity. Annu. Rev. Immunol. 1995, 13, 61–92. [Google Scholar] [CrossRef] [PubMed]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Agerberth, B.; Gunne, H.; Odeberg, J.; Kogner, P.; Boman, H.G.; Gudmundsson, G.H. FALL-39, a putative human peptide antibiotic, is cysteine-free and expressed in bone marrow and testis. Proc. Natl. Acad. Sci. USA 1995, 92, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Leitgeb, B.; Szekeres, A.; Manczinger, L.; Vagvolgyi, C.; Kredics, L. The history of alamethicin: A review of the most extensively studied peptaibol. Chem. Biodivers. 2007, 4, 1027–1051. [Google Scholar] [CrossRef] [PubMed]
- Rautenbach, M.; Troskie, A.M.; Vosloo, J.A. Antifungal peptides: To be or not to be membrane active. Biochimie 2016, 130, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Dubos, R.J.; Hotchkiss, R.D. The production of bactericidal substances by aerobic sporulating bacilli. J. Exp. Med. 1941, 73, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Kiss, G.; Michl, H. Öber das Giftsekret der Gelbbauchunke Bombina variegata L. Toxicon 1962, 1, 33–39. [Google Scholar] [CrossRef]
- Zasloff, M. Magainins, a class of antimicrobial peptides from Xenopus skin: Isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 5449–5453. [Google Scholar] [CrossRef] [PubMed]
- Pirtskhalava, M.; Gabrielian, A.; Cruz, P.; Griggs, H.L.; Squires, R.B.; Hurt, D.E.; Grigolava, M.; Chubinidze, M.; Gogoladze, G.; Vishnepolsky, B.; et al. DBAASP v.2: An enhanced database of structure and antimicrobial/cytotoxic activity of natural and synthetic peptides. Nucleic Acids Res. 2016, 44, D1104–D1112. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Sievert, D.M.; Hageman, J.C.; Boulton, M.L.; Tenover, F.C.; Downes, F.P.; Shah, S.; Rudrik, J.T.; Pupp, G.R.; Brown, W.J.; et al. Infection with vancomycin-resistant Staphylococcus aureus containing the vanA resistance gene. N. Engl. J. Med. 2003, 348, 1342–1347. [Google Scholar] [CrossRef] [PubMed]
- Yuksel, E.; Karakecili, A. Antibacterial activity on electrospun poly(lactide-co-glycolide) based membranes via Magainin II grafting. Mater. Sci. Eng. C Mater. Biol. Appl. 2014, 45, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Zou, R.; Zhu, Y.; Liu, B.; Yao, D.; Jiang, J.; Wu, J.; Tian, H. Magainin II modified polydiacetylene micelles for cancer therapy. Nanoscale 2014, 6, 14772–14783. [Google Scholar] [CrossRef] [PubMed]
- Reijmar, K.; Edwards, K.; Andersson, K.; Agmo Hernandez, V. characterizing and controlling the loading and release of cationic amphiphilic peptides onto and from PEG-stabilized lipodisks. Langmuir 2016, 32, 12091–12099. [Google Scholar] [CrossRef] [PubMed]
- Rollins-Smith, L.A.; Doersam, J.K.; Longcore, J.E.; Taylor, S.K.; Shamblin, J.C.; Carey, C.; Zasloff, M.A. Antimicrobial peptide defenses against pathogens associated with global amphibian declines. Dev. Comp. Immunol. 2002, 26, 63–72. [Google Scholar] [CrossRef]
- Sansom, M.S.P. The Biophysics of Peptide Models of Ion Channels. Prog. Biophys. Mol. Biol. 1991, 55, 139–235. [Google Scholar] [CrossRef]
- Bechinger, B. Structure and Functions of Channel-Forming Polypeptides: Magainins, Cecropins, Melittin and Alamethicin. J. Membr. Biol. 1997, 156, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, L.S.; Elmore, J.M.; Dang, U.T.; Wallace, M.J.; Marreddy, R.; Lee, R.B.; Tan, G.T.; Hurdle, J.G.; Lee, R.E.; Sun, D. Solid-Phase Synthesis and Antibacterial Activity of Cyclohexapeptide Wollamide B Analogs. ACS Comb. Sci. 2018, 20, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Yang, Y.; Uche, F.I.; Hart, S.R.; Li, W.W.; Yuan, C. Coupling Plant-Derived Cyclotides to Metal Surfaces: An Antibacterial and Antibiofilm Study. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Xue, Y.; Gao, W.; Li, J.; Zu, X.; Fu, D.; Bai, X.; Zuo, Y.; Hu, Z.; Zhang, F. Bacillaceae-derived peptide antibiotics since 2000. Peptides 2018, 101, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Laurencin, M.; Simon, M.; Fleury, Y.; Baudy-Floc’h, M.; Bondon, A.; Legrand, B. Selectivity Modulation and Structure of alpha/aza-beta(3) Cyclic Antimicrobial Peptides. Chemistry 2018. [Google Scholar] [CrossRef] [PubMed]
- Rautenbach, M.; Troskie, A.M.; Vosloo, J.A.; Dathe, M.E. Antifungal membranolytic activity of the tyrocidines against filamentous plant fungi. Biochimie 2016, 130, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Sychev, S.V.; Sukhanov, S.V.; Panteleev, P.V.; Shenkarev, Z.O.; Ovchinnikova, T.V. Marine antimicrobial peptide arenicin adopts a monomeric twisted beta-hairpin structure and forms low conductivity pores in zwitterionic lipid bilayers. Biopolymers 2017. [Google Scholar] [CrossRef]
- Salnikov, E.; Aisenbrey, C.; Balandin, S.V.; Zhmak, M.N.; Ovchinnikova, A.Y.; Bechinger, B. Structure and alignment of the membrane-associated antimicrobial peptide arenicin by oriented solid-state NMR spectroscopy. Biochemistry 2011, 50, 3784–3795. [Google Scholar] [CrossRef] [PubMed]
- Usachev, K.S.; Kolosova, O.A.; Klochkova, E.A.; Yulmetov, A.R.; Aganov, A.V.; Klochkov, V.V. Oligomerization of the antimicrobial peptide Protegrin-5 in a membrane-mimicking environment. Structural studies by high-resolution NMR spectroscopy. Eur. Biophys. J. 2017, 46, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Su, Y. Structure and dynamics of cationic membrane peptides and proteins: Insights from solid-state NMR. Protein Sci. 2011, 20, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, C.; Manjunath, G.B.; Akkapeddi, P.; Yarlagadda, V.; Hoque, J.; Uppu, D.S.S.M.; Konai, M.M.; Haldar, J. Small Molecular Antibacterial Peptoid Mimics: The Simpler the Better! J. Med. Chem. 2014, 57, 1428–1436. [Google Scholar] [CrossRef] [PubMed]
- Arnusch, C.J.; Albada, H.B.; van Vaardegem, M.; Liskamp, R.M.J.; Sahl, H.G.; Shadkchan, Y.; Osherov, N.; Shai, Y. Trivalent Ultrashort Lipopeptides are Potent pH Dependent Antifungal Agents. J. Med. Chem. 2012, 55, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Makovitzki, A.; Baram, J.; Shai, Y. Antimicrobial lipopolypeptides composed of palmitoyl Di- and tricationic peptides: In Vitro and In Vivo activities, self-assembly to nanostructures, and a plausible mode of action. Biochemistry 2008, 47, 10630–10636. [Google Scholar] [CrossRef] [PubMed]
- Patch, J.A.; Barron, A.E. Helical peptoid mimics of magainin-2 amide. J. Am. Chem. Soc. 2003, 125, 12092–12093. [Google Scholar] [CrossRef] [PubMed]
- Porter, E.A.; Weisblum, B.; Gellman, S.H. Mimicry of host-defense peptides by unnatural oligomers: Antimicrobial beta-peptides. J. Am. Chem. Soc. 2002, 124, 7324–7330. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, K.; DeGrado, W.F. Amphiphilic polymethacrylate derivatives as antimicrobial agents. J. Am. Chem. Soc. 2005, 127, 4128–4129. [Google Scholar] [CrossRef] [PubMed]
- Violette, A.; Fournel, S.; Lamour, K.; Chaloin, O.; Frisch, B.; Briand, J.P.; Monteil, H.; Guichard, G. Mimicking helical antibacterial peptides with nonpeptidic folding oligomers. Chem. Biol. 2006, 13, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Palermo, E.F.; Kuroda, K. Structural determinants of antimicrobial activity in polymers which mimic host defense peptides. Appl. Microbiol. Biotechnol. 2010, 87, 1605–1615. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.W.; DeGrado, W.F.; Tew, G.N. De novo designed synthetic mimics of antimicrobial peptides. Curr. Opin. Biotechnol. 2008, 19, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Rotem, S.; Mor, A. Antimicrobial peptide mimics for improved therapeutic properties. Biochim. Biophys. Acta 2009, 1788, 1582–1592. [Google Scholar] [CrossRef] [PubMed]
- Rank, L.A.; Walsh, N.M.; Liu, R.; Lim, F.Y.; Bok, J.W.; Huang, M.; Keller, N.P.; Gellman, S.H.; Hull, C.M. A Cationic Polymer that Shows High Antifungal Activity against Diverse Human Pathogens. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B. Insights into the mechanisms of action of host defence peptides from biophysical and structural investigations. J. Pept Sci. 2011, 17, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Lear, J.D.; Wasserman, Z.R.; DeGrado, W.F. Synthetic amphiphilic peptide models for protein ion channels. Science 1988, 240, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Killian, J.A.; de Planque, M.R.R.; van der Wel, P.C.A.; Salemink, I.; De Kruijff, B.; Greathouse, D.V.; Koeppe, R.E. Modulation of membrane structure and function by hydrophobic mismatch between proteins and lipids. Pure Appl. Chem. 1998, 70, 75–82. [Google Scholar]
- Harzer, U.; Bechinger, B. The alignment of lysine-anchored membrane peptides under conditions of hydrophobic mismatch: A CD, 15N and 31P solid-state NMR spectroscopy investigation. Biochemistry 2000, 39, 13106–13114. [Google Scholar] [CrossRef] [PubMed]
- Salnikov, E.; Aisenbrey, C.; Vidovic, V.; Bechinger, B. Solid-state NMR approaches to measure topological equilibria and dynamics of membrane polypeptides. Biochim. Biophys. Acta 2010, 1798, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B. The SMART Model: Soft Membranes Adapt and Respond, also Transiently, to External Stimuli. J. Pept. Sci. 2015, 21, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Habermann, E.; Jentsch, J. Sequenzanalyse Des Melittins Aus Den Tryptischen Und Peptischen Spatstucken. Hoppe Seyler’s Z. Physiol. Chem. 1967, 348, 37–50. [Google Scholar] [CrossRef]
- Boman, H.G. Antibacterial peptides: Basic facts and emerging concepts. J. Intern. Med. 2003, 254, 197–215. [Google Scholar] [CrossRef] [PubMed]
- Roversi, D.; Luca, V.; Aureli, S.; Park, Y.; Mangoni, M.L.; Stella, L. How Many AMP Molecules Kill a Bacterium? Spectroscopic Determination of PMAP-23 Binding to E. coli. ACS Chem. Biol. 2014, 9, 2003–2007. [Google Scholar] [CrossRef] [PubMed]
- Diamond, G.; Beckloff, N.; Weinberg, A.; Kisich, K.O. The roles of antimicrobial peptides in innate host defense. Curr. Pharm. Des. 2009, 15, 2377–2392. [Google Scholar] [CrossRef] [PubMed]
- Steinstraesser, L.; Kraneburg, U.; Jacobsen, F.; Al-Benna, S. Host defense peptides and their antimicrobial-immunomodulatory duality. Immunobiology 2011, 216, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Holzl, M.A.; Hofer, J.; Steinberger, P.; Pfistershammer, K.; Zlabinger, G.J. Host antimicrobial proteins as endogenous immunomodulators. Immunol. Lett. 2008, 119, 4–11. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, D.G.; Cudic, P.; Yu, M.K.; Behenna, D.C.; Kruger, R. Synergy and duality in peptide antibiotic mechanisms. Curr. Opin. Chem. Biol. 1999, 3, 672–680. [Google Scholar] [CrossRef]
- Kozlowska, J.; Vermeer, L.S.; Rogers, G.B.; Rehnnuma, N.; Amos, S.B.; Koller, G.; McArthur, M.; Bruce, K.D.; Mason, A.J. Combined systems approaches reveal highly plastic responses to antimicrobial peptide challenge in Escherichia coli. PLoS Pathog. 2014, 10, e1004104. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, M.H.; de Almeida, K.C.; Candido, E.S.; Murad, A.M.; Dias, S.C.; Franco, O.L. Comparative NanoUPLC-MS(E) analysis between magainin I-susceptible and -resistant Escherichia coli strains. Sci. Rep. 2017, 7, 4197. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B.; Zasloff, M.; Opella, S.J. Structure and Dynamics of the Antibiotic Peptide PGLa in Membranes by Multidimensional Solution and Solid-State NMR Spectroscopy. Biophys. J. 1998, 74, 981–987. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Murase, O.; Tokuda, H.; Funakoshi, S.; Fujii, N.; Miyajima, K. Orientational and Aggregational States of Magainin 2 in Phospholipid Bilayers. Biochemistry 1994, 33, 3342–3349. [Google Scholar] [CrossRef] [PubMed]
- Dathe, M.; Nikolenko, H.; Meyer, J.; Beyermann, M.; Bienert, M. Optimization of the antimicrobial activity of magainin peptides by modification of charge. FEBS Lett. 2001, 501, 146–150. [Google Scholar] [CrossRef]
- Wieprecht, T.; Apostolov, O.; Seelig, J. Binding of the antibacterial peptide magainin 2 amide to small and large unilamellar vesicles. Biophys. Chem. 2000, 85, 187–198. [Google Scholar] [CrossRef]
- Ludtke, S.; He, K.; Huang, H. Membrane thinning caused by magainin 2. Biochemistry 1995, 34, 16764–16769. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B.; Lohner, K. Detergent-like action of linear cationic membrane-active antibiotic peptides. Biochim. Biophys. Acta 2006, 1758, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Duclohier, H.; Molle, G.; Spach, G. Antimicrobial Peptide Magainin I from Xenopus Skin Forms Anion-Permeable Channels in Planar Lipid Bilayers. Biophys. J. 1989, 56, 1017–1021. [Google Scholar] [CrossRef]
- Cruciani, R.A.; Barker, J.L.; Zasloff, M.; Chen, H.C.; Colamonici, O. Antibiotic magainins exert cytolytic activity transformed cell lines through channel formation. Proc. Natl. Acad. Sci. USA 1991, 88, 3792–3796. [Google Scholar] [CrossRef] [PubMed]
- Christensen, B.; Fink, J.; Merrifield, R.B.; Mauzerall, D. Channel-forming properties of cecropins and related model compounds incorporated into planar lipid membranes. Proc. Natl. Acad. Sci. USA 1988, 85, 5072–5076. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Kawano, R. Channel Current Analysis for Pore-forming Properties of an Antimicrobial Peptide, Magainin 1, Using the Droplet Contact Method. Anal. Sci. 2016, 32, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Tieleman, D.P.; Hess, B.; Sansom, M.S. Analysis and evaluation of channel models: Simulations of alamethicin. Biophys. J. 2002, 83, 2393–2407. [Google Scholar] [CrossRef]
- Islam, M.Z.; Alam, J.M.; Tamba, Y.; Karal, M.A.S.; Yamazaki, M. The single GUV method for revealing the functions of antimicrobial, pore-forming toxin, and cell-penetrating peptides or proteins. Phys. Chem. Chem. Phys. 2014, 16, 15752–15767. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.M.; Cavenaugh, A.; Journigan, V.; Pokorny, A.; Almeida, P.F.F. A quantitative model for the all-or-none permeabilization of phospholipid vesicles by the antimicrobial peptide cecropin A. Biophys. J. 2008, 94, 1667–1680. [Google Scholar] [CrossRef] [PubMed]
- Tamba, Y.; Ariyama, H.; Levadny, V.; Yamazaki, M. Kinetic Pathway of Antimicrobial Peptide Magainin 2-Induced Pore Formation in Lipid Membranes. J. Phys. Chem. B 2010, 114, 12018–12026. [Google Scholar] [CrossRef] [PubMed]
- Barns, K.J.; Weisshaar, J.C. Real-time attack of LL-37 on single Bacillus subtilis cells. Biochim. Biophys. Acta 2013, 1828, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, N.; Bakshi, S.; Weisshaar, J.C. Localized permeabilization of E. coli membranes by the antimicrobial peptide Cecropin A. Biochemistry 2013, 52, 6584–6594. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Choi, H.; Weisshaar, J.C. Melittin-Induced Permeabilization, Re-sealing, and Re-permeabilization of E. coli Membranes. Biophys. J. 2018, 114, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Yang, Z.; Weisshaar, J.C. Oxidative stress induced in E. coli by the human antimicrobial peptide LL-37. PLoS Pathog. 2017, 13, e1006481. [Google Scholar] [CrossRef] [PubMed]
- Barns, K.J.; Weisshaar, J.C. Single-cell, time-resolved study of the effects of the antimicrobial peptide alamethicin on Bacillus subtilis. Biochim. Biophys. Acta 2016, 1858, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B. The structure, dynamics and orientation of antimicrobial peptides in membranes by multidimensional solid-state NMR spectroscopy. Biochim. Biophys. Acta 1999, 1462, 157–183. [Google Scholar] [CrossRef]
- Vacha, R.; Frenkel, D. Simulations suggest possible novel membrane pore structure. Langmuir 2014, 30, 1304–1310. [Google Scholar] [CrossRef] [PubMed]
- Aisenbrey, C.; Bechinger, B. Molecular Packing of Amphipathic Peptides on the Surface of Lipid Membranes. Langmuir 2014, 30, 10374–10383. [Google Scholar] [CrossRef] [PubMed]
- Leber, R.; Pachler, M.; Kabelka, I.; Svoboda, I.; Enkoller, D.; Vácha, R.; Lohner, K.; Pabst, G. Synergism of Antimicrobial Frog Peptides Couples to Membrane Intrinsic Curvature Strain. Biophys. J. 2018, in press. [Google Scholar]
- Marquette, A.; Lorber, B.; Bechinger, B. Reversible liposome association induced by LAH4: A peptide with potent antimicrobial and nucleic acid transfection activities. Biophys. J. 2010, 98, 2544–2553. [Google Scholar] [CrossRef] [PubMed]
- Marquette, A.; Salnikov, E.; Glattard, E.; Aisenbrey, C.; Bechinger, B. Magainin 2-PGLa interactions in membranes—Two peptides that exhibit synergistic enhancement of antimicrobial activity. Curr. Top. Med. Chem. 2015, 16, 65–75. [Google Scholar] [CrossRef]
- Klocek, G.; Schulthess, T.; Shai, Y.; Seelig, J. Thermodynamics of melittin binding to lipid bilayers. Aggregation and pore formation. Biochemistry 2009, 48, 2586–2596. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Baldwin, R.L. Mechanism of helix induction by trifluoroethanol: A framework for extrapolating the helix-forming properties of peptides from trifluoroethanol/water mixtures back to water. Biochemistry 1997, 36, 8413–8421. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, A.; Thennarasu, S.; Lee, D.K.; Tan, A.; Maloy, L. Solid-state NMR investigation of the membrane-disrupting mechanism of antimicrobial peptides MSI-78 and MSI-594 derived from magainin 2 and melittin. Biophys. J. 2006, 91, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Mason, A.J.; Moussaoui, W.; Abdelrhaman, T.; Boukhari, A.; Bertani, P.; Marquette, A.; Shooshtarizaheh, P.; Moulay, G.; Boehm, N.; Guerold, B.; et al. Structural determinants of antimicrobial and antiplasmodial activity and selectivity in histidine rich amphipathic cationic peptides. J. Biol. Chem. 2009, 284, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Sani, M.A.; Separovic, F. Antimicrobial Peptide Structures: From Model Membranes to Live Cells. Chemistry 2018, 24, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Hayden, R.M.; Goldberg, G.K.; Ferguson, B.M.; Schoeneck, M.W.; Libardo, M.D.; Mayeux, S.E.; Shrestha, A.; Bogardus, K.A.; Hammer, J.; Pryshchep, S.; et al. Complementary Effects of Host Defense Peptides Piscidin 1 and Piscidin 3 on DNA and Lipid Membranes: Biophysical Insights into Contrasting Biological Activities. J. Phys. Chem. B 2015, 119, 15235–15246. [Google Scholar] [CrossRef] [PubMed]
- Resende, J.M.; Verly, R.M.; Aisenbrey, C.; Amary, C.; Bertani, P.; Pilo-Veloso, D.; Bechinger, B. Membrane interactions of Phylloseptin-1, -2, and -3 peptides by oriented solid-state NMR spectroscopy. Biophys. J. 2014, 107, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Resende, J.M.; Moraes, C.M.; Munhoz, V.H.D.O.; Aisenbrey, C.; Verly, R.M.; Bertani, P.; Cesar, A.; Pilo-Veloso, D.; Bechinger, B. Membrane structure and conformational changes of the antibiotic heterodimeric peptide distinctin by solid-state NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2009, 106, 16639–16644. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B.; Gorr, S.U. Antimicrobial Peptides: Mechanisms of Action and Resistance. J. Dent. Res. 2017, 96, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Sansom, M.S. Alamethicin and related peptaibols—Model ion channels. Eur. Biophys. J. 1993, 22, 105–124. [Google Scholar] [CrossRef] [PubMed]
- North, C.L.; Barranger-Mathys, M.; Cafiso, D.S. Membrane orientation of the N-terminal segment of alamethicin determined by solid-state 15N-NMR. Biophys. J. 1995, 69, 2392–2397. [Google Scholar] [CrossRef]
- Bak, M.; Bywater, R.P.; Hohwy, M.; Thomsen, J.K.; Adelhorst, K.; Jakobsen, H.J.; Sorensen, O.W.; Nielsen, N.C. Conformation of alamethicin in oriented phospholipid bilayers determined by N-15 solid-state nuclear magnetic resonance. Biophys. J. 2001, 81, 1684–1698. [Google Scholar] [CrossRef]
- Salnikov, E.S.; Friedrich, H.; Li, X.; Bertani, P.; Reissmann, S.; Hertweck, C.; O’Neil, J.D.; Raap, J.; Bechinger, B. Structure and alignment of the membrane-associated peptaibols ampullosporin A and alamethicin by oriented 15N and 31P solid-state NMR spectroscopy. Biophys. J. 2009, 96, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Salnikov, E.S.; Raya, J.; De Zotti, M.; Zaitseva, E.; Peggion, C.; Ballano, G.; Toniolo, C.; Raap, J.; Bechinger, B. Alamethicin supramolecular organization in lipid membranes from 19F solid-state NMR. Biophys. J. 2016, 111, 2450–2459. [Google Scholar] [CrossRef] [PubMed]
- Milov, A.D.; Samoilova, R.I.; Tsvetkov, Y.D.; De Zotti, M.; Formaggio, F.; Toniolo, C.; Handgraaf, J.W.; Raap, J. Structure of Self-Aggregated Alamethicin in ePC Membranes Detected by Pulsed Electron-Electron Double Resonance and Electron Spin Echo Envelope Modulation Spectroscopies. Biophys. J. 2009, 96, 3197–3209. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Ludtke, S.J.; Heller, W.T.; Huang, H.W. Mechanism of alamethicin insertion into lipid bilayers. Biophys. J. 1996, 71, 2669–2679. [Google Scholar] [CrossRef]
- Avitabile, C.; D’Andrea, L.D.; Romanelli, A. Circular Dichroism studies on the interactions of antimicrobial peptides with bacterial cells. Sci. Rep. 2014, 4, 4293. [Google Scholar] [CrossRef] [PubMed]
- Salnikov, E.; Bechinger, B. Lipid-controlled peptide topology and interactions in bilayers: Structural insights into the synergistic enhancement of the antimicrobial activities of PGLa and magainin 2. Biophys. J. 2011, 100, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Tremouilhac, P.; Strandberg, E.; Wadhwani, P.; Ulrich, A.S. Synergistic transmembrane alignment of the antimicrobial heterodimer PGLa/magainin. J. Biol. Chem. 2006, 281, 32089–32094. [Google Scholar] [CrossRef] [PubMed]
- Tremouilhac, P.; Strandberg, E.; Wadhwani, P.; Ulrich, A.S. Conditions affecting the re-alignment of the antimicrobial peptide PGLa in membranes as monitored by solid state 2H-NMR. Biochim. Biophys. Acta 2006, 1758, 1330–1342. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, E.; Tiltak, D.; Ehni, S.; Wadhwani, P.; Ulrich, A.S. Lipid shape is a key factor for membrane interactions of amphipathic helical peptides. Biochim. Biophys. Acta 2012, 1818, 1764–1776. [Google Scholar] [CrossRef] [PubMed]
- Salnikov, E.S.; Mason, A.J.; Bechinger, B. Membrane order perturbation in the presence of antimicrobial peptides by 2H solid-state NMR spectroscopy. Biochimie 2009, 91, 743. [Google Scholar] [CrossRef]
- Bortolus, M.; Dalzini, A.; Toniolo, C.; Hahm, K.S.; Maniero, A.L. Interaction of hydrophobic and amphipathic antimicrobial peptides with lipid bicelles. J. Pept. Sci. 2014, 20, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Spano, J.; Park, E.K.; Wi, S. Evidence of pores and thinned lipid bilayers induced in oriented lipid membranes interacting with the antimicrobial peptides, magainin-2 and aurein-3.3. Biochim. Biophys. Acta 2009, 1788, 1482–1496. [Google Scholar] [CrossRef] [PubMed]
- Hallock, K.J.; Lee, D.K.; Omnaas, J.; Mosberg, H.I.; Ramamoorthy, A. Membrane composition determines pardaxin’s mechanism of lipid bilayer disruption. Biophys. J. 2002, 83, 1004–1013. [Google Scholar] [CrossRef]
- Grage, S.L.; Afonin, S.; Kara, S.; Buth, G.; Ulrich, A.S. Membrane Thinning and Thickening Induced by Membrane-Active Amphipathic Peptides. Front. Cell Dev. Biol. 2016, 4, 65. [Google Scholar] [CrossRef] [PubMed]
- Harmouche, N.; Pachler, M.; Lohner, K.; Pabst, G.; Bechinger, B. Lipid-mediated interactions between the amphipathic antimicrobial peptides magainin 2 and PGLa in phospholipid bilayers. Preparation submitted for publication. 2018. [Google Scholar]
- Chen, F.Y.; Lee, M.T.; Huang, H.W. Evidence for membrane thinning effect as the mechanism for Peptide-induced pore formation. Biophys. J. 2003, 84, 3751–3758. [Google Scholar] [CrossRef]
- Mecke, A.; Lee, D.K.; Ramamoorthy, A.; Orr, B.G.; Banaszak Holl, M.M. Membrane thinning due to antimicrobial peptide binding: An atomic force microscopy study of MSI-78 in lipid bilayers. Biophys. J. 2005, 89, 4043–4050. [Google Scholar] [CrossRef] [PubMed]
- Farrotti, A.; Bocchinfuso, G.; Palleschi, A.; Rosato, N.; Salnikov, E.S.; Voievoda, N.; Bechinger, B.; Stella, L. Molecular Dynamics Methods to Predict Peptide Location in Membranes: LAH4 as a Stringent Test Case. Biochim. Biophys. Acta 2015, 1848, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Pino-Angeles, A.; Leveritt, J.M., III; Lazaridis, T. Pore Structure and Synergy in Antimicrobial Peptides of the Magainin Family. PLoS Comput. Biol. 2016, 12, e1004570. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kabelka, I.; Vacha, R. Optimal conditions for opening of membrane pore by amphiphilic peptides. J. Chem. Phys. 2015, 143, 243115. [Google Scholar] [CrossRef] [PubMed]
- Amos, S.T.; Vermeer, L.S.; Ferguson, P.M.; Kozlowska, J.; Davy, M.; Bui, T.T.; Drake, A.F.; Lorenz, C.D.; Mason, A.J. Antimicrobial Peptide Potency is Facilitated by Greater Conformational Flexibility when Binding to Gram-negative Bacterial Inner Membranes. Sci. Rep. 2016, 6, 37639. [Google Scholar] [CrossRef] [PubMed]
- Smart, M.; Rajagopal, A.; Liu, W.K.; Ha, B.Y. Opposing effects of cationic antimicrobial peptides and divalent cations on bacterial lipopolysaccharides. Phys. Rev. E 2017, 96. [Google Scholar] [CrossRef] [PubMed]
- Russ, W.P.; Engelman, D.M. The GxxxG motif: A framework for transmembrane helix-helix association. J. Mol. Biol. 2000, 296, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Ulmschneider, J.P.; Smith, J.C.; Ulmschneider, M.B.; Ulrich, A.S.; Strandberg, E. Reorientation and Dimerization of the Membrane-Bound Antimicrobial Peptide PGLa from Microsecond All-Atom MD Simulations. Biophys. J. 2012, 103, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Israelachvili, J.N.; Marcelja, S.; Horn, R.G. Physical principles of membrane organization. Q. Rev. Biophys. 1980, 13, 121–200. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B. Rationalizing the membrane interactions of cationic amphipathic antimicrobial peptides by their molecular shape. Curr. Opin. Colloid Interface Sci. 2009, 14, 349–355. [Google Scholar] [CrossRef]
- Paterson, D.J.; Tassieri, M.; Reboud, J.; Wilson, R.; Cooper, J.M. Lipid topology and electrostatic interactions underpin lytic activity of linear cationic antimicrobial peptides in membranes. Proc. Natl. Acad. Sci. USA 2017, 114, E8324–E8332. [Google Scholar] [CrossRef] [PubMed]
- Ludtke, S.J.; He, K.; Heller, W.T.; Harroun, T.A.; Yang, L.; Huang, H.W. Membrane pores induced by magainin. Biochemistry 1996, 35, 13723–13728. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K. Magainins as paradigm for the mode of action of pore forming polypeptides. Biochim. Biophys. Acta 1998, 1376, 391–400. [Google Scholar] [CrossRef]
- Shai, Y. Mechanism of the binding, insertion, and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective lytic peptides. Biochim. Biophys. Acta 1999, 1462, 55–70. [Google Scholar] [CrossRef]
- Jenssen, H.; Hamill, P.; Hancock, R.E. Peptide antimicrobial agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Aisenbrey, C.; Harmouche, N.; Raya, J.; Bertani, P.; Voievoda, N.; Süss, R.; Bechinger, B. pH-dependent membrane interactions of the histidine-rich cell penetrating peptide LAH4-L1. Biophys. J. 2017, 113, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.T.J.; Hale, J.D.; Elliot, M.; Hancock, R.E.W.; Straus, S.K. Effect of Membrane Composition on Antimicrobial Peptides Aurein 2.2 and 2.3 From Australian Southern Bell Frogs. Biophys. J. 2009, 96, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.T.J.; Hale, J.D.; Elliott, M.; Hancock, R.E.W.; Straus, S.K. The importance of bacterial membrane composition in the structure and function of aurein 2.2 and selected variants. Biochim. Biophys. Acta 2011, 1808, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.W.; Wu, Y. Lipid-alamethicin interactions influence alamethicin orientation. Biophys. J. 1991, 60, 1079–1087. [Google Scholar] [CrossRef]
- Bechinger, B. Towards membrane protein design: pH-sensitive topology of histidine-containing polypeptides. J. Mol. Biol. 1996, 263, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.W. Molecular mechanism of antimicrobial peptides: The origin of cooperativity. Biochim. Biophys. Acta 2006, 1758, 1292–1302. [Google Scholar] [CrossRef] [PubMed]
- Hall, K.; Lee, T.H.; Mechler, A.I.; Swann, M.J.; Aguilar, M.I. Real-time measurement of membrane conformational states induced by antimicrobial peptides: Balance between recovery and lysis. Sci. Rep. 2014, 4, 5479. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.M.; Waring, A.J.; Separovic, F.; Lee, K.Y.C. Antimicrobial Peptides Share a Common Interaction Driven by Membrane Line Tension Reduction. Biophys. J. 2016, 111, 2176–2189. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B. Membrane-lytic peptides. Crit. Rev. Plant Sci. 2004, 23, 271–292. [Google Scholar] [CrossRef]
- Lohner, K. New strategies for novel antibiotics: Peptides targeting bacterial cell membranes. Gen. Physiol. Biophys. 2009, 28, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Wenk, M.; Seelig, J. Magainin 2 amide interaction with lipid membranes: Calorimetric detection of peptide binding and pore formation. Biochemistry 1998, 37, 3909–3916. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Harada, M.; Funakoshi, S.; Fujii, N.; Miyajima, K. Physicochemical Determinants for the Interactions of Magainins 1 and 2 with Acidic Lipid Bilayers. Biochim. Biophys. Acta 1991, 1063, 162–170. [Google Scholar] [CrossRef]
- Klocek, G.; Seelig, J. Melittin interaction with sulfated cell surface sugars. Biochemistry 2008, 47, 2841–2849. [Google Scholar] [CrossRef] [PubMed]
- Wieprecht, T.; Beyermann, M.; Seelig, J. Binding of antibacterial magainin peptides to electrically neutral membranes: Thermodynamics and structure. Biochemistry 1999, 38, 10377–10378. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, M.; Chiriac, A.I.; Otto, A.; Zweytick, D.; May, C.; Schumacher, C.; Gust, R.; Albada, H.B.; Penkova, M.; Kramer, U.; et al. Small cationic antimicrobial peptides delocalize peripheral membrane proteins. Proc. Natl. Acad. Sci. USA 2014, 111, E1409–E1418. [Google Scholar] [CrossRef] [PubMed]
- Perrone, B.; Miles, A.J.; Salnikov, E.S.; Wallace, B.; Bechinger, B. Lipid-interactions of the LAH4, a peptide with antimicrobial and nucleic transfection activities. Eur. Biophys. J. 2014, 43, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Mason, A.J.; Martinez, A.; Glaubitz, C.; Danos, O.; Kichler, A.; Bechinger, B. The antibiotic and DNA-transfecting peptide LAH4 selectively associates with, and disorders, anionic lipids in mixed membranes. FASEB J. 2006, 20, 320–322. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Voievoda, N. Biophysical Investigations of the Membrane and Nucleic Acids Interactions of the Transfection Peptide LAH4-L1. Ph.D. Thesis, University of Strasbourg, Strasbourg, France, 2014. [Google Scholar]
- Kindrachuk, J.; Napper, S. Structure-activity relationships of multifunctional host defence peptides. Mini Rev. Med. Chem. 2010, 10, 596–614. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.P.; Zhou, L.; Lakshminarayanan, R.; Beuerman, R.W. Multivalent Antimicrobial Peptides as Therapeutics: Design Principles and Structural Diversities. Int. J. Pept. Res. Ther. 2010, 16, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Hadley, E.B.; Hancock, R.E. Strategies for the Discovery and Advancement of Novel Cationic Antimicrobial Peptides. Curr. Top. Med. Chem. 2010, 10, 1872–1881. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, F. Cationic amphiphilic peptides with cancer-selective toxicity. Eur. J. Pharmacol. 2009, 625, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Oyston, P.C.; Fox, M.A.; Richards, S.J.; Clark, G.C. Novel peptide therapeutics for treatment of infections. J. Med. Microbiol. 2009, 58 (Pt 8), 977–987. [Google Scholar] [CrossRef]
- Ahn, M.; Gunasekaran, P.; Rajasekaran, G.; Kim, E.Y.; Lee, S.J.; Bang, G.; Cho, K.; Hyun, J.K.; Lee, H.J.; Jeon, Y.H.; et al. Pyrazole derived ultra-short antimicrobial peptidomimetics with potent anti-biofilm activity. Eur. J. Med. Chem. 2017, 125, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.; Shao, C.; Wang, J.; Shan, A.; Xu, L.; Dong, N.; Li, Z. Short, multiple-stranded beta-hairpin peptides have antimicrobial potency with high selectivity and salt resistance. Acta Biomater. 2016, 30, 78–93. [Google Scholar] [CrossRef] [PubMed]
- Acar, J.F. Antibiotic synergy and antagonism. Med. Clin. N. Am. 2000, 84, 1391–1406. [Google Scholar] [CrossRef]
- Kim, E.Y.; Rajasekaran, G.; Shin, S.Y. LL-37-derived short antimicrobial peptide KR-12-a5 and its d-amino acid substituted analogs with cell selectivity, anti-biofilm activity, synergistic effect with conventional antibiotics, and anti-inflammatory activity. Eur. J. Med. Chem. 2017, 136, 428–441. [Google Scholar] [CrossRef] [PubMed]
- Payne, J.E.; Dubois, A.V.; Ingram, R.J.; Weldon, S.; Taggart, C.C.; Elborn, J.S.; Tunney, M.M. Activity of innate antimicrobial peptides and ivacaftor against clinical cystic fibrosis respiratory pathogens. Int. J. Antimicrob. Agents 2017, 50, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Sakoulas, G.; Kumaraswamy, M.; Kousha, A.; Nizet, V. Interaction of Antibiotics with Innate Host Defense Factors against Salmonella enterica Serotype Newport. mSphere 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Bolosov, I.A.; Kalashnikov, A.A.; Panteleev, P.V.; Ovchinnikova, T.V. Analysis of Synergistic Effects of Antimicrobial Peptide Arenicin-1 and Conventional Antibiotics. Bull. Exp. Biol. Med. 2017, 162, 765–768. [Google Scholar] [CrossRef] [PubMed]
- Walkenhorst, W.F.; Sundrud, J.N.; Laviolette, J.M. Additivity and synergy between an antimicrobial peptide and inhibitory ions. Biochim. Biophys. Acta Biomembr. 2014, 1838, 2234–2242. [Google Scholar] [CrossRef] [PubMed]
- Giacometti, A.; Cirioni, O.; Del Prete, M.S.; Paggi, A.M.; D’Errico, M.M.; Scalise, G. Combination studies between polycationic peptides and clinically used antibiotics against Gram-positive and Gram-negative bacteria. Peptides 2000, 21, 1155–1160. [Google Scholar] [CrossRef]
- Mor, A.; Hani, K.; Nicolas, P. The vertebrate peptide antibiotics dermaseptins have overlapping structural features but target specific microorganisms. J. Biol. Chem. 1994, 269, 31635–31641. [Google Scholar] [PubMed]
- Vaz Gomes, A.; de Waal, A.; Berden, J.A.; Westerhoff, H.V. Electric Potentiation, Cooperativity, and Synergism of Magainin Peptides in Protein-Free Liposomes. Biochemistry 1993, 32, 5365–5372. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Hirakura, Y.; Matsuzaki, K. Bacteria-selective synergism between the antimicrobial peptides alpha-helical magainin 2 and cyclic beta-sheet tachyplesin I: Toward cocktail therapy. Biochemistry 2001, 40, 14330–14335. [Google Scholar] [CrossRef] [PubMed]
- Westerhoff, H.V.; Zasloff, M.; Rosner, J.L.; Hendler, R.W.; de Waal, A.; Vaz, G.; Jongsma, P.M.; Riethorst, A.; Juretic, D. Functional synergism of the magainins PGLa and magainin-2 in Escherichia coli, tumor cells and liposomes. Eur. J. Biochem. 1995, 228, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Mitani, Y.; Akada, K.; Murase, O.; Yoneyama, S.; Zasloff, M.; Miyajima, K. Mechanism of synergism between antimicrobial peptides magainin 2 and PGLa. Biochemistry 1998, 37, 15144–15153. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Huynh, Q.; Barlehner, D.; Heerklotz, H. Additive and Synergistic Membrane Permeabilization by Antimicrobial (Lipo)Peptides and Detergents. Biophys. J. 2014, 106, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, E.; Zerweck, J.; Wadhwani, P.; Ulrich, A.S. Synergistic Insertion of Antimicrobial Magainin-Family Peptides in Membranes Depends on the Lipid Spontaneous Curvature. Biophys. J. 2013, 104, L09–L11. [Google Scholar] [CrossRef] [PubMed]
- Salnikov, E.S.; Aisenbrey, C.; Aussenac, F.; Ouari, O.; Sarrouj, H.; Reiter, C.; Tordo, P.; Engelke, F.; Bechinger, B. Membrane topologies of the PGLa antimicrobial peptide and a transmembrane anchor sequence by Dynamic Nuclear Polarization/solid-state NMR spectroscopy. Sci. Rep. 2016, 6, 20895. [Google Scholar] [CrossRef] [PubMed]
- Glattard, E.; Salnikov, E.S.; Aisenbrey, C.; Bechinger, B. Investigations of the synergistic enhancement of antimicrobial activity in mixtures of magainin 2 and PGLa. Biophys. Chem. 2016, 210, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, E.; Horn, D.; Reisser, S.; Zerweck, J.; Wadhwani, P.; Ulrich, A.S. 2H-NMR and MD Simulations Reveal Membrane-Bound Conformation of Magainin 2 and Its Synergy with PGLa. Biophys. J. 2016, 111, 2149–2161. [Google Scholar] [CrossRef] [PubMed]
- Nishida, M.; Imura, Y.; Yamamoto, M.; Kobayashi, S.; Yano, Y.; Matsuzaki, K. Interaction of a magainin-PGLa hybrid peptide with membranes: Insight into the mechanism of synergism. Biochemistry 2007, 46, 14284–14290. [Google Scholar] [CrossRef] [PubMed]
- Zerweck, J.; Strandberg, E.; Kukharenko, O.; Reichert, J.; Burck, J.; Wadhwani, P.; Ulrich, A.S. Molecular mechanism of synergy between the antimicrobial peptides PGLa and magainin 2. Sci. Rep. 2017, 7, 13153. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, E.; Zerweck, J.; Horn, D.; Pritz, G.; Berditsch, M.; Bürck, J.; Wadhwani, P.; Ulrich, A.S. Influence of hydrophobic residues on the activity of the antimicrobial peptide magainin 2 and its synergy with PGLa. J. Pept. Sci. 2015, 21, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Benz, R.; Hancock, R.E. Influence of proline residues on the antibacterial and synergistic activities of alpha-helical peptides. Biochemistry 1999, 38, 8102–8111. [Google Scholar] [CrossRef] [PubMed]
- Han, E.; Lee, H. Synergistic effects of magainin 2 and PGLa on their heterodimer formation, aggregation, and insertion into the bilayer. RSC Adv. 2015, 5, 2047–2055. [Google Scholar] [CrossRef]
- Zerweck, J.; Strandberg, E.; Burck, J.; Reichert, J.; Wadhwani, P.; Kukharenko, O.; Ulrich, A.S. Homo- and heteromeric interaction strengths of the synergistic antimicrobial peptides PGLa and magainin 2 in membranes. Eur. Biophys. J. 2016, 45, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Wieprecht, T.; Apostolov, O.; Beyermann, M.; Seelig, J. Membrane binding and pore formation of the antibacterial peptide PGLa: Thermodynamic and mechanistic aspects. Biochemistry 2000, 39, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Wieprecht, T.; Apostolov, O.; Beyermann, M.; Seelig, J. Thermodynamics of the alpha-helix-coil transition of amphipathic peptides in a membrane environment: Implications for the peptide-membrane binding equilibrium. J. Mol. Biol. 1999, 294, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Wieprecht, T.; Beyermann, M.; Seelig, J. Thermodynamics of the coil-alpha-helix transition of amphipathic peptides in a membrane environment: The role of vesicle curvature. Biophys. Chem. 2002, 96, 191–201. [Google Scholar] [CrossRef]
- Vermeer, L.S.; Marquette, A.; Schoup, M.; Fenard, D.; Galy, A.; Bechinger, B. Simultaneous Analysis of Secondary Structure and Light Scattering from Circular Dichroism Titrations: Application to Vectofusin-1. Sci. Rep. 2016, 6, 39450. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, J.R.; Andresen, T.L. Thermodynamic profiling of peptide membrane interactions by isothermal titration calorimetry: A search for pores and micelles. Biophys. J. 2011, 101, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Kodama, H.; Kondo, M.; Wakamatsu, K.; Takeda, A.; Tachi, T.; Matsuzaki, K. Effects of peptide dimerization on pore formation: Antiparallel disulfide-dimerized magainin 2 analogue. Biopolymers 2001, 58, 437–446. [Google Scholar] [CrossRef]
- Dempsey, C.E.; Ueno, S.; Avison, M.B. Enhanced membrane permeabilization and antibacterial activity of a disulfide-dimerized magainin analogue. Biochemistry 2003, 42, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Lorenzon, E.N.; Santos-Filho, N.A.; Ramos, M.A.; Bauab, T.M.; Camargo, I.L.; Cilli, E.M. C-terminal Lysine-Linked Magainin 2 with Increased Activity Against Multidrug-Resistant Bacteria. Protein Pept. Lett. 2016, 23, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Verly, R.M.; Resende, J.M.; Junior, E.F.C.; de Magalhães, M.T.C.; Guimarães, C.F.C.R.; Munhoz, V.H.O.; Bemquerer, M.P.; Almeida, F.C.L.; Santoro, M.M.; Piló-Veloso, D.; et al. Structure and membrane interactions of the homodimeric antibiotic peptide homotarsinin. Sci. Rep. 2017, 7, 40854. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B.; Kim, Y.; Chirlian, L.E.; Gesell, J.; Neumann, J.M.; Montal, M.; Tomich, J.; Zasloff, M.; Opella, S.J. Orientations of amphipathic helical peptides in membrane bilayers determined by solid-state NMR spectroscopy. J. Biomol. NMR 1991, 1, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Pouny, Y.; Rapaport, D.; Mor, A.; Nicolas, P.; Shai, Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogues with phospholipid membranes. Biochemistry 1992, 31, 12416–12423. [Google Scholar] [CrossRef] [PubMed]
- Vogt, T.C.B.; Bechinger, B. The interactions of histidine-containing amphipathic helical peptide antibiotics with lipid bilayers: The effects of charges and pH. J. Biol. Chem. 1999, 274, 29115–29121. [Google Scholar] [CrossRef] [PubMed]
- Leontiadou, H.; Mark, A.E.; Marrink, S.J. Antimicrobial peptides in action. J. Am. Chem. Soc. 2006, 128, 12156–12161. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| magainin 2 | GIGKF LHSAK KFGKA FVGEI MNS |
| PGLa | GMASK AGAIA GKIAK VALKA L-NH2 |
| cecropin A | KWKLF KKIEK VGQNI RDGII KAGPA VAVVG QATQI AK-NH2 |
| LL37 | LLGDF FRKSK EKIGK EFKRI VQRIK DFLRN LVPRT ES |
| melittin | GIGAV LKVLT TGLPA LISWI KRKRQ Q-NH2 |
| LAH4 | KKALL ALALH HLAHL ALHLA LALKK A-NH2 |
| alamethicin (F50/7) | Ac-Aib-Pro-Aib-Ala-Aib-Aib-Gln-Aib-Val-Aib-Gly-Leu-Aib-Pro-Val-Aib-Aib-Gln-Gln-Phl |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marquette, A.; Bechinger, B. Biophysical Investigations Elucidating the Mechanisms of Action of Antimicrobial Peptides and Their Synergism. Biomolecules 2018, 8, 18. https://doi.org/10.3390/biom8020018
Marquette A, Bechinger B. Biophysical Investigations Elucidating the Mechanisms of Action of Antimicrobial Peptides and Their Synergism. Biomolecules. 2018; 8(2):18. https://doi.org/10.3390/biom8020018
Chicago/Turabian StyleMarquette, Arnaud, and Burkhard Bechinger. 2018. "Biophysical Investigations Elucidating the Mechanisms of Action of Antimicrobial Peptides and Their Synergism" Biomolecules 8, no. 2: 18. https://doi.org/10.3390/biom8020018
APA StyleMarquette, A., & Bechinger, B. (2018). Biophysical Investigations Elucidating the Mechanisms of Action of Antimicrobial Peptides and Their Synergism. Biomolecules, 8(2), 18. https://doi.org/10.3390/biom8020018