DOT1L and H3K79 Methylation in Transcription and Genomic Stability


Abstract
1. Introduction
2. Dot1/DOT1L Activity
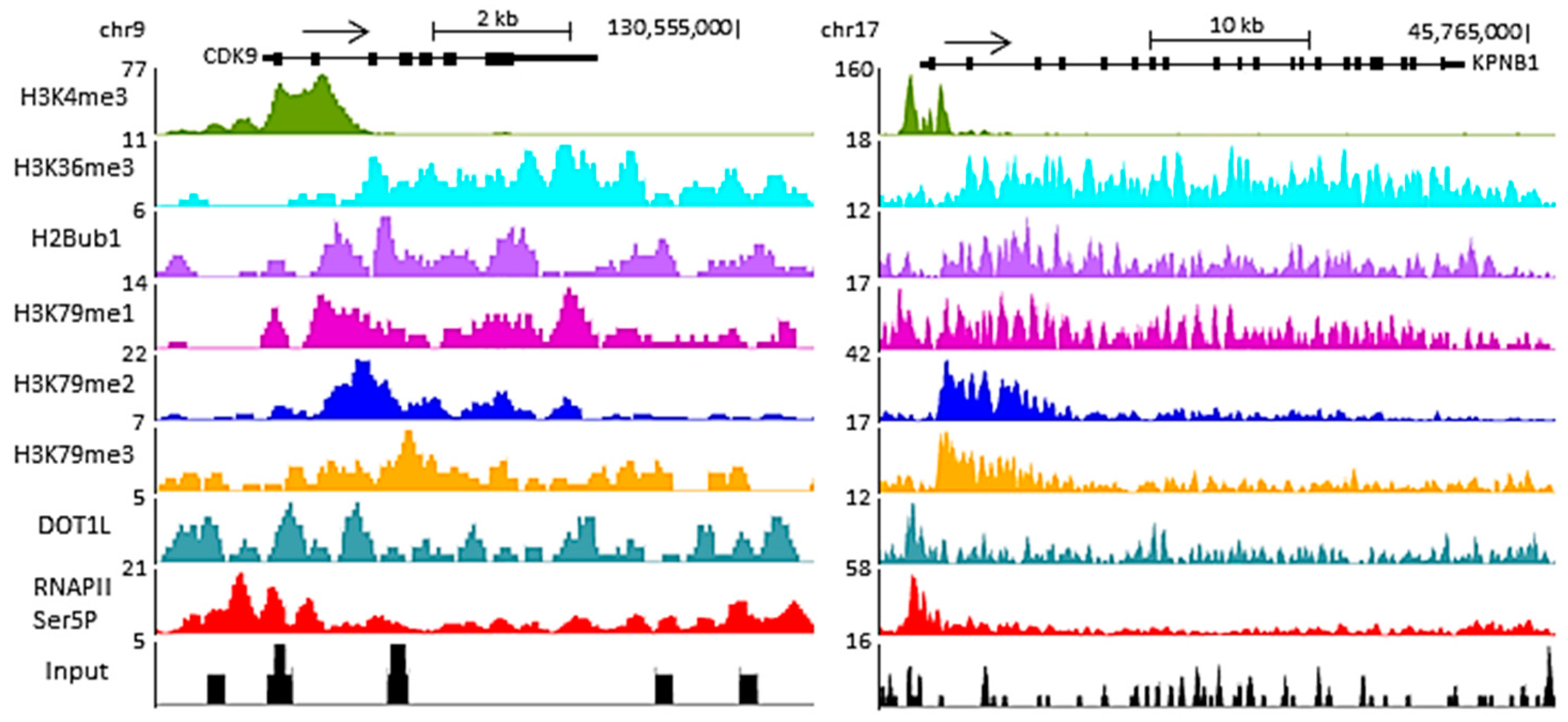
3. H3K79 Methylation and Active Transcription
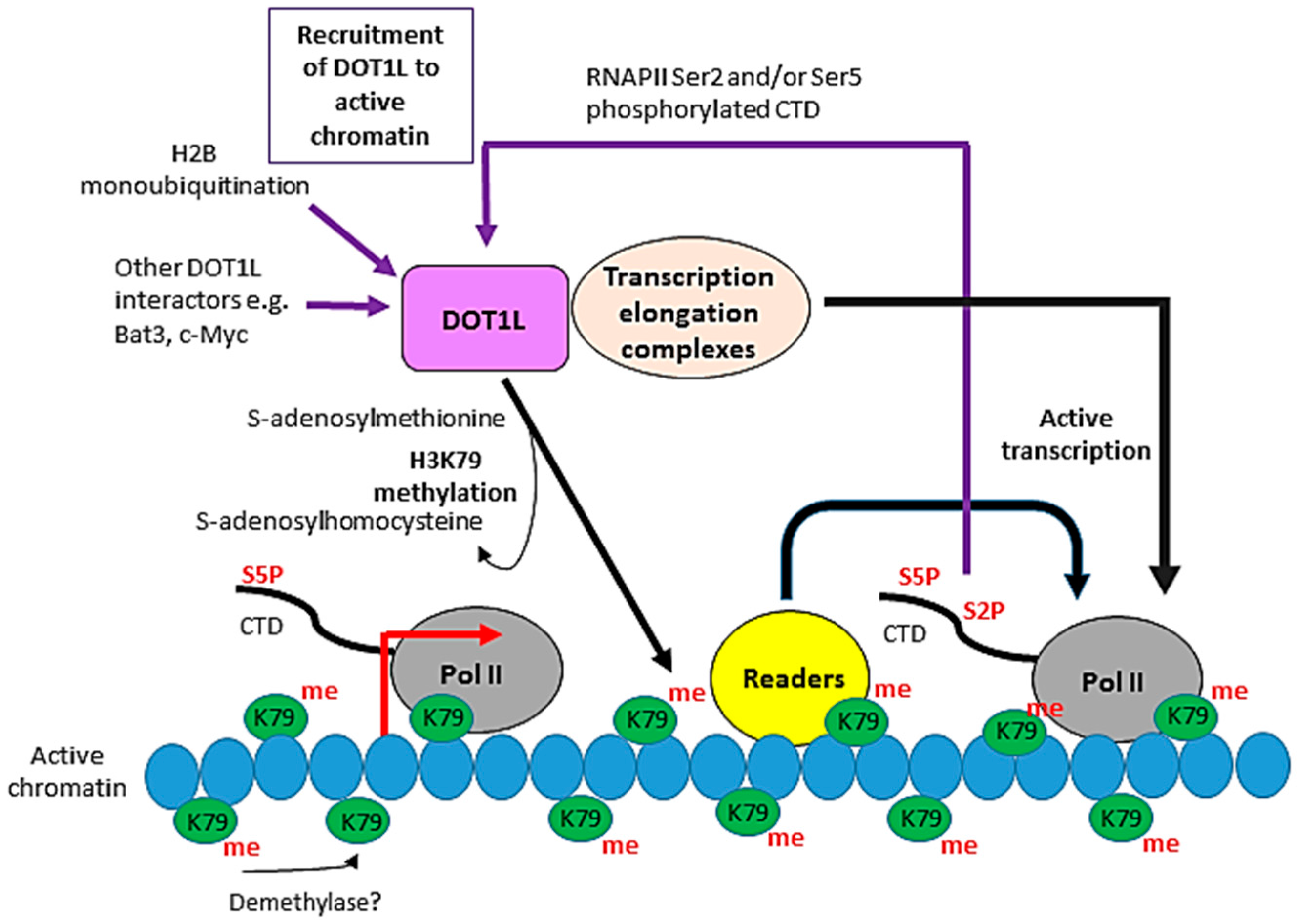
4. DOT1L in Transcriptional Elongation
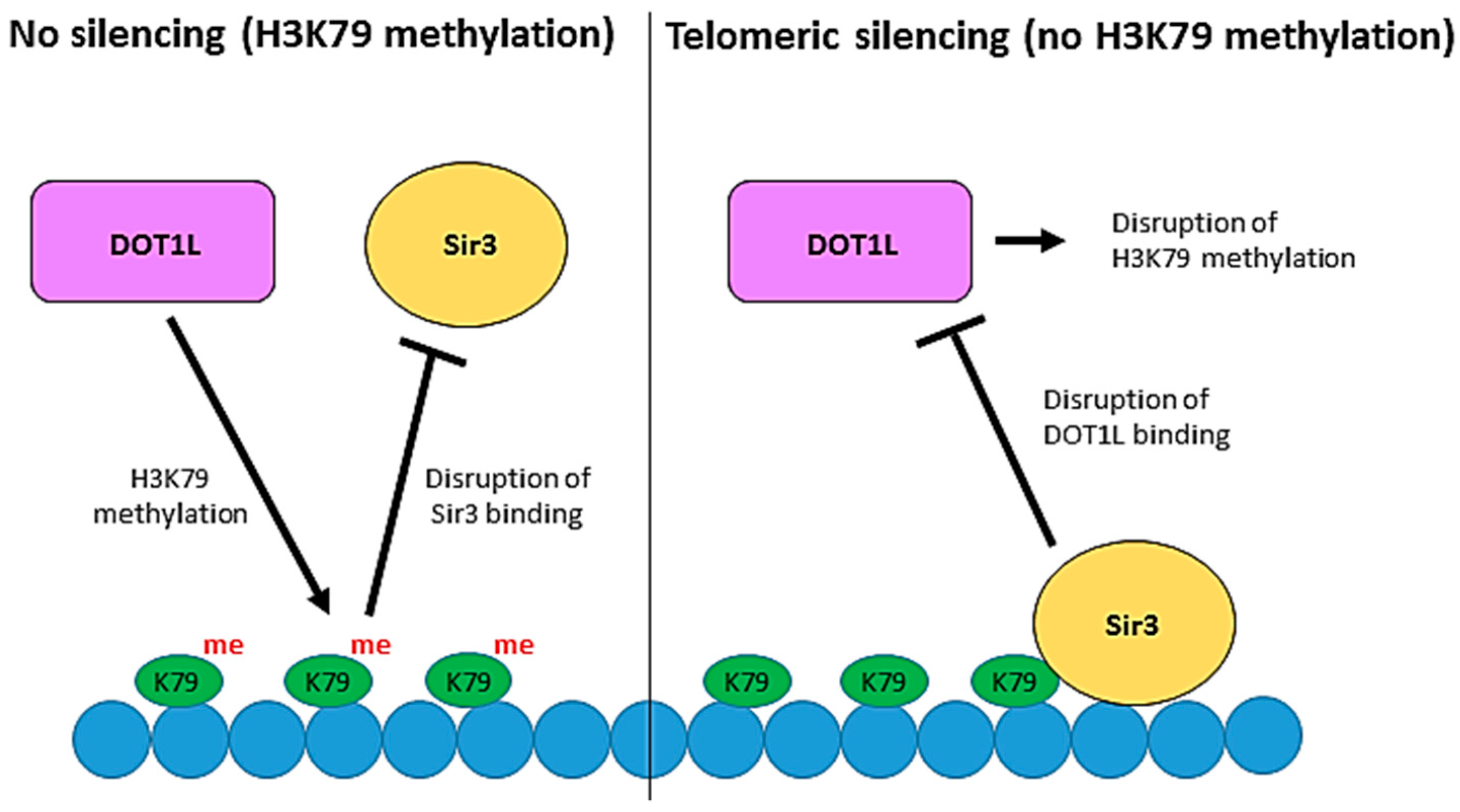
5. Dot1 and Telomeric Silencing in Saccharomyces Cerevisiae
6. Dot1/DOT1L and the DNA Damage Response
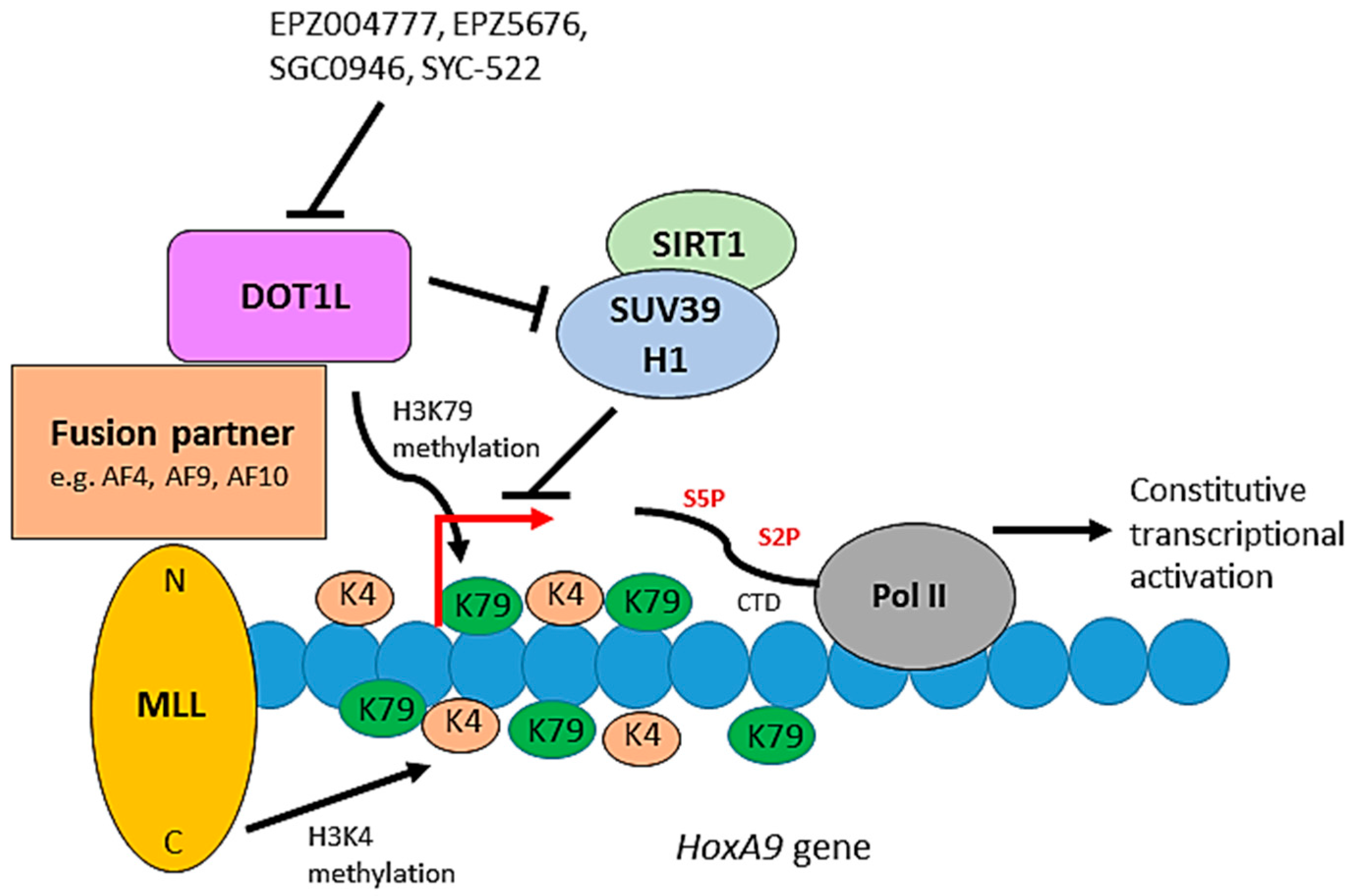
7. H3K79 Methylation and Leukemia
8. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Van Holde, K.E.; Allen, J.R.; Tatchell, K.; Weischet, W.O.; Lohr, D. DNA-histone interactions in nucleosomes. Biophys. J. 1980, 32, 271–282. [Google Scholar] [CrossRef]
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 a resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, R.D.; Lorch, Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 1999, 98, 285–294. [Google Scholar] [CrossRef]
- Zhang, K.; Dent, S.Y. Histone modifying enzymes and cancer: Going beyond histones. J. Cell. Biochem. 2005, 96, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Histone methylation in transcriptional control. Curr. Opin. Genet. Dev. 2002, 12, 198–209. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Oshimura, M.; Barrett, J.C. Epigenetic mechanisms in human disease. Cancer Res. 2002, 62, 6784–6787. [Google Scholar] [PubMed]
- Handel, A.E.; Ebers, G.C.; Ramagopalan, S.V. Epigenetics: Molecular mechanisms and implications for disease. Trends Mol. Med. 2010, 16, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Laible, G.; Dorn, R.; Reuter, G. Set domain proteins modulate chromatin domains in eu- and heterochromatin. Cell. Mol. Life Sci. 1998, 54, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.S.; Kahana, A.; Wolf, A.J.; Meisinger, L.L.; Peterson, S.E.; Goggin, C.; Mahowald, M.; Gottschling, D.E. Identification of high-copy disruptors of telomeric silencing in Saccharomyces cerevisiae. Genetics 1998, 150, 613–632. [Google Scholar] [PubMed]
- Feng, Q.; Wang, H.; Ng, H.H.; Erdjument-Bromage, H.; Tempst, P.; Struhl, K.; Zhang, Y. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a set domain. Cur. Biol. 2002, 12, 1052–1058. [Google Scholar] [CrossRef]
- Van Leeuwen, F.; Gafken, P.R.; Gottschling, D.E. Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell 2002, 109, 745–756. [Google Scholar] [CrossRef]
- Lacoste, N.; Utley, R.T.; Hunter, J.M.; Poirier, G.G.; Cote, J. Disruptor of telomeric silencing-1 is a chromatin-specific histone H3 methyltransferase. J. Biol. Chem. 2002, 277, 30421–30424. [Google Scholar] [CrossRef] [PubMed]
- Schubert, H.L.; Blumenthal, R.M.; Cheng, X. Many paths to methyltransfer: A chronicle of convergence. Trends Biochem. Sci. 2003, 28, 329–335. [Google Scholar] [CrossRef]
- Ng, H.H.; Feng, Q.; Wang, H.; Erdjument-Bromage, H.; Tempst, P.; Zhang, Y.; Struhl, K. Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association. Genes Dev. 2002, 16, 1518–1527. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Feng, Q.; Li, Z.; Zhang, Y.; Xu, R.M. Structure of the catalytic domain of human Dot1l, a non-set domain nucleosomal histone methyltransferase. Cell 2003, 112, 711–723. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Zhang, Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011, 25, 1345–1358. [Google Scholar] [CrossRef] [PubMed]
- Frederiks, F.; Tzouros, M.; Oudgenoeg, G.; van Welsem, T.; Fornerod, M.; Krijgsveld, J.; van Leeuwen, F. Nonprocessive methylation by Dot1 leads to functional redundancy of histone H3K79 methylation states. Nat. Struct. Mol. Biol. 2008, 15, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.; Su, H.; Bhat, A.; Lei, H.; Bajko, J.; Hevi, S.; Baltus, G.A.; Kadam, S.; Zhai, H.; Valdez, R.; et al. The histone H3K79 methyltransferase DOT1L is essential for mammalian development and heterochromatin structure. PLoS Genet. 2008, 4, e1000190. [Google Scholar] [CrossRef] [PubMed]
- Shanower, G.A.; Muller, M.; Blanton, J.L.; Honti, V.; Gyurkovics, H.; Schedl, P. Characterization of the grappa gene, the Drosophila histone H3 lysine 79 methyltransferase. Genetics 2005, 169, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, M.D.; Zhang, K.; Grunstein, M. Histone H2b ubiquitylation controls processive methylation but not monomethylation by Dot1 and Set1. Mol. Cell 2005, 19, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Tomson, B.N.; Arndt, K.M. The many roles of the conserved eukaryotic Paf1 complex in regulating transcription, histone modifications, and disease states. Biochem. Biophys. Acta 2013, 1829, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Briggs, S.D.; Xiao, T.; Sun, Z.W.; Caldwell, J.A.; Shabanowitz, J.; Hunt, D.F.; Allis, C.D.; Strahl, B.D. Gene silencing: Trans-histone regulatory pathway in chromatin. Nature 2002, 418, 498. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.H.; Xu, R.M.; Zhang, Y.; Struhl, K. Ubiquitination of histone H2b by Rad6 is required for efficient dot1-mediated methylation of histone H3 lysine 79. J. Biol. Chem. 2002, 277, 34655–34657. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.W.; Allis, C.D. Ubiquitination of histone H2b regulates H3 methylation and gene silencing in yeast. Nature 2002, 418, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Tang, Z.; Fu, X.; Yin, J.; Liang, Y.; Li, C.; Li, H.; Tian, Q.; Roeder, R.G.; Wang, G. The mediator subunit med23 couples H2b mono-ubiquitination to transcriptional control and cell fate determination. EMBO J. 2015, 34, 2885–2902. [Google Scholar] [CrossRef] [PubMed]
- McGinty, R.K.; Kim, J.; Chatterjee, C.; Roeder, R.G.; Muir, T.W. Chemically ubiquitylated histone H2b stimulates HDOT1L-mediated intranucleosomal methylation. Nature 2008, 453, 812–816. [Google Scholar] [CrossRef] [PubMed]
- Izzo, A.; Schneider, R. Chatting histone modifications in mammals. Brief. Funct. Genom. 2010, 9, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Barth, T.K.; Imhof, A. Fast signals and slow marks: The dynamics of histone modifications. Trends Biochem. Sci. 2010, 35, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Kallin, E.M.; Zhang, Y. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 2006, 7, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Zhang, Y. Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Cloos, P.A.; Christensen, J.; Agger, K.; Helin, K. Erasing the methyl mark: Histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008, 22, 1115–1140. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Tanasa, B.; Tyurina, O.V.; Zhou, T.Y.; Gassmann, R.; Liu, W.T.; Ohgi, K.A.; Benner, C.; Garcia-Bassets, I.; Aggarwal, A.K.; et al. Phf8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature 2010, 466, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.H.; Sarkissian, M.; Hu, G.Q.; Wang, Z.; Bhattacharjee, A.; Gordon, D.B.; Gonzales, M.; Lan, F.; Ongusaha, P.P.; Huarte, M.; et al. Histone H4K20/H3K9 demethylase Phf8 regulates zebrafish brain and craniofacial development. Nature 2010, 466, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Milne, T.A.; Martin, M.E.; Brock, H.W.; Slany, R.K.; Hess, J.L. Leukemogenic MLL fusion proteins bind across a broad region of the Hox A9 locus, promoting transcription and multiple histone modifications. Cancer Res. 2005, 65, 11367–11374. [Google Scholar] [CrossRef] [PubMed]
- Schulze, J.M.; Jackson, J.; Nakanishi, S.; Gardner, J.M.; Hentrich, T.; Haug, J.; Johnston, M.; Jaspersen, S.L.; Kobor, M.S.; Shilatifard, A. Linking cell cycle to histone modifications: Sbf and H2b monoubiquitination machinery and cell-cycle regulation of H3K79 dimethylation. Mol. Cell 2009, 35, 626–641. [Google Scholar] [CrossRef] [PubMed]
- Ooga, M.; Inoue, A.; Kageyama, S.; Akiyama, T.; Nagata, M.; Aoki, F. Changes in H3K79 methylation during preimplantation development in mice. Biol. Reprod. 2008, 78, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.H.; Ciccone, D.N.; Morshead, K.B.; Oettinger, M.A.; Struhl, K. Lysine-79 of histone H3 is hypomethylated at silenced loci in yeast and mammalian cells: A potential mechanism for position-effect variegation. Proc. Natl. Acad. Sci. USA 2003, 100, 1820–1825. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Schubeler, D.; MacAlpine, D.M.; Scalzo, D.; Wirbelauer, C.; Kooperberg, C.; van Leeuwen, F.; Gottschling, D.E.; O’Neill, L.P.; Turner, B.M.; Delrow, J.; et al. The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes Dev. 2004, 18, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Schuettengruber, B.; Chourrout, D.; Vervoort, M.; Leblanc, B.; Cavalli, G. Genome regulation by polycomb and trithorax proteins. Cell 2007, 128, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Steger, D.J.; Lefterova, M.I.; Ying, L.; Stonestrom, A.J.; Schupp, M.; Zhuo, D.; Vakoc, A.L.; Kim, J.E.; Chen, J.; Lazar, M.A.; et al. DOT1L/kmt4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol. Cell. Biol. 2008, 28, 2825–2839. [Google Scholar] [CrossRef] [PubMed]
- Jonkers, I.; Kwak, H.; Lis, J.T. Genome-wide dynamics of pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. eLife 2014, 3, e02407. [Google Scholar] [CrossRef] [PubMed]
- Veloso, A.; Kirkconnell, K.S.; Magnuson, B.; Biewen, B.; Paulsen, M.T.; Wilson, T.E.; Ljungman, M. Rate of elongation by RNA polymerase II is associated with specific gene features and epigenetic modifications. Genome Res. 2014, 24, 896–905. [Google Scholar] [CrossRef] [PubMed]
- McKittrick, E.; Gafken, P.R.; Ahmad, K.; Henikoff, S. Histone H3.3 is enriched in covalent modifications associated with active chromatin. Proc. Natl. Acad. Sci. USA 2004, 101, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Hake, S.B.; Garcia, B.A.; Duncan, E.M.; Kauer, M.; Dellaire, G.; Shabanowitz, J.; Bazett-Jones, D.P.; Allis, C.D.; Hunt, D.F. Expression patterns and post-translational modifications associated with mammalian histone H3 variants. J. Biol. Chem. 2006, 281, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Takatsuka, S.; Akashi, H.; Yamamoto, E.; Nojima, M.; Maruyama, R.; Kai, M.; Yamano, H.O.; Sasaki, Y.; Tokino, T.; et al. Genome-wide profiling of chromatin signatures reveals epigenetic regulation of microRNA genes in colorectal cancer. Cancer Res. 2011, 71, 5646–5658. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Jung, I.; Lee, H.; Kang, K.; Kim, M.; Jeong, K.; Kwon, C.S.; Han, Y.M.; Kim, Y.S.; Kim, D.; et al. Human histone H3K79 methyltransferase DOT1L protein binds actively transcribing RNA polymerase II to regulate gene expression. J. Biol. Chem. 2012, 287, 39698–39709. [Google Scholar] [CrossRef] [PubMed]
- Corden, J.L. Tails of RNA polymerase II. Trends Biochem. Sci. 1990, 15, 383–387. [Google Scholar] [CrossRef]
- Zaborowska, J.; Egloff, S.; Murphy, S. The pol II CTD: New twists in the tail. Nat. Struct. Mol. Biol. 2016, 23, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Phatnani, H.P.; Greenleaf, A.L. Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 2006, 20, 2922–2936. [Google Scholar] [CrossRef] [PubMed]
- Wakeman, T.P.; Wang, Q.; Feng, J.; Wang, X.F. Bat3 facilitates H3K79 dimethylation by DOT1L and promotes DNA damage-induced 53bp1 foci at G1/G2 cell-cycle phases. EMBO J. 2012, 31, 2169–2181. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.H.; Park, J.H.; Choi, H.J.; Park, M.K.; Won, H.Y.; Park, Y.J.; Lee, C.H.; Oh, S.H.; Song, Y.S.; Kim, H.S.; et al. DOT1L cooperates with the c-myc-p300 complex to epigenetically derepress CDH1 transcription factors in breast cancer progression. Nat. Commun. 2015, 6, 7821. [Google Scholar] [CrossRef] [PubMed]
- Marson, A.; Levine, S.S.; Cole, M.F.; Frampton, G.M.; Brambrink, T.; Johnstone, S.; Guenther, M.G.; Johnston, W.K.; Wernig, M.; Newman, J.; et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell 2008, 134, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Huff, J.T.; Plocik, A.M.; Guthrie, C.; Yamamoto, K.R. Reciprocal intronic and exonic histone modification regions in humans. Nat. Struct. Mol. Biol. 2010, 17, 1495–1499. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.; Kim, S.K.; Kim, M.; Han, Y.M.; Kim, Y.S.; Kim, D.; Lee, D. H2b monoubiquitylation is a 5′-enriched active transcription mark and correlates with exon-intron structure in human cells. Genome Res. 2012, 22, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Kouskouti, A.; Talianidis, I. Histone modifications defining active genes persist after transcriptional and mitotic inactivation. EMBO J. 2005, 24, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Krogan, N.J.; Dover, J.; Wood, A.; Schneider, J.; Heidt, J.; Boateng, M.A.; Dean, K.; Ryan, O.W.; Golshani, A.; Johnston, M.; et al. The Paf1 complex is required for histone H3 methylation by compass and DOT1P: Linking transcriptional elongation to histone methylation. Mol. Cell 2003, 11, 721–729. [Google Scholar] [CrossRef]
- Yokoyama, A.; Lin, M.; Naresh, A.; Kitabayashi, I.; Cleary, M.L. A higher-order complex containing AF4 and ENL family proteins with p-Tefb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell 2010, 17, 198–212. [Google Scholar] [CrossRef] [PubMed]
- Bitoun, E.; Oliver, P.L.; Davies, K.E. The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum. Mol. Genet. 2007, 16, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Biswas, D.; Milne, T.A.; Basrur, V.; Kim, J.; Elenitoba-Johnson, K.S.; Allis, C.D.; Roeder, R.G. Function of leukemogenic mixed lineage leukemia 1 (MLL) fusion proteins through distinct partner protein complexes. Proc. Natl. Acad. Sci. USA 2011, 108, 15751–15756. [Google Scholar] [CrossRef] [PubMed]
- Mohan, M.; Herz, H.M.; Takahashi, Y.H.; Lin, C.; Lai, K.C.; Zhang, Y.; Washburn, M.P.; Florens, L.; Shilatifard, A. Linking H3K79 trimethylation to wnt signaling through a novel Dot1-containing complex (DotCom). Genes Dev. 2010, 24, 574–589. [Google Scholar] [CrossRef] [PubMed]
- Mueller, D.; Bach, C.; Zeisig, D.; Garcia-Cuellar, M.P.; Monroe, S.; Sreekumar, A.; Zhou, R.; Nesvizhskii, A.; Chinnaiyan, A.; Hess, J.L.; et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 2007, 110, 4445–4454. [Google Scholar] [CrossRef] [PubMed]
- Mueller, D.; Garcia-Cuellar, M.P.; Bach, C.; Buhl, S.; Maethner, E.; Slany, R.K. Misguided transcriptional elongation causes mixed lineage leukemia. PLoS Biol. 2009, 7, e1000249. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Smith, E.R.; Takahashi, H.; Lai, K.C.; Martin-Brown, S.; Florens, L.; Washburn, M.P.; Conaway, J.W.; Conaway, R.C.; Shilatifard, A. Aff4, a component of the Ell/p-Tefb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol. Cell 2010, 37, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.L.; Sinha, A.; Verzi, M.; Bernt, K.M.; Armstrong, S.A.; Shivdasani, R.A. DOT1L-mediated H3K79 methylation in chromatin is dispensable for Wnt pathway-specific and other intestinal epithelial functions. Mol. Cell. Biol. 2013, 33, 1735–1745. [Google Scholar] [CrossRef] [PubMed]
- Leach, B.I.; Kuntimaddi, A.; Schmidt, C.R.; Cierpicki, T.; Johnson, S.A.; Bushweller, J.H. Leukemia fusion target AF9 is an intrinsically disordered transcriptional regulator that recruits multiple partners via coupled folding and binding. Struct. 2013, 21, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Simon, M.D.; Chodaparambil, J.V.; Hansen, J.C.; Shokat, K.M.; Luger, K. The effect of H3K79 dimethylation and H4K20 trimethylation on nucleosome and chromatin structure. Nat. Struct. Mol. Biol. 2008, 15, 1122–1124. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Min, J. Structure and function of the nucleosome-binding PWWP domain. Trends Biochem. Sci. 2014, 39, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Sabra, M.; Texier, P.; El Maalouf, J.; Lomonte, P. The tudor protein survival motor neuron (SMN) is a chromatin-binding protein that interacts with methylated lysine 79 of histone H3. J. Cell Sci. 2013, 126, 3664–3677. [Google Scholar] [CrossRef] [PubMed]
- Huyen, Y.; Zgheib, O.; Ditullio, R.A., Jr.; Gorgoulis, V.G.; Zacharatos, P.; Petty, T.J.; Sheston, E.A.; Mellert, H.S.; Stavridi, E.S.; Halazonetis, T.D. Methylated lysine 79 of histone H3 targets 53bp1 to DNA double-strand breaks. Nature 2004, 432, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Alpatov, R.; Lesch, B.J.; Nakamoto-Kinoshita, M.; Blanco, A.; Chen, S.; Stutzer, A.; Armache, K.J.; Simon, M.D.; Xu, C.; Ali, M.; et al. A chromatin-dependent role of the fragile X mental retardation protein FMRP in the DNA damage response. Cell 2014, 157, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Altaf, M.; Utley, R.T.; Lacoste, N.; Tan, S.; Briggs, S.D.; Cote, J. Interplay of chromatin modifiers on a short basic patch of histone H4 tail defines the boundary of telomeric heterochromatin. Mol. Cell 2007, 28, 1002–1014. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.W.; Koche, R.P.; Sinha, A.U.; Deshpande, A.J.; Zhu, N.; Eng, R.; Doench, J.G.; Xu, H.; Chu, S.H.; Qi, J.; et al. DOT1L inhibits Sirt1-mediated epigenetic silencing to maintain leukemic gene expression in MLL-rearranged leukemia. Nat. Med. 2015, 21, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Norris, A.; Boeke, J.D. Silent information regulator 3: The goldilocks of the silencing complex. Genes Dev. 2010, 24, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Fingerman, I.M.; Li, H.C.; Briggs, S.D. A charge-based interaction between histone H4 and Dot1 is required for H3K79 methylation and telomere silencing: Identification of a new trans-histone pathway. Genes Dev. 2007, 21, 2018–2029. [Google Scholar] [CrossRef] [PubMed]
- Rossmann, M.P.; Luo, W.; Tsaponina, O.; Chabes, A.; Stillman, B. A common telomeric gene silencing assay is affected by nucleotide metabolism. Mol. Cell 2011, 42, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.H.; Schulze, J.M.; Jackson, J.; Hentrich, T.; Seidel, C.; Jaspersen, S.L.; Kobor, M.S.; Shilatifard, A. Dot1 and histone H3K79 methylation in natural telomeric and Hm silencing. Mol. Cell 2011, 42, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Wysocki, R.; Javaheri, A.; Allard, S.; Sha, F.; Cote, J.; Kron, S.J. Role of Dot1-dependent histone H3 methylation in G1 and S phase DNA damage checkpoint functions of Rad9. Mol. Cell. Biol. 2005, 25, 8430–8443. [Google Scholar] [CrossRef] [PubMed]
- Botuyan, M.V.; Lee, J.; Ward, I.M.; Kim, J.E.; Thompson, J.R.; Chen, J.; Mer, G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53bp1 and Crb2 in DNA repair. Cell 2006, 127, 1361–1373. [Google Scholar] [CrossRef] [PubMed]
- Tong, Q.; Cui, G.; Botuyan, M.V.; Rothbart, S.B.; Hayashi, R.; Musselman, C.A.; Singh, N.; Appella, E.; Strahl, B.D.; Mer, G.; et al. Structural plasticity of methyllysine recognition by the tandem tudor domain of 53bp1. Structure 2015, 23, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53bp1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014, 15, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Giannattasio, M.; Lazzaro, F.; Plevani, P.; Muzi-Falconi, M. The DNA damage checkpoint response requires histone H2b ubiquitination by Rad6-Bre1 and H3 methylation by Dot1. J. Biol. Chem. 2005, 280, 9879–9886. [Google Scholar] [CrossRef] [PubMed]
- Game, J.C.; Williamson, M.S.; Spicakova, T.; Brown, J.M. The Rad6/Bre1 histone modification pathway in saccharomyces confers radiation resistance through a Rad51-dependent process that is independent of Rad18. Genetics 2006, 173, 1951–1968. [Google Scholar] [CrossRef] [PubMed]
- Chernikova, S.B.; Dorth, J.A.; Razorenova, O.V.; Game, J.C.; Brown, J.M. Deficiency in Bre1 impairs homologous recombination repair and cell cycle checkpoint response to radiation damage in mammalian cells. Radiat. Res. 2010, 174, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Toh, G.W.; O’Shaughnessy, A.M.; Jimeno, S.; Dobbie, I.M.; Grenon, M.; Maffini, S.; O’Rorke, A.; Lowndes, N.F. Histone H2a phosphorylation and H3 methylation are required for a novel Rad9 DSB repair function following checkpoint activation. DNA Repair 2006, 5, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Grenon, M.; Costelloe, T.; Jimeno, S.; O’Shaughnessy, A.; Fitzgerald, J.; Zgheib, O.; Degerth, L.; Lowndes, N.F. Docking onto chromatin via the Saccharomyces cerevisiae Rad9 tudor domain. Yeast 2007, 24, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Lazzaro, F.; Sapountzi, V.; Granata, M.; Pellicioli, A.; Vaze, M.; Haber, J.E.; Plevani, P.; Lydall, D.; Muzi-Falconi, M. Histone methyltransferase Dot1 and Rad9 inhibit single-stranded DNA accumulation at DSBs and uncapped telomeres. EMBO J. 2008, 27, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Conde, F.; Refolio, E.; Cordon-Preciado, V.; Cortes-Ledesma, F.; Aragon, L.; Aguilera, A.; San-Segundo, P.A. The Dot1 histone methyltransferase and the Rad9 checkpoint adaptor contribute to cohesin-dependent double-strand break repair by sister chromatid recombination in Saccharomyces cerevisiae. Genetics 2009, 182, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Bostelman, L.J.; Keller, A.M.; Albrecht, A.M.; Arat, A.; Thompson, J.S. Methylation of histone H3 lysine-79 by DPT1P plays multiple roles in the response to UV damage in Saccharomyces cerevisiae. DNA Repair 2007, 6, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Prakash, S.; Prakash, L. Nucleotide excision repair in yeast. Mutat. Res. 2000, 451, 13–24. [Google Scholar] [CrossRef]
- Conde, F.; San-Segundo, P.A. Role of Dot1 in the response to alkylating DNA damage in Saccharomyces cerevisiae: Regulation of DNA damage tolerance by the error-prone polymerases polzeta/Rev1. Genetics 2008, 179, 1197–1210. [Google Scholar] [CrossRef] [PubMed]
- Conde, F.; Ontoso, D.; Acosta, I.; Gallego-Sanchez, A.; Bueno, A.; San-Segundo, P.A. Regulation of tolerance to DNA alkylating damage by Dot1 and Rad53 in Saccharomyces cerevisiae. DNA Repair 2010, 9, 1038–1049. [Google Scholar] [CrossRef] [PubMed]
- Levesque, N.; Leung, G.P.; Fok, A.K.; Schmidt, T.I.; Kobor, M.S. Loss of H3 K79 trimethylation leads to suppression of Rtt107-dependent DNA damage sensitivity through the translesion synthesis pathway. J. Biol. Chem. 2010, 285, 35113–35122. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Kakadia, P.M.; Chen, Y.; Li, Y.Q.; Deshpande, A.J.; Buske, C.; Zhang, K.L.; Zhang, Y.; Xu, G.L.; Bohlander, S.K. Global reduction of the epigenetic H3K79 methylation mark and increased chromosomal instability in Calm-AF10-positive leukemias. Blood 2009, 114, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Oksenych, V.; Zhovmer, A.; Ziani, S.; Mari, P.O.; Eberova, J.; Nardo, T.; Stefanini, M.; Giglia-Mari, G.; Egly, J.M.; Coin, F. Histone methyltransferase DOT1L drives recovery of gene expression after a genotoxic attack. PLoS Genet. 2013, 9, e1003611. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Armstrong, S.A. MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer 2007, 7, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Feng, Q.; Lin, Y.; Jiang, Q.; Li, Y.; Coffield, V.M.; Su, L.; Xu, G.; Zhang, Y. HDOT1L links histone methylation to leukemogenesis. Cell 2005, 121, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Ayton, P.M.; Cleary, M.L. Molecular mechanisms of leukemogenesis mediated by MLL fusion proteins. Oncogene 2001, 20, 5695–5707. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Chory, E.J.; Wernimont, A.K.; Tempel, W.; Scopton, A.; Federation, A.; Marineau, J.J.; Qi, J.; Barsyte-Lovejoy, D.; Yi, J.; et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat. Commun. 2012, 3, 1288. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; Tallman, M.S. Mixed lineage rearranged leukaemia: Pathogenesis and targeting DOT1L. Curr. Opin. Hematol. 2015, 22, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Basavapathruni, A.; Olhava, E.J.; Daigle, S.R.; Therkelsen, C.A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Nonclinical pharmacokinetics and metabolism of Epz-5676, a novel DOT1L histone methyltransferase inhibitor. Biopharm. Drug Dispos. 2014, 35, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Deng, L.; Song, Y.; Redell, M. DOT1L inhibition sensitizes MLL-rearranged aml to chemotherapy. PLoS ONE 2014, 9, e98270. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.M.; Karemaker, I.D.; van Leeuwen, F. The emerging roles of DOT1L in leukemia and normal development. Leukemia 2014, 28, 2131–2138. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Hofmann, J.; Burmeister, T.; Groger, D.; Park, T.S.; Emerenciano, M.; Pombo de Oliveira, M.; Renneville, A.; Villarese, P.; Macintyre, E.; et al. The MLL recombinome of acute leukemias in 2013. Leukemia 2013, 27, 2165–2176. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Protein Components | Reference |
|---|---|---|
| AEP | AFF1/4, ENL, P-TEFb | [60] |
| AF4-associated | AFF1, AF9, DOT1L, ENL, MLLT10, P-TEFb | [61] |
| AF9-associated | AF9, DOT1L (mutually exclusive with AF4/AFF4, AF9, P-TEFb) | [62] |
| DotCom | AF9, CTNNB1, DOT1L, ENL, MLLT6, MLLT10, SKP1, TRRAP | [63] |
| EAP | AFF1/3/4, BCOR, CBX8, DOT1L, ENL, P-TEFb, RING1 | [64] |
| EAP core | AFF1, DOT1L, ENL, P-TEFb | [65] |
| SEC | AFF1/4, AF9, EAF1/2, ELL1/2/3, ENL, P-TEFb | [66] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wood, K.; Tellier, M.; Murphy, S. DOT1L and H3K79 Methylation in Transcription and Genomic Stability. Biomolecules 2018, 8, 11. https://doi.org/10.3390/biom8010011
Wood K, Tellier M, Murphy S. DOT1L and H3K79 Methylation in Transcription and Genomic Stability. Biomolecules. 2018; 8(1):11. https://doi.org/10.3390/biom8010011
Chicago/Turabian StyleWood, Katherine, Michael Tellier, and Shona Murphy. 2018. "DOT1L and H3K79 Methylation in Transcription and Genomic Stability" Biomolecules 8, no. 1: 11. https://doi.org/10.3390/biom8010011
APA StyleWood, K., Tellier, M., & Murphy, S. (2018). DOT1L and H3K79 Methylation in Transcription and Genomic Stability. Biomolecules, 8(1), 11. https://doi.org/10.3390/biom8010011