
Friends Turned Foes: Angiogenic Growth Factors beyond Angiogenesis

Abstract
1. Introduction
2. Vascular Endothelial Growth Factor
3. Role of Vascular Endothelial Growth Factors beyond Angiogenesis
3.1. Rheumatoid Arthritis and Osteoarthritis
3.2. Diabetes Mellitus and Associated Complications
3.3. Chronic Obstructive Pulmonary Disease
3.4. Endometriosis, Preeclampsia, and Ovarian Hyperstimulation Syndrome
3.5. Psoriasis
3.6. Neurodegenerative Disorders
3.7. Organ Fibrosis via Endothelial-to-Mesenchymal Transition
4. Fibroblast Growth Factor
5. Role of Fibroblast Growth Factors beyond Angiogenesis
5.1. Non-Alcoholic Fatty Liver Disease
5.2. Vascular Calcification
5.3. Cardiac Hypertrophy
5.4. Atherosclerosis
5.5. Chronic Kidney Disease (CKD)
5.6. Lung Disease
5.7. Cutaneous Inflammation
5.8. Alzheimer’s Disease
5.9. Corneal Fibrosis
6. Angiopoietins
7. Role of Angiopoietins beyond Angiogenesis
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Papetti, M.; Herman, I.M. Mechanisms of normal and tumor-derived angiogenesis. Am. J. Physiol. Cell Physiol. 2002, 282, C947–C970. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.S.; Lee, J.; Ferrara, N. Targeting the tumour vasculature: Insights from physiological angiogenesis. Nat. Rev. Cancer 2010, 10, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Distler, J.H.; Hirth, A.; Kurowska-Stolarska, M.; Gay, R.E.; Gay, S.; Distler, O. Angiogenic and angiostatic factors in the molecular control of angiogenesis. Q. J. Nucl. Med. 2003, 47, 149–161. [Google Scholar] [PubMed]
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; Van Oosterom, A.T.; De Bruijn, E.A. Vascular endothelial growth factor and angiogenesis. Pharmacol. Rev. 2004, 56, 549–580. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.J.; Bates, D.O. VEGF-A splicing: The key to anti-angiogenic therapeutics? Nat. Rev. Cancer 2008, 8, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Flamme, I.; von Reutern, M.; Drexler, H.C.; Syed-Ali, S.; Risau, W. Overexpression of vascular endothelial growth factor in the avian embryo induces hypervascularization and increased vascular permeability without alterations of embryonic pattern formation. Dev. Biol. 1995, 171, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Sanyal, S.; Mukhopadhyay, D. Tyrosine residues 951 and 1059 of vascular endothelial growth factor receptor-2 (KDR) are essential for vascular permeability factor/vascular endothelial growth factor-induced endothelium migration and proliferation, respectively. J. Biol. Chem. 2001, 276, 32714–32719. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. Vascular endothelial growth factor receptor-1 (VEGFR-1/Flt-1): A dual regulator for angiogenesis. Angiogenesis 2006, 9, 225–230, discussion 231. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.H.; Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 464–478. [Google Scholar] [CrossRef] [PubMed]
- Joukov, V.; Pajusola, K.; Kaipainen, A.; Chilov, D.; Lahtinen, I.; Kukk, E.; Saksela, O.; Kalkkinen, N.; Alitalo, K. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J. 1996, 15, 290–298. [Google Scholar] [PubMed]
- Achen, M.G.; Jeltsch, M.; Kukk, E.; Makinen, T.; Vitali, A.; Wilks, A.F.; Alitalo, K.; Stacker, S.A. Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc. Natl. Acad. Sci. USA 1998, 95, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Herzog, B.; Pellet-Many, C.; Britton, G.; Hartzoulakis, B.; Zachary, I.C. VEGF binding to NRP1 is essential for VEGF stimulation of endothelial cell migration, complex formation between NRP1 and VEGFR2, and signaling via FAK Tyr407 phosphorylation. Mol. Biol Cell 2011, 22, 2766–2776. [Google Scholar] [CrossRef] [PubMed]
- Roth, L.; Prahst, C.; Ruckdeschel, T.; Savant, S.; Westrom, S.; Fantin, A.; Riedel, M.; Heroult, M.; Ruhrberg, C.; Augustin, H.G. Neuropilin-1 mediates vascular permeability independently of vascular endothelial growth factor receptor-2 activation. Sci. Signal. 2016, 9, ra42. [Google Scholar] [CrossRef] [PubMed]
- Gluzman-Poltorak, Z.; Cohen, T.; Herzog, Y.; Neufeld, G. Neuropilin-2 is a receptor for the vascular endothelial growth factor (VEGF) forms VEGF-145 and VEGF-165 [corrected]. J. Biol. Chem. 2000, 275, 18040–18045. [Google Scholar] [CrossRef] [PubMed]
- Evans, I.M.; Yamaji, M.; Britton, G.; Pellet-Many, C.; Lockie, C.; Zachary, I.C.; Frankel, P. Neuropilin-1 signaling through p130Cas tyrosine phosphorylation is essential for growth factor-dependent migration of glioma and endothelial cells. Mol. Cell. Biol. 2011, 31, 1174–1185. [Google Scholar] [CrossRef] [PubMed]
- Pellet-Many, C.; Frankel, P.; Evans, I.M.; Herzog, B.; Junemann-Ramirez, M.; Zachary, I.C. Neuropilin-1 mediates PDGF stimulation of vascular smooth muscle cell migration and signalling via p130Cas. Biochem. J. 2011, 435, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Migdal, M.; Huppertz, B.; Tessler, S.; Comforti, A.; Shibuya, M.; Reich, R.; Baumann, H.; Neufeld, G. Neuropilin-1 is a placenta growth factor-2 receptor. J. Biol. Chem. 1998, 273, 22272–22278. [Google Scholar] [CrossRef] [PubMed]
- Gaur, P.; Bielenberg, D.R.; Samuel, S.; Bose, D.; Zhou, Y.; Gray, M.J.; Dallas, N.A.; Fan, F.; Xia, L.; Lu, J.; et al. Role of class 3 semaphorins and their receptors in tumor growth and angiogenesis. Clin. Cancer Res. 2009, 15, 6763–6770. [Google Scholar] [CrossRef] [PubMed]
- Autiero, M.; Waltenberger, J.; Communi, D.; Kranz, A.; Moons, L.; Lambrechts, D.; Kroll, J.; Plaisance, S.; De Mol, M.; Bono, F.; et al. Role of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat. Med. 2003, 9, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Zampeli, E.; Vlachoyiannopoulos, P.G.; Tzioufas, A.G. Treatment of rheumatoid arthritis: Unraveling the conundrum. J. Autoimmun. 2015, 65, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, E.; van Caam, A.; van der Kraan, P.M. Obesity and osteoarthritis, more than just wear and tear: Pivotal roles for inflamed adipose tissue and dyslipidaemia in obesity-induced osteoarthritis. Rheumatology 2015, 54, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Burr, D.B.; Gallant, M.A. Bone remodelling in osteoarthritis. Nat. Rev. Rheumatol. 2012, 8, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.A. Angiogenesis and arthritis. Rheumatology 1999, 38, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Pufe, T.; Petersen, W.; Tillmann, B.; Mentlein, R. The splice variants VEGF121 and VEGF189 of the angiogenic peptide vascular endothelial growth factor are expressed in osteoarthritic cartilage. Arthritis Rheumatol. 2001, 44, 1082–1088. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, J.; Wan, L.; Sun, Y.; Wang, F.; Qi, Y.; Huang, C. Up-regulated expressions of HIF-1alpha, VEGF and CD34 promote synovial angiogenesis in rats with adjuvant arthritis. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2015, 31, 1053–1056. [Google Scholar] [PubMed]
- Raatz, Y.; Ibrahim, S.; Feldmann, M.; Paleolog, E.M. Gene expression profiling and functional analysis of angiogenic markers in murine collagen-induced arthritis. Arthritis Res. Ther. 2012, 14, R169. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Hosoda, Y.; Hirose, S.; Okada, Y.; Ikeda, E. Expression of vascular endothelial growth factor isoforms and their receptors Flt-1, KDR, and neuropilin-1 in synovial tissues of rheumatoid arthritis. J. Pathol. 2000, 191, 426–433. [Google Scholar] [CrossRef]
- Ballara, S.; Taylor, P.C.; Reusch, P.; Marme, D.; Feldmann, M.; Maini, R.N.; Paleolog, E.M. Raised serum vascular endothelial growth factor levels are associated with destructive change in inflammatory arthritis. Arthritis Rheumatol. 2001, 44, 2055–2064. [Google Scholar] [CrossRef]
- Lee, S.S.; Joo, Y.S.; Kim, W.U.; Min, D.J.; Min, J.K.; Park, S.H.; Cho, C.S.; Kim, H.Y. Vascular endothelial growth factor levels in the serum and synovial fluid of patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 2001, 19, 321–324. [Google Scholar] [PubMed]
- Kim, H.Y.; Park, S.Y.; Lee, S.W.; Lee, H.R.; Lee, W.S.; Rhim, B.Y.; Hong, K.W.; Kim, C.D. Inhibition of HMGB1-induced angiogenesis by cilostazol via SIRT1 activation in synovial fibroblasts from rheumatoid arthritis. PLoS ONE 2014, 9, e104743. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Zhang, Y.; Liu, C.; Guo, W.; Li, X.; Su, X.; Wan, H.; Sun, Y.; Lin, N. Anti-angiogenic effect of triptolide in rheumatoid arthritis by targeting angiogenic cascade. PLoS ONE 2013, 8, e77513. [Google Scholar] [CrossRef] [PubMed]
- Barranco, C. Osteoarthritis: Animal data show VEGF blocker inhibits post-traumatic OA. Nat. Rev. Rheumatol. 2014, 10, 638. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Sato, M.; Kobayashi, M.; Yokoyama, M.; Tani, Y.; Mochida, J. Bevacizumab, an anti-vascular endothelial growth factor antibody, inhibits osteoarthritis. Arthritis Res. Ther. 2014, 16, 427. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Modena, V.; Sciascia, S.; Roccatello, D. Rheumatoid arthritis: Biological therapy other than anti-TNF. Int. Immunopharmacol. 2015, 27, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, X.; Wang, J.; Wang, Z.; Jiang, W.; Reed, E.; Zhang, Y.; Liu, Y.; Li, Q.Q. Thalidomide down-regulates the expression of VEGF and bFGF in cisplatin-resistant human lung carcinoma cells. Anticancer Res. 2003, 23, 2481–2487. [Google Scholar] [PubMed]
- Kim, H.S.; Oh, J.M.; Jin, D.H.; Yang, K.H.; Moon, E.Y. Paclitaxel induces vascular endothelial growth factor expression through reactive oxygen species production. Pharmacology 2008, 81, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, R.; Vermeulen, L.C.; Jiang, Z.; Lau, E.; Kolesar, J.M. Gemcitabine and paclitaxel suppress the production of vascular endothelial growth factor induced by deferoxamine in human non-small cell lung cancer A549 cells. Exp. Ther. Med. 2010, 1, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Jarosova, K.; Cieslak, D.; Alper, J.; Kivitz, A.; Hough, D.R.; Maes, P.; Pineda, L.; Chen, M.; Zaidi, F. Apremilast in Patients With Active Rheumatoid Arthritis: A Phase II, Multicenter, Randomized, Double-Blind, Placebo-Controlled, Parallel-Group Study. Arthritis Rheumatol. 2015, 67, 1703–1710. [Google Scholar] [CrossRef] [PubMed]
- Kurose, A.; Yoshida, W.; Yoshida, M.; Sawai, T. Effects of paclitaxel on cultured synovial cells from patients with rheumatoid arthritis. Cytometry 2001, 44, 349–354. [Google Scholar] [CrossRef]
- Wauke, K.; Nagashima, M.; Ishiwata, T.; Asano, G.; Yoshino, S. Expression and localization of vascular endothelial growth factor-C in rheumatoid arthritis synovial tissue. J. Rheumatol. 2002, 29, 34–38. [Google Scholar] [PubMed]
- Paavonen, K.; Mandelin, J.; Partanen, T.; Jussila, L.; Li, T.F.; Ristimaki, A.; Alitalo, K.; Konttinen, Y.T. Vascular endothelial growth factors C and D and their VEGFR-2 and 3 receptors in blood and lymphatic vessels in healthy and arthritic synovium. J. Rheumatol. 2002, 29, 39–45. [Google Scholar] [PubMed]
- Kelly, S.; Bombardieri, M.; Humby, F.; Ng, N.; Marrelli, A.; Riahi, S.; DiCicco, M.; Mahto, A.; Zou, L.; Pyne, D.; et al. Angiogenic gene expression and vascular density are reflected in ultrasonographic features of synovitis in early Rheumatoid Arthritis: An observational study. Arthritis Res. Ther. 2015, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Roodhart, J.M.; Langenberg, M.H.; Witteveen, E.; Voest, E.E. The molecular basis of class side effects due to treatment with inhibitors of the VEGF/VEGFR pathway. Curr. Clin. Pharmacol. 2008, 3, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Kamba, T.; McDonald, D.M. Mechanisms of adverse effects of anti-VEGF therapy for cancer. Br. J. Cancer 2007, 96, 1788–1795. [Google Scholar] [CrossRef] [PubMed]
- Eremina, V.; Jefferson, J.A.; Kowalewska, J.; Hochster, H.; Haas, M.; Weisstuch, J.; Richardson, C.; Kopp, J.B.; Kabir, M.G.; Backx, P.H.; et al. VEGF inhibition and renal thrombotic microangiopathy. N. Engl. J. Med. 2008, 358, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hao, L.; Zhang, S.; Ji, Y.; Zhang, Y.; Lu, X.; Shi, B.; Pei, H.; Wang, Y.; Chen, D.; et al. Genetic repression of mouse VEGF expression regulates coagulation cascade. IUBMB Life 2010, 62, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Facemire, C.S.; Nixon, A.B.; Griffiths, R.; Hurwitz, H.; Coffman, T.M. Vascular endothelial growth factor receptor 2 controls blood pressure by regulating nitric oxide synthase expression. Hypertension 2009, 54, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Mansoor, S.; Sharma, A.; Sapkal, A.; Sheth, J.; Falatoonzadeh, P.; Kuppermann, B.; Kenney, M. Diabetic retinopathy and VEGF. Open Ophthalmol. J. 2013, 7, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.L.; Mao, X.O.; Greenberg, D.A. Vascular endothelial growth factor: Direct neuroprotective effect in in vitro ischemia. Proc. Natl. Acad. Sci. USA 2000, 97, 10242–10247. [Google Scholar] [CrossRef] [PubMed]
- Beazley-Long, N.; Hua, J.; Jehle, T.; Hulse, R.P.; Dersch, R.; Lehrling, C.; Bevan, H.; Qiu, Y.; Lagreze, W.A.; Wynick, D.; et al. VEGF-A165b is an endogenous neuroprotective splice isoform of vascular endothelial growth factor A in vivo and in vitro. Am. J. Pathol. 2013, 183, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Kaidonis, G.; Burdon, K.P.; Gillies, M.C.; Abhary, S.; Essex, R.W.; Chang, J.H.; Pal, B.; Pefkianaki, M.; Daniell, M.; Lake, S.; et al. Common Sequence Variation in the VEGFC Gene Is Associated with Diabetic Retinopathy and Diabetic Macular Edema. Ophthalmology 2015, 122, 1828–1836. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.N.; Raij, L.; Mundel, P. Role of angiotensin II in the development of nephropathy and podocytopathy of diabetes. Curr. Diabetes Rev. 2011, 7, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Veron, D.; Aggarwal, P.K.; Velazquez, H.; Kashgarian, M.; Moeckel, G.; Tufro, A. Podocyte-specific VEGF-a gain of function induces nodular glomerulosclerosis in eNOS null mice. J. Am. Soc. Nephrol. 2014, 25, 1814–1824. [Google Scholar] [CrossRef] [PubMed]
- Coward, R.J.; Welsh, G.I.; Koziell, A.; Hussain, S.; Lennon, R.; Ni, L.; Tavare, J.M.; Mathieson, P.W.; Saleem, M.A. Nephrin is critical for the action of insulin on human glomerular podocytes. Diabetes 2007, 56, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Reddy, G.R.; Kotlyarevska, K.; Ransom, R.F.; Menon, R.K. The podocyte and diabetes mellitus: Is the podocyte the key to the origins of diabetic nephropathy? Curr. Opin. Nephrol. Hypertens. 2008, 17, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Tufro, A.; Veron, D. VEGF and podocytes in diabetic nephropathy. Semin. Nephrol. 2012, 32, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, K.; Makita, Z.; Yamagishi, S.; Atsumi, T.; Miyoshi, H.; Obara, S.; Ishida, M.; Ishikawa, S.; Yasumura, K.; Koike, T. Suppression of transforming growth factor beta and vascular endothelial growth factor in diabetic nephropathy in rats by a novel advanced glycation end product inhibitor, OPB-9195. Diabetologia 1999, 42, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Chou, E.; Suzuma, I.; Way, K.J.; Opland, D.; Clermont, A.C.; Naruse, K.; Suzuma, K.; Bowling, N.L.; Vlahos, C.J.; Aiello, L.P.; et al. Decreased cardiac expression of vascular endothelial growth factor and its receptors in insulin-resistant and diabetic States: A possible explanation for impaired collateral formation in cardiac tissue. Circulation 2002, 105, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Schrijvers, B.F.; Flyvbjerg, A.; Tilton, R.G.; Lameire, N.H.; De Vriese, A.S. A neutralizing VEGF antibody prevents glomerular hypertrophy in a model of obese type 2 diabetes, the Zucker diabetic fatty rat. Nephrol. Dial. Transplant. 2006, 21, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Martin-Timon, I.; Sevillano-Collantes, C.; Segura-Galindo, A.; Del Canizo-Gomez, F.J. Type 2 diabetes and cardiovascular disease: Have all risk factors the same strength? World J. Diabetes 2014, 5, 444–470. [Google Scholar] [CrossRef] [PubMed]
- Nichols, G.A.; Gullion, C.M.; Koro, C.E.; Ephross, S.A.; Brown, J.B. The incidence of congestive heart failure in type 2 diabetes: An update. Diabetes Care 2004, 27, 1879–1884. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.S.; Uchida, S.; Masuo, O.; Cejna, M.; Park, J.S.; Gwon, H.C.; Kirchmair, R.; Bahlman, F.; Walter, D.; Curry, C.; et al. Progressive attenuation of myocardial vascular endothelial growth factor expression is a seminal event in diabetic cardiomyopathy: Restoration of microvascular homeostasis and recovery of cardiac function in diabetic cardiomyopathy after replenishment of local vascular endothelial growth factor. Circulation 2005, 111, 2073–2085. [Google Scholar] [PubMed]
- Han, B.; Baliga, R.; Huang, H.; Giannone, P.J.; Bauer, J.A. Decreased cardiac expression of vascular endothelial growth factor and redox imbalance in murine diabetic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H829–H835. [Google Scholar] [CrossRef] [PubMed]
- Shida, T.; Nozawa, T.; Sobajima, M.; Ihori, H.; Matsuki, A.; Inoue, H. Fluvastatin-induced reduction of oxidative stress ameliorates diabetic cardiomyopathy in association with improving coronary microvasculature. Heart Vessels 2014, 29, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; He, X.; Hou, X.; Li, L.; Chen, J.X. Apelin gene therapy increases myocardial vascular density and ameliorates diabetic cardiomyopathy via upregulation of sirtuin 3. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H585–H597. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.K.; Cheng, R.; Nguyen, T.; Fan, T.; Kariyawasam, A.P.; Liu, Y.; Osmond, D.H.; George, S.R.; O’Dowd, B.F. Characterization of apelin, the ligand for the APJ receptor. J. Neurochem. 2000, 74, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.Y.; Lee, C.C.; Hsieh, M.F.; Chen, C.H.; Chou, K.M. Clinical association of circulating VEGF-B levels with hyperlipidemia and target organ damage in type 2 diabetic patients. J. Biol. Regul. Homeost. Agents 2014, 28, 225–236. [Google Scholar] [PubMed]
- Schurgin, S.; Rich, S.; Mazzone, T. Increased prevalence of significant coronary artery calcification in patients with diabetes. Diabetes Care 2001, 24, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Snell-Bergeon, J.K.; Budoff, M.J.; Hokanson, J.E. Vascular calcification in diabetes: Mechanisms and implications. Curr. Diabetes Rep. 2013, 13, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Yadav, B.K.; Hong, Y.; Shin, B.S. Correlation of VEGF genetic polymorphisms and lipid profile to aortic calcification. Gene 2014, 550, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Mikhaylova, L.; Malmquist, J.; Nurminskaya, M. Regulation of in vitro vascular calcification by BMP4, VEGF and Wnt3a. Calcif. Tissue Int. 2007, 81, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Liebow, A.A. Pulmonary emphysema with special reference to vascular changes. Am. Rev. Respir. Dis. 1959, 80(Part 2), 67–93. [Google Scholar] [PubMed]
- Koyama, S.; Sato, E.; Haniuda, M.; Numanami, H.; Nagai, S.; Izumi, T. Decreased level of vascular endothelial growth factor in bronchoalveolar lavage fluid of normal smokers and patients with pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2002, 166, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, Y.; Tuder, R.M.; Cool, C.D.; Lynch, D.A.; Flores, S.C.; Voelkel, N.F. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am. J. Respir. Crit. Care Med. 2001, 163 Pt 1, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, Y.; Tuder, R.M.; Taraseviciene-Stewart, L.; Le Cras, T.D.; Abman, S.; Hirth, P.K.; Waltenberger, J.; Voelkel, N.F. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J. Clin. Investig. 2000, 106, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, H.; Yoshikawa, J. Elevated oxidative stress and reciprocal reduction of vascular endothelial growth factor levels with severity of COPD. Chest 2005, 128, 3191–3197. [Google Scholar] [CrossRef] [PubMed]
- Imesch, P.; Fink, D. Endometriosis Update 2016. Praxis 2016, 105, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Oosterlynck, D.J.; Meuleman, C.; Sobis, H.; Vandeputte, M.; Koninckx, P.R. Angiogenic activity of peritoneal fluid from women with endometriosis. Fertil. Steril. 1993, 59, 778–782. [Google Scholar] [CrossRef]
- Shifren, J.L.; Tseng, J.F.; Zaloudek, C.J.; Ryan, I.P.; Meng, Y.G.; Ferrara, N.; Jaffe, R.B.; Taylor, R.N. Ovarian steroid regulation of vascular endothelial growth factor in the human endometrium: implications for angiogenesis during the menstrual cycle and in the pathogenesis of endometriosis. J. Clin. Endocrinol. Metab. 1996, 81, 3112–3118. [Google Scholar] [PubMed]
- McLaren, J.; Prentice, A.; Charnock-Jones, D.S.; Smith, S.K. Vascular endothelial growth factor (VEGF) concentrations are elevated in peritoneal fluid of women with endometriosis. Hum. Reprod. 1996, 11, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Phipps, E.; Prasanna, D.; Brima, W.; Jim, B. Preeclampsia: Updates in Pathogenesis, Definitions, and Guidelines. Clin. J. Am. Soc. Nephrol. 2016, 11, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Liberis, A.; Stanulov, G.; Ali, E.C.; Hassan, A.; Pagalos, A.; Kontomanolis, E.N. Pre-eclampsia and the vascular endothelial growth factor: A new aspect. Clin. Exp. Obstet. Gynecol. 2016, 43, 9–13. [Google Scholar] [PubMed]
- Gibson, J.L.; Lyall, F.; Boswell, F.; Young, A.; Maccuish, A.C.; Greer, I.A. Circulating cell adhesion molecule concentrations in diabetic women during pregnancy. Obstet. Gynecol. 1997, 90, 874–879. [Google Scholar] [CrossRef]
- Clark, C.J.; Boswell, F.; Greer, I.A.; Lyall, F. Treatment of endothelial cells with serum from women with preeclampsia: Effect on neutrophil adhesion. J. Soc. Gynecol. Investig. 1997, 4, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Lyall, F.; Greer, I.A.; Boswell, F.; Fleming, R. Suppression of serum vascular endothelial growth factor immunoreactivity in normal pregnancy and in pre-eclampsia. Br. J. Obstet. Gynaecol. 1997, 104, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Roberts, W.G.; Palade, G.E. Neovasculature induced by vascular endothelial growth factor is fenestrated. Cancer Res. 1997, 57, 765–772. [Google Scholar] [PubMed]
- Helske, S.; Vuorela, P.; Carpen, O.; Hornig, C.; Weich, H.; Halmesmaki, E. Expression of vascular endothelial growth factor receptors 1, 2 and 3 in placentas from normal and complicated pregnancies. Mol. Hum. Reprod. 2001, 7, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.E.; Min, J.Y.; Merchan, J.; Lim, K.H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Tsatsaris, V.; Goffin, F.; Munaut, C.; Brichant, J.F.; Pignon, M.R.; Noel, A.; Schaaps, J.P.; Cabrol, D.; Frankenne, F.; Foidart, J.M. Overexpression of the soluble vascular endothelial growth factor receptor in preeclamptic patients: Pathophysiological consequences. J. Clin. Endocrinol. Metab. 2003, 88, 5555–5563. [Google Scholar] [CrossRef] [PubMed]
- Souders, C.A.; Maynard, S.E.; Yan, J.; Wang, Y.; Boatright, N.K.; Sedan, J.; Balyozian, D.; Cheslock, P.S.; Molrine, D.C.; Simas, T.A. Circulating Levels of sFlt1 Splice Variants as Predictive Markers for the Development of Preeclampsia. Int. J. Mol. Sci. 2015, 16, 12436–12453. [Google Scholar] [CrossRef] [PubMed]
- Amosco, M.D.; Villar, V.A.; Naniong, J.M.; David-Bustamante, L.M.; Jose, P.A.; Palmes-Saloma, C.P. VEGF-A and VEGFR1 SNPs associate with preeclampsia in a Philippine population. Clin. Exp. Hypertens. 2016, 38, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Escudero, C.; Celis, C.; Saez, T.; San Martin, S.; Valenzuela, F.J.; Aguayo, C.; Bertoglia, P.; Roberts, J.M.; Acurio, J. Increased placental angiogenesis in late and early onset pre-eclampsia is associated with differential activation of vascular endothelial growth factor receptor 2. Placenta 2014, 35, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Smith, V.; Osianlis, T.; Vollenhoven, B. Prevention of Ovarian Hyperstimulation Syndrome: A Review. Obstet. Gynecol. Int. 2015, 2015, 514159. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, L.; Humaidan, P. Ovarian hyperstimulation syndrome in the 21st century: The role of gonadotropin-releasing hormone agonist trigger and kisspeptin. Curr. Opin. Obstet. Gynecol. 2015, 27, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Christenson, L.K.; Stouffer, R.L.; Burry, K.A.; Patton, P.E. Vascular endothelial growth factor levels in serum and follicular fluid of patients undergoing in vitro fertilization. Fertil. Steril. 1997, 68, 305–311. [Google Scholar] [CrossRef]
- Artini, P.G.; Fasciani, A.; Monti, M.; Luisi, S.; D’Ambrogio, G.; Genazzani, A.R. Changes in vascular endothelial growth factor levels and the risk of ovarian hyperstimulation syndrome in women enrolled in an in vitro fertilization program. Fertil. Steril. 1998, 70, 560–564. [Google Scholar] [CrossRef]
- Ferrara, N.; Chen, H.; Davis-Smyth, T.; Gerber, H.P.; Nguyen, T.N.; Peers, D.; Chisholm, V.; Hillan, K.J.; Schwall, R.H. Vascular endothelial growth factor is essential for corpus luteum angiogenesis. Nat. Med. 1998, 4, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Nouri, K.; Haslinger, P.; Szabo, L.; Sator, M.; Schreiber, M.; Schneeberger, C.; Pietrowski, D. Polymorphisms of VEGF and VEGF receptors are associated with the occurrence of ovarian hyperstimulation syndrome (OHSS)-a retrospective case-control study. J. Ovarian Res. 2014, 7, 54. [Google Scholar] [CrossRef] [PubMed]
- Scotti, L.; Abramovich, D.; Pascuali, N.; Irusta, G.; Meresman, G.; Tesone, M.; Parborell, F. Local VEGF inhibition prevents ovarian alterations associated with ovarian hyperstimulation syndrome. J. Steroid Biochem. Mol. Biol. 2014, 144 Pt B, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Cenksoy, C.; Cenksoy, P.O.; Erdem, O.; Sancak, B.; Gursoy, R. A potential novel strategy, inhibition of vasopressin-induced VEGF secretion by relcovaptan, for decreasing the incidence of ovarian hyperstimulation syndrome in the hyperstimulated rat model. Eur. J. Obstet. Gynecol. Reprod. Biol. 2014, 174, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Naredi, N.; Talwar, P.; Sandeep, K. VEGF antagonist for the prevention of ovarian hyperstimulation syndrome: Current status. Med. J. Armed Forces India 2014, 70, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Bronckers, I.M.; Paller, A.S.; van Geel, M.J.; van de Kerkhof, P.C.; Seyger, M.M. Psoriasis in Children and Adolescents: Diagnosis, Management and Comorbidities. Paediatr. Drugs 2015, 17, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Detmar, M.; Brown, L.F.; Claffey, K.P.; Yeo, K.T.; Kocher, O.; Jackman, R.W.; Berse, B.; Dvorak, H.F. Overexpression of vascular permeability factor/vascular endothelial growth factor and its receptors in psoriasis. J. Exp. Med. 1994, 180, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Bhushan, M.; McLaughlin, B.; Weiss, J.B.; Griffiths, C.E. Levels of endothelial cell stimulating angiogenesis factor and vascular endothelial growth factor are elevated in psoriasis. Br. J. Dermatol. 1999, 141, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.P.; Li, B.; Hylton, D.; Detmar, M.; Yancopoulos, G.D.; Rudge, J.S. Transgenic delivery of VEGF to mouse skin leads to an inflammatory condition resembling human psoriasis. Blood 2003, 102, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Kunstfeld, R.; Hirakawa, S.; Hong, Y.K.; Schacht, V.; Lange-Asschenfeldt, B.; Velasco, P.; Lin, C.; Fiebiger, E.; Wei, X.; Wu, Y.; et al. Induction of cutaneous delayed-type hypersensitivity reactions in VEGF-A transgenic mice results in chronic skin inflammation associated with persistent lymphatic hyperplasia. Blood 2004, 104, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Sauder, D.N.; Dekoven, J.; Champagne, P.; Croteau, D.; Dupont, E. Neovastat (AE-941), an inhibitor of angiogenesis: Randomized phase I/II clinical trial results in patients with plaque psoriasis. J. Am. Acad. Dermatol. 2002, 47, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Canavese, M.; Altruda, F.; Ruzicka, T.; Schauber, J. Vascular endothelial growth factor (VEGF) in the pathogenesis of psoriasis—A possible target for novel therapies? J. Dermatol. Sci. 2010, 58, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, D.; Storkebaum, E.; Morimoto, M.; Del-Favero, J.; Desmet, F.; Marklund, S.L.; Wyns, S.; Thijs, V.; Andersson, J.; van Marion, I.; et al. VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nat. Genet. 2003, 34, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Storkebaum, E.; Carmeliet, P. VEGF: A critical player in neurodegeneration. J. Clin. Investig. 2004, 113, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Oosthuyse, B.; Moons, L.; Storkebaum, E.; Beck, H.; Nuyens, D.; Brusselmans, K.; Van Dorpe, J.; Hellings, P.; Gorselink, M.; Heymans, S.; et al. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat. Genet. 2001, 28, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Storkebaum, E.; Lambrechts, D.; Dewerchin, M.; Moreno-Murciano, M.P.; Appelmans, S.; Oh, H.; Van Damme, P.; Rutten, B.; Man, W.Y.; De Mol, M.; et al. Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat. Neurosci. 2005, 8, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Xu, W.; Luo, C.; Gozal, D.; Liu, R. VEGF-induced activation of the PI3-K/Akt pathway reduces mutant SOD1-mediated motor neuron cell death. Brain Res. Mol. Brain Res. 2003, 111, 155–164. [Google Scholar] [CrossRef]
- Sondell, M.; Kanje, M. Postnatal expression of VEGF and its receptor Flk-1 in peripheral ganglia. Neuroreport 2001, 12, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Sondell, M.; Lundborg, G.; Kanje, M. Vascular endothelial growth factor has neurotrophic activity and stimulates axonal outgrowth, enhancing cell survival and Schwann cell proliferation in the peripheral nervous system. J. Neurosci. 1999, 19, 5731–5740. [Google Scholar] [PubMed]
- Svensson, B.; Peters, M.; Konig, H.G.; Poppe, M.; Levkau, B.; Rothermundt, M.; Arolt, V.; Kogel, D.; Prehn, J.H. Vascular endothelial growth factor protects cultured rat hippocampal neurons against hypoxic injury via an antiexcitotoxic, caspase-independent mechanism. J. Cereb. Blood Flow Metab. 2002, 22, 1170–1175. [Google Scholar] [CrossRef] [PubMed]
- Krum, J.M.; Mani, N.; Rosenstein, J.M. Angiogenic and astroglial responses to vascular endothelial growth factor administration in adult rat brain. Neuroscience 2002, 110, 589–604. [Google Scholar] [CrossRef]
- Forstreuter, F.; Lucius, R.; Mentlein, R. Vascular endothelial growth factor induces chemotaxis and proliferation of microglial cells. J. Neuroimmunol. 2002, 132, 93–98. [Google Scholar] [CrossRef]
- Schratzberger, P.; Schratzberger, G.; Silver, M.; Curry, C.; Kearney, M.; Magner, M.; Alroy, J.; Adelman, L.S.; Weinberg, D.H.; Ropper, A.H.; et al. Favorable effect of VEGF gene transfer on ischemic peripheral neuropathy. Nat. Med. 2000, 6, 405–413. [Google Scholar] [PubMed]
- Piera-Velazquez, S.; Li, Z.; Jimenez, S.A. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 2011, 179, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Shore, E.M.; Lounev, V.Y.; Kaplan, F.S.; Kalluri, R.; Olsen, B.R. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat. Med. 2010, 16, 1400–1406. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Lovren, F.; Pan, Y.; Quan, A.; Ramadan, A.; Matkar, P.N.; Ehsan, M.; Sandhu, P.; Mantella, L.E.; Gupta, N.; et al. The essential autophagy gene ATG7 modulates organ fibrosis via regulation of endothelial-to-mesenchymal transition. J. Biol. Chem. 2015, 290, 2547–2559. [Google Scholar] [CrossRef] [PubMed]
- Markwald, R.R.; Fitzharris, T.P.; Smith, W.N. Sturctural analysis of endocardial cytodifferentiation. Dev. Biol. 1975, 42, 160–180. [Google Scholar] [CrossRef]
- Li, J.; Qu, X.; Bertram, J.F. Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am. J. Pathol. 2009, 175, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Nataraj, D.; Ernst, A.; Kalluri, R. Idiopathic pulmonary fibrosis is associated with endothelial to mesenchymal transition. Am. J. Respir. Cell Mol. Biol. 2010, 43, 129–130. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef] [PubMed]
- Paruchuri, S.; Yang, J.H.; Aikawa, E.; Melero-Martin, J.M.; Khan, Z.A.; Loukogeorgakis, S.; Schoen, F.J.; Bischoff, J. Human pulmonary valve progenitor cells exhibit endothelial/mesenchymal plasticity in response to vascular endothelial growth factor-A and transforming growth factor-beta2. Circ. Res. 2006, 99, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Srivastava, S.P.; Kanasaki, M.; He, J.; Kitada, M.; Nagai, T.; Nitta, K.; Takagi, S.; Kanasaki, K.; Koya, D. Interactions of DPP-4 and integrin beta1 influences endothelial-to-mesenchymal transition. Kidney Int. 2015, 88, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Sopova, K.; Gatsiou, K.; Stellos, K.; Laske, C. Dysregulation of neurotrophic and haematopoietic growth factors in Alzheimer’s disease: From pathophysiology to novel treatment strategies. Curr. Alzheimer Res. 2014, 11, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Augustin, H.G. Translating angiogenesis research into the clinic: The challenges ahead. Br. J. Radiol. 2003, 76, S3–S10. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zhou, H.; Lu, J.; Qu, Y.; Yu, D.; Tong, Y. Vascular endothelial growth factor: An attractive target in the treatment of hypoxic/ischemic brain injury. Neural Regen. Res. 2016, 11, 174–179. [Google Scholar] [PubMed]
- Rasmussen, H.S.; Rasmussen, C.S.; Macko, J. VEGF gene therapy for coronary artery disease and peripheral vascular disease. Cardiovasc. Radiat. Med. 2002, 3, 114–117. [Google Scholar] [CrossRef]
- Yano, K.; Liaw, P.C.; Mullington, J.M.; Shih, S.C.; Okada, H.; Bodyak, N.; Kang, P.M.; Toltl, L.; Belikoff, B.; Buras, J.; et al. Vascular endothelial growth factor is an important determinant of sepsis morbidity and mortality. J. Exp. Med. 2006, 203, 1447–1458. [Google Scholar] [CrossRef] [PubMed]
- Van der Flier, M.; van Leeuwen, H.J.; van Kessel, K.P.; Kimpen, J.L.; Hoepelman, A.I.; Geelen, S.P. Plasma vascular endothelial growth factor in severe sepsis. Shock 2005, 23, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Han, L.; Xu, X.; Tang, H.; Wang, H.; Wei, B. Serum biomarkers VEGF-C and IL-6 are associated with severe human Peripheral Artery Stenosis. J. Inflamm. 2015, 12, 50. [Google Scholar] [CrossRef] [PubMed]
- Shing, Y.; Folkman, J.; Sullivan, R.; Butterfield, C.; Murray, J.; Klagsbrun, M. Heparin affinity: Purification of a tumor-derived capillary endothelial cell growth factor. Science 1984, 223, 1296–1299. [Google Scholar] [CrossRef] [PubMed]
- Dow, J.K.; deVere White, R.W. Fibroblast growth factor 2: Its structure and property, paracrine function, tumor angiogenesis, and prostate-related mitogenic and oncogenic functions. Urology 2000, 55, 800–806. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Bar-Shavit, R.; Ishai-Michaeli, R.; Bashkin, P.; Fuks, Z. Extracellular sequestration and release of fibroblast growth factor: A regulatory mechanism? Trends Biochem. Sci. 1991, 16, 268–271. [Google Scholar] [CrossRef]
- Cross, M.J.; Claesson-Welsh, L. FGF and VEGF function in angiogenesis: Signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol. Sci. 2001, 22, 201–207. [Google Scholar] [CrossRef]
- Johnson, D.E.; Williams, L.T. Structural and functional diversity in the FGF receptor multigene family. Adv. Cancer Res. 1993, 60, 1–41. [Google Scholar] [PubMed]
- Deng, C.X.; Wynshaw-Boris, A.; Shen, M.M.; Daugherty, C.; Ornitz, D.M.; Leder, P. Murine FGFR-1 is required for early postimplantation growth and axial organization. Genes Dev. 1994, 8, 3045–3057. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Weinstein, M.; Li, C.; Naski, M.; Cohen, R.I.; Ornitz, D.M.; Leder, P.; Deng, C. Fibroblast growth factor receptor 2 (FGFR2)-mediated reciprocal regulation loop between FGF8 and FGF10 is essential for limb induction. Development 1998, 125, 753–765. [Google Scholar] [PubMed]
- Lee, S.H.; Schloss, D.J.; Swain, J.L. Maintenance of vascular integrity in the embryo requires signaling through the fibroblast growth factor receptor. J. Biol. Chem. 2000, 275, 33679–33687. [Google Scholar] [CrossRef] [PubMed]
- Dono, R.; Texido, G.; Dussel, R.; Ehmke, H.; Zeller, R. Impaired cerebral cortex development and blood pressure regulation in FGF-2-deficient mice. EMBO J. 1998, 17, 4213–4225. [Google Scholar] [CrossRef] [PubMed]
- Seghezzi, G.; Patel, S.; Ren, C.J.; Gualandris, A.; Pintucci, G.; Robbins, E.S.; Shapiro, R.L.; Galloway, A.C.; Rifkin, D.B.; Mignatti, P. Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: An autocrine mechanism contributing to angiogenesis. J. Cell Biol. 1998, 141, 1659–1673. [Google Scholar] [CrossRef] [PubMed]
- Teven, C.M.; Farina, E.M.; Rivas, J.; Reid, R.R. Fibroblast growth factor (FGF) signaling in development and skeletal diseases. Genes Dis. 2014, 1, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Williams, T. Metabolic Syndrome: Nonalcoholic Fatty Liver Disease. FP Essent. 2015, 435, 24–29. [Google Scholar] [PubMed]
- Ahmed, A.; Wong, R.J.; Harrison, S.A. Nonalcoholic Fatty Liver Disease Review: Diagnosis, Treatment, and Outcomes. Clin. Gastroenterol. Hepatol. 2015, 13, 2062–2070. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Cruz, A.; Gomez-Miranda, L.M.; Diaz Ramirez, G.; Carvali Meza, N.Y.; Bacardi-Gascon, M. Adiposity as a risk factor of non alcoholic fat disease; systematic review. Nutr. Hosp. 2014, 29, 771–775. [Google Scholar] [PubMed]
- Yilmaz, Y.; Younossi, Z.M. Obesity-associated nonalcoholic fatty liver disease. Clin. Liver Dis. 2014, 18, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Gruben, N.; Shiri-Sverdlov, R.; Koonen, D.P.; Hofker, M.H. Nonalcoholic fatty liver disease: A main driver of insulin resistance or a dangerous liaison? Biochim. Biophys. Acta 2014, 1842, 2329–2343. [Google Scholar] [CrossRef] [PubMed]
- Hanaka, H.; Hamada, T.; Ito, M.; Nakashima, H.; Tomita, K.; Seki, S.; Kobayashi, Y.; Imaki, J. Fibroblast growth factor-5 participates in the progression of hepatic fibrosis. Exp. Anim. 2014, 63, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, Y.; Eren, F. Identification of a support vector machine-based biomarker panel with high sensitivity and specificity for nonalcoholic steatohepatitis. Clin. Chim. Acta 2012, 414, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Chan, H.L.; Wong, G.L.; Choi, P.C.; Chan, A.W.; Chan, H.Y.; Chim, A.M.; Yeung, D.K.; Chan, F.K.; Woo, J.; et al. Non-invasive diagnosis of non-alcoholic steatohepatitis by combined serum biomarkers. J. Hepatol. 2012, 56, 1363–1370. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Xia, M.; Chang, X.; Xu, Q.; Bian, H.; Zeng, M.; Rao, S.; Yao, X.; Tu, Y.; Jia, W.; et al. Circulating fibroblast growth factor 21 levels are closely associated with hepatic fat content: A cross-sectional study. PLoS ONE 2011, 6, e24895. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lloyd, D.J.; Hale, C.; Stanislaus, S.; Chen, M.; Sivits, G.; Vonderfecht, S.; Hecht, R.; Li, Y.S.; Lindberg, R.A.; et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 2009, 58, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Takahashi, S.; Zhang, Y.; Krausz, K.W.; Smith, P.B.; Patterson, A.D.; Gonzalez, F.J. Role of fibroblast growth factor 21 in the early stage of NASH induced by methionine- and choline-deficient diet. Biochim. Biophys. Acta 2015, 1852, 1242–1252. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Lv, D.; Zhao, Y.; Chen, X.; Song, M.; Liu, J.; Bei, Y.; Wang, F.; Yang, W.; Yang, C. miR-149 controls non-alcoholic fatty liver by targeting FGF-21. J. Cell Mol. Med. 2016, 20, 1603–1608. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Bei, Y.; Liu, J.; Dimitrova-Shumkovska, J.; Kuang, D.; Zhou, Q.; Li, J.; Yang, Y.; Xiang, Y.; Wang, F.; et al. miR-212 downregulation contributes to the protective effect of exercise against non-alcoholic fatty liver via targeting FGF-21. J. Cell Mol. Med. 2016, 20, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Alisi, A.; Ceccarelli, S.; Panera, N.; Prono, F.; Petrini, S.; De Stefanis, C.; Pezzullo, M.; Tozzi, A.; Villani, A.; Bedogni, G.; et al. Association between Serum Atypical Fibroblast Growth Factors 21 and 19 and Pediatric Nonalcoholic Fatty Liver Disease. PLoS ONE 2013, 8, e67160. [Google Scholar] [CrossRef] [PubMed]
- Allison, M.A.; Criqui, M.H.; Wright, C.M. Patterns and risk factors for systemic calcified atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Wayhs, R.; Zelinger, A.; Raggi, P. High coronary artery calcium scores pose an extremely elevated risk for hard events. J. Am. Coll. Cardiol. 2002, 39, 225–230. [Google Scholar] [CrossRef]
- Demer, L.L.; Tintut, Y. Vascular calcification: Pathobiology of a multifaceted disease. Circulation 2008, 117, 2938–2948. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.A.; Larsson, A.; Lind, L.; Larsson, T.E. Circulating fibroblast growth factor-23 is associated with vascular dysfunction in the community. Atherosclerosis 2009, 205, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Ozkok, A.; Kekik, C.; Karahan, G.E.; Sakaci, T.; Ozel, A.; Unsal, A.; Yildiz, A. FGF-23 associated with the progression of coronary artery calcification in hemodialysis patients. BMC Nephrol. 2013, 14, 241. [Google Scholar] [CrossRef] [PubMed]
- Asicioglu, E.; Kahveci, A.; Arikan, H.; Koc, M.; Tuglular, S.; Ozener, C.I. Fibroblast growth factor-23 levels are associated with vascular calcifications in peritoneal dialysis patients. Nephron Clin. Pract. 2013, 124, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Peng, C.; Huang, W.; Zhang, J.; Xia, M.; Zhang, Y.; Ling, W. Circulating fibroblast growth factor 23 is associated with angiographic severity and extent of coronary artery disease. PLoS ONE 2013, 8, e72545. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Cui, Q.Q.; Ning, J.P.; Fu, S.S.; Liao, X.H. High-Flux Hemodialysis Benefits Hemodialysis Patients by Reducing Serum FGF-23 Levels and Reducing Vascular Calcification. Med. Sci. Monit. 2015, 21, 3467–3473. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, K.; Nagano, N.; Nitta, K. Klotho/FGF23 Axis in CKD. Contrib. Nephrol. 2015, 185, 56–65. [Google Scholar] [PubMed]
- Zhu, D.; Mackenzie, N.C.; Millan, J.L.; Farquharson, C.; MacRae, V.E. A protective role for FGF-23 in local defence against disrupted arterial wall integrity? Mol. Cell. Endocrinol. 2013, 372, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Fujita, S.; Morita, H.; Okamoto, Y.; Sohmiya, K.; Hoshiga, M.; Ishizaka, N. Association between circulating fibroblast growth factor 23, alpha-Klotho, and the left ventricular ejection fraction and left ventricular mass in cardiology inpatients. PLoS ONE 2013, 8, e73184. [Google Scholar] [CrossRef]
- Wyatt, C.M.; Drueke, T.B. Fibroblast growth factor receptor 4: The missing link between chronic kidney disease and FGF23-induced left ventricular hypertrophy? Kidney Int. 2016, 89, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Leifheit-Nestler, M.; Grosse Siemer, R.; Flasbart, K.; Richter, B.; Kirchhoff, F.; Ziegler, W.H.; Klintschar, M.; Becker, J.U.; Erbersdobler, A.; Aufricht, C.; et al. Induction of cardiac FGF23/FGFR4 expression is associated with left ventricular hypertrophy in patients with chronic kidney disease. Nephrol. Dial. Transplant. 2016, 31, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Grabner, A.; Amaral, A.P.; Schramm, K.; Singh, S.; Sloan, A.; Yanucil, C.; Li, J.; Shehadeh, L.A.; Hare, J.M.; David, V.; et al. Activation of Cardiac Fibroblast Growth Factor Receptor 4 Causes Left Ventricular Hypertrophy. Cell Metab. 2015, 22, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.Y.; Ma, H.X. Significant roles of anti-aging protein klotho and fibroblast growth factor 23 in cardiovascular disease. J. Geriatr. Cardiol. 2015, 12, 439–447. [Google Scholar] [PubMed]
- Van Ballegooijen, A.J.; Visser, M.; Kestenbaum, B.; Siscovick, D.S.; de Boer, I.H.; Gottdiener, J.S.; deFilippi, C.R.; Brouwer, I.A. Relation of vitamin D and parathyroid hormone to cardiac biomarkers and to left ventricular mass (from the Cardiovascular Health Study). Am. J. Cardiol. 2013, 111, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Tang, W.; Zhou, J.; Stubbs, J.R.; Luo, Q.; Pi, M.; Quarles, L.D. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J. Am. Soc. Nephrol. 2006, 17, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Donate-Correa, J.; Muros-de-Fuentes, M.; Mora-Fernandez, C.; Navarro-Gonzalez, J.F. FGF23/Klotho axis: Phosphorus, mineral metabolism and beyond. Cytokine Growth Factor Rev. 2012, 23, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Virag, J.A.; Rolle, M.L.; Reece, J.; Hardouin, S.; Feigl, E.O.; Murry, C.E. Fibroblast growth factor-2 regulates myocardial infarct repair: Effects on cell proliferation, scar contraction, and ventricular function. Am. J. Pathol. 2007, 171, 1431–1440. [Google Scholar] [CrossRef] [PubMed]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.C.; Sloan, A.; Isakova, T.; Gutierrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 induces left ventricular hypertrophy. J. Clin. Investig. 2011, 121, 4393–4408. [Google Scholar] [CrossRef] [PubMed]
- Itoh, N.; Ohta, H. Pathophysiological roles of FGF signaling in the heart. Front. Physiol. 2013, 4, 247. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ibrahimi, O.A.; Olsen, S.K.; Umemori, H.; Mohammadi, M.; Ornitz, D.M. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J. Biol. Chem. 2006, 281, 15694–15700. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.Y.; Sontag, D.P.; Detillieux, K.A.; Cattini, P.A. FGF-16 is released from neonatal cardiac myocytes and alters growth-related signaling: A possible role in postnatal development. Am. J. Physiol. Cell Physiol. 2008, 294, C1242–C1249. [Google Scholar] [CrossRef] [PubMed]
- Planavila, A.; Redondo, I.; Hondares, E.; Vinciguerra, M.; Munts, C.; Iglesias, R.; Gabrielli, L.A.; Sitges, M.; Giralt, M.; van Bilsen, M.; et al. Fibroblast growth factor 21 protects against cardiac hypertrophy in mice. Nat. Commun. 2013, 4, 2019. [Google Scholar] [CrossRef] [PubMed]
- Faul, C. Fibroblast growth factor 23 and the heart. Curr. Opin. Nephrol. Hypertens. 2012, 21, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Santiago, J.J.; McNaughton, L.J.; Koleini, N.; Ma, X.; Bestvater, B.; Nickel, B.E.; Fandrich, R.R.; Wigle, J.T.; Freed, D.H.; Arora, R.C.; et al. High molecular weight fibroblast growth factor-2 in the human heart is a potential target for prevention of cardiac remodeling. PLoS ONE 2014, 9, e97281. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, B.J.; Molkentin, J.D. Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem. Biophys. Res. Commun. 2004, 322, 1178–1191. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.H.; Khang-Loon, H. Carotid atherosclerosis. Definition, pathogenesis, and clinical significance. Neuroimaging Clin. N. Am. 1996, 6, 801–810. [Google Scholar] [PubMed]
- Chow, W.S.; Xu, A.; Woo, Y.C.; Tso, A.W.; Cheung, S.C.; Fong, C.H.; Tse, H.F.; Chau, M.T.; Cheung, B.M.; Lam, K.S. Serum fibroblast growth factor-21 levels are associated with carotid atherosclerosis independent of established cardiovascular risk factors. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2454–2459. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Crasto, C.; Strait, J.; Sun, K.; Schaumberg, D.A.; Ferrucci, L. Elevated serum fibroblast growth factor 21 is associated with hypertension in community-dwelling adults. J. Hum. Hypertens. 2013, 27, 397–399. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.J.; Hong, H.C.; Choi, H.Y.; Yoo, H.J.; Cho, G.J.; Hwang, T.G.; Baik, S.H.; Choi, D.S.; Kim, S.M.; Choi, K.M. Effects of a three-month combined exercise programme on fibroblast growth factor 21 and fetuin-A levels and arterial stiffness in obese women. Clin. Endocrinol. 2011, 75, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Qin, L.; Baeyens, N.; Li, G.; Afolabi, T.; Budatha, M.; Tellides, G.; Schwartz, M.A.; Simons, M. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J. Clin. Investig. 2015, 125, 4514–4528. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Qin, L.; Li, G.; Tellides, G.; Simons, M. Smooth muscle FGF/TGFbeta cross talk regulates atherosclerosis progression. EMBO Mol. Med. 2016, 8, 712–728. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Wang, C.; Liu, L.; Li, Y.; Li, X.; Cai, J.; Wang, H. Effects of fibroblast growth factor 21 on cell damage in vitro and atherosclerosis in vivo. Can. J. Physiol. Pharmacol. 2014, 92, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Llaurado, G.; Megia, A.; Cano, A.; Gimenez-Palop, O.; Simon, I.; Gonzalez-Sastre, M.; Berlanga, E.; Fernandez-Veledo, S.; Vendrell, J.; Gonzalez-Clemente, J.M. FGF-23/Vitamin D Axis in Type 1 Diabetes: The Potential Role of Mineral Metabolism in Arterial Stiffness. PLoS ONE 2015, 10, e0140222. [Google Scholar] [CrossRef] [PubMed]
- Dimas, G.; Iliadis, F.; Tegos, T.; Spiroglou, S.; Kanellos, I.; Karamouzis, I.; Savopoulos, C.; Hatzitolios, A.; Grekas, D. 4a.05: Circulating Fgf-23 as an Independent Correlate of Hypertension and Atherosclerosis in Early Stages of Ckd. J. Hypertens. 2015, 33 (Suppl. S1), e50. [Google Scholar] [CrossRef] [PubMed]
- Akin, F.; Celik, O.; Altun, I.; Ayca, B.; Diker, V.O.; Satilmis, S.; Sahin, C. Relationship of fibroblast growth factor 23 and fetuin—A to coronary atherosclerosis. J. Diabetes Complicat. 2015, 29, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Feng, S.; Han, O.Y.; Shen, H.Y.; Jin, D.H.; Shi, Y.B. Role of fibroblast growth factor-23 in the pathogenesis of atherosclerosis in peritoneal dialysis patients. Genet. Mol. Res. 2015, 14, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Misialek, J.R.; Eckfeldt, J.H.; Selvin, E.; Coresh, J.; Chen, L.Y.; Soliman, E.Z.; Agarwal, S.K.; Lutsey, P.L. Circulating fibroblast growth factor-23 and the incidence of atrial fibrillation: The Atherosclerosis Risk in Communities study. J. Am. Heart Assoc. 2014, 3, e001082. [Google Scholar] [CrossRef] [PubMed]
- Mathew, J.S.; Sachs, M.C.; Katz, R.; Patton, K.K.; Heckbert, S.R.; Hoofnagle, A.N.; Alonso, A.; Chonchol, M.; Deo, R.; Ix, J.H.; et al. Fibroblast growth factor-23 and incident atrial fibrillation: The Multi-Ethnic Study of Atherosclerosis (MESA) and the Cardiovascular Health Study (CHS). Circulation 2014, 130, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Kestenbaum, B.; Sachs, M.C.; Hoofnagle, A.N.; Siscovick, D.S.; Ix, J.H.; Robinson-Cohen, C.; Lima, J.A.; Polak, J.F.; Blondon, M.; Ruzinski, J.; et al. Fibroblast growth factor-23 and cardiovascular disease in the general population: The Multi-Ethnic Study of Atherosclerosis. Circ. Heart Fail. 2014, 7, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Urakawa, I.; Yamazaki, Y.; Shimada, T.; Iijima, K.; Hasegawa, H.; Okawa, K.; Fujita, T.; Fukumoto, S.; Yamashita, T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006, 444, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Fliser, D.; Kollerits, B.; Neyer, U.; Ankerst, D.P.; Lhotta, K.; Lingenhel, A.; Ritz, E.; Kronenberg, F.; Group, M.S.; Kuen, E.; et al. Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: The Mild to Moderate Kidney Disease (MMKD) Study. J. Am. Soc. Nephrol. 2007, 18, 2600–2608. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Xie, H.; Yang, W.; Xie, D.; Anderson, A.H.; Scialla, J.; Wahl, P.; Gutierrez, O.M.; Steigerwalt, S.; He, J.; et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA 2011, 305, 2432–2439. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, O.M.; Mannstadt, M.; Isakova, T.; Rauh-Hain, J.A.; Tamez, H.; Shah, A.; Smith, K.; Lee, H.; Thadhani, R.; Juppner, H.; et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N. Engl. J. Med. 2008, 359, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Pavik, I.; Jaeger, P.; Ebner, L.; Wagner, C.A.; Petzold, K.; Spichtig, D.; Poster, D.; Wuthrich, R.P.; Russmann, S.; Serra, A.L. Secreted Klotho and FGF23 in chronic kidney disease Stage 1 to 5: A sequence suggested from a cross-sectional study. Nephrol. Dial. Transplant. 2013, 28, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Floege, J.; Kriz, W.; Schulze, M.; Susani, M.; Kerjaschki, D.; Mooney, A.; Couser, W.G.; Koch, K.M. Basic fibroblast growth factor augments podocyte injury and induces glomerulosclerosis in rats with experimental membranous nephropathy. J. Clin. Investig. 1995, 96, 2809–2819. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Nie, L.; He, T.; Yang, K.; Xiao, T.; Wang, S.; Huang, Y.; Zhang, J.; Wang, J.; Sharma, K.; et al. Klotho suppresses renal tubulo-interstitial fibrosis by controlling basic fibroblast growth factor-2 signalling. J. Pathol. 2014, 234, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Oehmichen, J.; Van Aken, H.; Reuter, S.; Pavenstadt, H.J.; Meersch, M.; Unruh, M.; Zarbock, A. FGF23 signaling impairs neutrophil recruitment and host defense during CKD. J. Clin. Investig. 2016, 126, 962–974. [Google Scholar] [CrossRef] [PubMed]
- Rossini, M.; Cheunsuchon, B.; Donnert, E.; Ma, L.J.; Thomas, J.W.; Neilson, E.G.; Fogo, A.B. Immunolocalization of fibroblast growth factor-1 (FGF-1), its receptor (FGFR-1), and fibroblast-specific protein-1 (FSP-1) in inflammatory renal disease. Kidney Int. 2005, 68, 2621–2628. [Google Scholar] [CrossRef] [PubMed]
- Silswal, N.; Touchberry, C.D.; Daniel, D.R.; McCarthy, D.L.; Zhang, S.; Andresen, J.; Stubbs, J.R.; Wacker, M.J. FGF23 directly impairs endothelium-dependent vasorelaxation by increasing superoxide levels and reducing nitric oxide bioavailability. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E426–E436. [Google Scholar] [CrossRef] [PubMed]
- Hindricks, J.; Ebert, T.; Bachmann, A.; Kralisch, S.; Lossner, U.; Kratzsch, J.; Stolzenburg, J.U.; Dietel, A.; Beige, J.; Anders, M.; et al. Serum levels of fibroblast growth factor-21 are increased in chronic and acute renal dysfunction. Clin. Endocrinol. 2014, 80, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Hines, E.A.; Sun, X. Tissue crosstalk in lung development. J. Cell. Biochem. 2014, 115, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Volckaert, T.; Dill, E.; Campbell, A.; Tiozzo, C.; Majka, S.; Bellusci, S.; De Langhe, S.P. Parabronchial smooth muscle constitutes an airway epithelial stem cell niche in the mouse lung after injury. J. Clin. Investig. 2011, 121, 4409–4419. [Google Scholar] [CrossRef] [PubMed]
- Coffey, E.; Newman, D.R.; Sannes, P.L. Expression of fibroblast growth factor 9 in normal human lung and idiopathic pulmonary fibrosis. J. Histochem. Cytochem. 2013, 61, 671–679. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, B.; Korfei, M.; Henneke, I.; Sibinska, Z.; Tian, X.; Hezel, S.; Dilai, S.; Wasnick, R.; Schneider, B.; Wilhelm, J.; et al. Increased FGF1-FGFRc expression in idiopathic pulmonary fibrosis. Respir. Res. 2015, 16, 83. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.; Becerril, C.; Montano, M.; Garcia-De-Alba, C.; Ramirez, R.; Checa, M.; Pardo, A.; Selman, M. FGF-1 reverts epithelial-mesenchymal transition induced by TGF-{beta}1 through MAPK/ERK kinase pathway. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L222–L231. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, S.; Scotton, C.J.; McNulty, K.; Nye, E.; Stamp, G.; Laurent, G.; Bonnet, D.; Janes, S.M. Bone marrow stem cells expressing keratinocyte growth factor via an inducible lentivirus protects against bleomycin-induced pulmonary fibrosis. PLoS ONE 2009, 4, e8013. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Stoilov, I.; Elton, T.J.; Mecham, R.P.; Ornitz, D.M. Fibroblast growth factor 2 is required for epithelial recovery, but not for pulmonary fibrosis, in response to bleomycin. Am. J. Respir. Cell Mol. Biol. 2015, 52, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Joannes, A.; Brayer, S.; Besnard, V.; Marchal-Somme, J.; Jaillet, M.; Mordant, P.; Mal, H.; Borie, R.; Crestani, B.; Mailleux, A.A. FGF9 and FGF18 in idiopathic pulmonary fibrosis promote survival and migration and inhibit myofibroblast differentiation of human lung fibroblasts in vitro. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L615–L629. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shi, C.; Meng, X.; Zhang, K.; Li, X.; Wang, C.; Xiang, Z.; Hu, K.; Han, X. Inhibition of Wnt/beta-catenin signaling suppresses bleomycin-induced pulmonary fibrosis by attenuating the expression of TGF-beta1 and FGF-2. Exp. Mol. Pathol. 2016, 101, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Zhang, G.; Ji, Y.; Zhua, H.; Lv, C.; Jiang, W. Protective role of gambogic acid in experimental pulmonary fibrosis in vitro and in vivo. Phytomedicine 2016, 23, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, H.V.; Maher, T.M. Nintedanib in idiopathic pulmonary fibrosis. Drugs Today 2015, 51, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Zhang, L.; Kang, Z.; Jiang, W.; Lv, C. Ponatinib ameliorates pulmonary fibrosis by suppressing TGF-beta1/Smad3 pathway. Pulm. Pharmacol. Ther. 2015, 34, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kumral, A.; Iscan, B.; Tuzun, F.; Micili, S.C.; Arslan, M.K.; Tugyan, K.; Duman, N.; Ozkan, H. Bacillus Calmette-Guerin vaccination: A novel therapeutic approach to preventing hyperoxic lung injury. J. Matern. Fetal Neonatal Med. 2015, 28, 1950–1956. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ji, Y.; Kang, Z.; Lv, C.; Jiang, W. Protocatechuic aldehyde ameliorates experimental pulmonary fibrosis by modulating HMGB1/RAGE pathway. Toxicol. Appl. Pharmacol. 2015, 283, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Meyer, M.; Muller, A.K.; Bohm, F.; Grose, R.; Dauwalder, T.; Verrey, F.; Kopf, M.; Partanen, J.; Bloch, W.; et al. Fibroblast growth factor receptors 1 and 2 in keratinocytes control the epidermal barrier and cutaneous homeostasis. J. Cell Biol. 2010, 188, 935–952. [Google Scholar] [CrossRef] [PubMed]
- Jameson, J.; Ugarte, K.; Chen, N.; Yachi, P.; Fuchs, E.; Boismenu, R.; Havran, W.L. A role for skin gammadelta T cells in wound repair. Science 2002, 296, 747–749. [Google Scholar] [CrossRef] [PubMed]
- Gay, D.; Kwon, O.; Zhang, Z.; Spata, M.; Plikus, M.V.; Holler, P.D.; Ito, M.; Yang, Z.; Treffeisen, E.; Kim, C.D.; et al. Fgf9 from dermal gammadelta T cells induces hair follicle neogenesis after wounding. Nat. Med. 2013, 19, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Werner, S.; Peters, K.G.; Longaker, M.T.; Fuller-Pace, F.; Banda, M.J.; Williams, L.T. Large induction of keratinocyte growth factor expression in the dermis during wound healing. Proc. Natl. Acad. Sci. USA 1992, 89, 6896–6900. [Google Scholar] [CrossRef] [PubMed]
- Casey-Sawicki, K.; Zhang, M.; Kim, S.; Zhang, A.; Zhang, S.B.; Zhang, Z.; Singh, R.; Yang, S.; Swarts, S.; Vidyasagar, S.; et al. A basic fibroblast growth factor analog for protection and mitigation against acute radiation syndromes. Health Phys. 2014, 106, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Samy, R.P.; Kandasamy, M.; Gopalakrishnakone, P.; Stiles, B.G.; Rowan, E.G.; Becker, D.; Shanmugam, M.K.; Sethi, G.; Chow, V.T. Wound healing activity and mechanisms of action of an antibacterial protein from the venom of the eastern diamondback rattlesnake (Crotalus adamanteus). PLoS ONE 2014, 9, e80199. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.J.; Watson, J.J.; Dawbarn, D. The neurotrophins and their role in Alzheimer’s disease. Curr. Neuropharmacol. 2011, 9, 559–573. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Mattson, M.P. Glucose deprivation elicits neurofibrillary tangle-like antigenic changes in hippocampal neurons: Prevention by NGF and bFGF. Exp. Neurol. 1992, 117, 114–123. [Google Scholar] [CrossRef]
- Johnson-Farley, N.N.; Patel, K.; Kim, D.; Cowen, D.S. Interaction of FGF-2 with IGF-1 and BDNF in stimulating Akt, ERK, and neuronal survival in hippocampal cultures. Brain Res. 2007, 1154, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Kiprianova, I.; Schindowski, K.; von Bohlen und Halbach, O.; Krause, S.; Dono, R.; Schwaninger, M.; Unsicker, K. Enlarged infarct volume and loss of BDNF mRNA induction following brain ischemia in mice lacking FGF-2. Exp. Neurol. 2004, 189, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Pinilla, F.; Cummings, B.J.; Cotman, C.W. Induction of basic fibroblast growth factor in Alzheimer’s disease pathology. Neuroreport 1990, 1, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Sasaki, H.; Katagiri, T.; Sasaki, H.; Koiwai, K.; Youki, H.; Totsuka, S.; Ishii, T. The binding of basic fibroblast growth factor to Alzheimer’s neurofibrillary tangles and senile plaques. Neurosci. Lett. 1991, 122, 33–36. [Google Scholar] [CrossRef]
- Stieber, A.; Mourelatos, Z.; Gonatas, N.K. In Alzheimer’s disease the Golgi apparatus of a population of neurons without neurofibrillary tangles is fragmented and atrophic. Am. J. Pathol. 1996, 148, 415–426. [Google Scholar] [PubMed]
- Hanneken, A.; Frautschy, S.; Galasko, D.; Baird, A. A fibroblast growth factor binding protein in human cerebral spinal fluid. Neuroreport 1995, 6, 886–888. [Google Scholar] [CrossRef] [PubMed]
- Cummings, B.J.; Su, J.H.; Cotman, C.W. Neuritic involvement within bFGF immunopositive plaques of Alzheimer’s disease. Exp. Neurol. 1993, 124, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Takami, K.; Matsuo, A.; Terai, K.; Walker, D.G.; McGeer, E.G.; McGeer, P.L. Fibroblast growth factor receptor-1 expression in the cortex and hippocampus in Alzheimer’s disease. Brain Res. 1998, 802, 89–97. [Google Scholar] [CrossRef]
- Gong, C.X.; Lidsky, T.; Wegiel, J.; Zuck, L.; Grundke-Iqbal, I.; Iqbal, K. Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in Alzheimer’s disease. J. Biol. Chem. 2000, 275, 5535–5544. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Liu, F.; Gong, C.X.; Grundke-Iqbal, I. Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Burack, M.A.; Halpain, S. Site-specific regulation of Alzheimer-like tau phosphorylation in living neurons. Neuroscience 1996, 72, 167–184. [Google Scholar] [CrossRef]
- Butt, A.M.; Dinsdale, J. Fibroblast growth factor 2 mediated disruption of myelin-forming oligodendrocytes in vivo is associated with increased tau immunoreactivity. Neurosci. Lett. 2005, 375, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Tatebayashi, Y.; Lee, M.H.; Li, L.; Iqbal, K.; Grundke-Iqbal, I. The dentate gyrus neurogenesis: A therapeutic target for Alzheimer’s disease. Acta Neuropathol. 2003, 105, 225–232. [Google Scholar] [PubMed]
- Tatebayashi, Y.; Haque, N.; Tung, Y.C.; Iqbal, K.; Grundke-Iqbal, I. Role of tau phosphorylation by glycogen synthase kinase-3beta in the regulation of organelle transport. J. Cell Sci. 2004, 117 Pt 9, 1653–1663. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.P.; Chen, T.; Yin, N.N.; Han, Y.M.; Yuan, F.; Duan, Y.J.; Shen, F.; Zhang, Y.H.; Chen, Z.B. Puerarin Ameliorates D-Galactose Induced Enhanced Hippocampal Neurogenesis and Tau Hyperphosphorylation in Rat Brain. J. Alzheimers Dis. 2016, 51, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Ogino, R.; Murayama, N.; Noshita, T.; Takemoto, N.; Toba, T.; Oka, T.; Narii, N.; Yoshida, S.; Ueno, N.; Inoue, T. SUN11602 has basic fibroblast growth factor-like activity and attenuates neuronal damage and cognitive deficits in a rat model of Alzheimer’s disease induced by amyloid beta and excitatory amino acids. Brain Res. 2014, 1585, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.W.; Lahiri, D.K. Regulation of promoter activity of the APP gene by cytokines and growth factors: Implications in Alzheimer’s disease. Ann. N.Y. Acad. Sci. 2002, 973, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Rossner, S.; Ueberham, U.; Schliebs, R.; Perez-Polo, J.R.; Bigl, V. Neurotrophin binding to the p75 neurotrophin receptor is necessary but not sufficient to mediate NGF-effects on APP secretion in PC-12 cells. J. Neural Transm. Suppl. 1998, 54, 279–285. [Google Scholar] [PubMed]
- Araujo, D.M.; Cotman, C.W. Beta-amyloid stimulates glial cells in vitro to produce growth factors that accumulate in senile plaques in Alzheimer’s disease. Brain Res. 1992, 569, 141–145. [Google Scholar] [CrossRef]
- Carlson, G.A.; Borchelt, D.R.; Dake, A.; Turner, S.; Danielson, V.; Coffin, J.D.; Eckman, C.; Meiners, J.; Nilsen, S.P.; Younkin, S.G.; et al. Genetic modification of the phenotypes produced by amyloid precursor protein overexpression in transgenic mice. Hum. Mol. Genet. 1997, 6, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Mashayekhi, F.; Hadavi, M.; Vaziri, H.R.; Naji, M. Increased acidic fibroblast growth factor concentrations in the serum and cerebrospinal fluid of patients with Alzheimer’s disease. J. Clin. Neurosci. 2010, 17, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Joyce, N.C. Proliferative capacity of the corneal endothelium. Prog. Retin. Eye Res. 2003, 22, 359–389. [Google Scholar] [CrossRef]
- Kay, E.P.; Gu, X.; Ninomiya, Y.; Smith, R.E. Corneal endothelial modulation: A factor released by leukocytes induces basic fibroblast growth factor that modulates cell shape and collagen. Investig. Ophthalmol. Vis. Sci. 1993, 34, 663–672. [Google Scholar]
- Waring, G.O., III. Posterior collagenous layer of the cornea. Ultrastructural classification of abnormal collagenous tissue posterior to Descemet’s membrane in 30 cases. Arch. Ophthalmol. 1982, 100, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.G.; Kay, E.P. Two populations of p27 use differential kinetics to phosphorylate Ser-10 and Thr-187 via phosphatidylinositol 3-Kinase in response to fibroblast growth factor-2 stimulation. J. Biol. Chem. 2007, 282, 6444–6454. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.G.; Kay, E.P. Involvement of two distinct ubiquitin E3 ligase systems for p27 degradation in corneal endothelial cells. Investig. Ophthalmol. Vis. Sci. 2008, 49, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.K.; Kay, E.P. Regulatory role of FGF-2 on type I collagen expression during endothelial mesenchymal transformation. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4495–4503. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.G.; Kay, E.P. Common and distinct pathways for cellular activities in FGF-2 signaling induced by IL-1beta in corneal endothelial cells. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Song, J.S.; Lee, J.G.; Kay, E.P. Induction of FGF-2 synthesis by IL-1beta in aqueous humor through P13-kinase and p38 in rabbit corneal endothelium. Investig. Ophthalmol. Vis. Sci. 2010, 51, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, X.; Ma, J.; Tian, H.; Jiao, Y.; Zhang, R.; Huang, Z.; Xiao, J.; Zhao, B.; Qian, H.; et al. Effects of keratinocyte growth factor-2 on corneal epithelial wound healing in a rabbit model of carbon dioxide laser injury. Biol. Pharm. Bull. 2010, 33, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Yancopoulos, G.D.; Davis, S.; Gale, N.W.; Rudge, J.S.; Wiegand, S.J.; Holash, J. Vascular-specific growth factors and blood vessel formation. Nature 2000, 407, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Suri, C.; Jones, P.F.; Patan, S.; Bartunkova, S.; Maisonpierre, P.C.; Davis, S.; Sato, T.N.; Yancopoulos, G.D. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell 1996, 87, 1171–1180. [Google Scholar] [CrossRef]
- Wu, X.; Liu, N. The role of Ang/Tie signaling in lymphangiogenesis. Lymphology 2010, 43, 59–72. [Google Scholar] [PubMed]
- Davis, S.; Aldrich, T.H.; Jones, P.F.; Acheson, A.; Compton, D.L.; Jain, V.; Ryan, T.E.; Bruno, J.; Radziejewski, C.; Maisonpierre, P.C.; et al. Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell 1996, 87, 1161–1169. [Google Scholar] [CrossRef]
- Maisonpierre, P.C.; Suri, C.; Jones, P.F.; Bartunkova, S.; Wiegand, S.J.; Radziejewski, C.; Compton, D.; McClain, J.; Aldrich, T.H.; Papadopoulos, N.; et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997, 277, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Partanen, J.; Dumont, D.J. Functions of Tie1 and Tie2 receptor tyrosine kinases in vascular development. Curr. Top. Microbiol. Immunol. 1999, 237, 159–172. [Google Scholar] [PubMed]
- DeBusk, L.M.; Chen, Y.; Nishishita, T.; Chen, J.; Thomas, J.W.; Lin, P.C. Tie2 receptor tyrosine kinase, a major mediator of tumor necrosis factor alpha-induced angiogenesis in rheumatoid arthritis. Arthritis Rheumatol. 2003, 48, 2461–2471. [Google Scholar] [CrossRef] [PubMed]
- Takahara, K.; Iioka, T.; Furukawa, K.; Uchida, T.; Nakashima, M.; Tsukazaki, T.; Shindo, H. Autocrine/paracrine role of the angiopoietin-1 and -2/Tie2 system in cell proliferation and chemotaxis of cultured fibroblastic synoviocytes in rheumatoid arthritis. Hum. Pathol. 2004, 35, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Scott, B.B.; Zaratin, P.F.; Gilmartin, A.G.; Hansbury, M.J.; Colombo, A.; Belpasso, C.; Winkler, J.D.; Jackson, J.R. TNF-alpha modulates angiopoietin-1 expression in rheumatoid synovial fibroblasts via the NF-kappa B signalling pathway. Biochem. Biophys. Res. Commun. 2005, 328, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Hashiramoto, A.; Sakai, C.; Yoshida, K.; Tsumiyama, K.; Miura, Y.; Shiozawa, K.; Nose, M.; Komai, K.; Shiozawa, S. Angiopoietin 1 directly induces destruction of the rheumatoid joint by cooperative, but independent, signaling via ERK/MAPK and phosphatidylinositol 3-kinase/Akt. Arthritis Rheumatol. 2007, 56, 2170–2179. [Google Scholar] [CrossRef] [PubMed]
- Kurosaka, D.; Hirai, K.; Nishioka, M.; Miyamoto, Y.; Yoshida, K.; Noda, K.; Ukichi, T.; Yanagimachi, M.; Furuya, K.; Takahashi, E.; et al. Clinical significance of serum levels of vascular endothelial growth factor, angiopoietin-1, and angiopoietin-2 in patients with rheumatoid arthritis. J. Rheumatol. 2010, 37, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Krausz, S.; Garcia, S.; Ambarus, C.A.; de Launay, D.; Foster, M.; Naiman, B.; Iverson, W.; Connor, J.R.; Sleeman, M.A.; Coyle, A.J.; et al. Angiopoietin-2 promotes inflammatory activation of human macrophages and is essential for murine experimental arthritis. Ann. Rheum. Dis. 2012, 71, 1402–1410. [Google Scholar] [CrossRef] [PubMed]
- Kanakaraj, P.; Puffer, B.A.; Yao, X.T.; Kankanala, S.; Boyd, E.; Shah, R.R.; Wang, G.; Patel, D.; Krishnamurthy, R.; Kaithamana, S.; et al. Simultaneous targeting of TNF and Ang2 with a novel bispecific antibody enhances efficacy in an in vivo model of arthritis. mAbs 2012, 4, 600–613. [Google Scholar] [CrossRef] [PubMed]
- Tasaki, Y.; Shimizu, M.; Inoue, N.; Mizuta, M.; Nakagishi, Y.; Wada, T.; Yachie, A. Disruption of vascular endothelial homeostasis in systemic juvenile idiopathic arthritis-associated macrophage activation syndrome: The dynamic roles of angiopoietin-1 and -2. Cytokine 2016, 80, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wolfram, J.A.; Diaconu, D.; Hatala, D.A.; Rastegar, J.; Knutsen, D.A.; Lowther, A.; Askew, D.; Gilliam, A.C.; McCormick, T.S.; Ward, N.L. Keratinocyte but not endothelial cell-specific overexpression of Tie2 leads to the development of psoriasis. Am. J. Pathol. 2009, 174, 1443–1458. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Xie, H.; Ji, J.; Zhou, X.; Goltzman, D.; Miao, D. Defective female reproductive function in 1,25(OH)2D-deficient mice results from indirect effect mediated by extracellular calcium and/or phosphorus. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E928–E935. [Google Scholar] [CrossRef] [PubMed]
- Tal, R.; Seifer, D.B.; Grazi, R.V.; Malter, H.E. Angiopoietin-1 and angiopoietin-2 are altered in polycystic ovarian syndrome (PCOS) during controlled ovarian stimulation. Vasc. Cell 2013, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Scotti, L.; Abramovich, D.; Pascuali, N.; Durand, L.H.; Irusta, G.; de Zuniga, I.; Tesone, M.; Parborell, F. Inhibition of angiopoietin-1 (ANGPT1) affects vascular integrity in ovarian hyperstimulation syndrome (OHSS). Reprod. Fertil. Dev. 2016, 28, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Hilbert, T.; Klaschik, S. The angiopoietin/TIE receptor system: Focusing its role for ischemia-reperfusion injury. Cytokine Growth Factor Rev. 2015, 26, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Novotny, N.M.; Lahm, T.; Markel, T.A.; Crisostomo, P.R.; Wang, M.; Wang, Y.; Tan, J.; Meldrum, D.R. Angiopoietin-1 in the treatment of ischemia and sepsis. Shock 2009, 31, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chopp, M. Vascular endothelial growth factor and angiopoietins in focal cerebral ischemia. Trends Cardiovasc. Med. 2002, 12, 62–66. [Google Scholar] [CrossRef]
- Thorin-Trescases, N.; Thorin, E. Angiopoietin-like-2: A multifaceted protein with physiological and pathophysiological properties. Expert Rev. Mol. Med. 2014, 16, e17. [Google Scholar] [CrossRef] [PubMed]
- Kadomatsu, T.; Endo, M.; Miyata, K.; Oike, Y. Diverse roles of ANGPTL2 in physiology and pathophysiology. Trends Endocrinol. Metab. 2014, 25, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.J.; Teo, Z.; Sng, M.K.; Zhu, P.; Tan, N.S. Emerging roles of angiopoietin-like 4 in human cancer. Mol. Cancer Res. 2012, 10, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Goh, Y.Y.; Chin, H.F.; Kersten, S.; Tan, N.S. Angiopoietin-like 4: A decade of research. Biosci. Rep. 2012, 32, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Kadomatsu, T.; Tabata, M.; Oike, Y. Angiopoietin-like proteins: Emerging targets for treatment of obesity and related metabolic diseases. FEBS J. 2011, 278, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Oike, Y.; Tabata, M. Angiopoietin-like proteins—Potential therapeutic targets for metabolic syndrome and cardiovascular disease. Circ. J. 2009, 73, 2192–2197. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.C.; Chiang, W.C.; Tsai, M.H.; Chou, Y.H.; Pan, S.Y.; Chang, Y.T.; Yeh, P.Y.; Chen, Y.T.; Chiang, C.K.; Chen, Y.M.; et al. Angiopoietin-2-induced arterial stiffness in CKD. J. Am. Soc. Nephrol. 2014, 25, 1198–1209. [Google Scholar] [CrossRef] [PubMed]
- Joussen, A.M.; Poulaki, V.; Tsujikawa, A.; Qin, W.; Qaum, T.; Xu, Q.; Moromizato, Y.; Bursell, S.E.; Wiegand, S.J.; Rudge, J.; et al. Suppression of diabetic retinopathy with angiopoietin-1. Am. J. Pathol. 2002, 160, 1683–1693. [Google Scholar] [CrossRef]
- Hammes, H.P.; Lin, J.; Wagner, P.; Feng, Y.; Vom Hagen, F.; Krzizok, T.; Renner, O.; Breier, G.; Brownlee, M.; Deutsch, U. Angiopoietin-2 causes pericyte dropout in the normal retina: Evidence for involvement in diabetic retinopathy. Diabetes 2004, 53, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Pfister, F.; Wang, Y.; Schreiter, K.; vom Hagen, F.; Altvater, K.; Hoffmann, S.; Deutsch, U.; Hammes, H.P.; Feng, Y. Retinal overexpression of angiopoietin-2 mimics diabetic retinopathy and enhances vascular damages in hyperglycemia. Acta Diabetol. 2010, 47, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, S.; Srinivasan, R.; Maestas, J.; McGuire, P.G.; Das, A. A potential role for angiopoietin 2 in the regulation of the blood-retinal barrier in diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3784–3791. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Yun, J.H.; Kim, J.H.; Kim, K.W.; Cho, C.H.; Kim, J.H. Angiopoietin 2 induces pericyte apoptosis via alpha3beta1 integrin signaling in diabetic retinopathy. Diabetes 2014, 63, 3057–3068. [Google Scholar] [CrossRef] [PubMed]
- Cahoon, J.M.; Rai, R.R.; Carroll, L.S.; Uehara, H.; Zhang, X.; O’Neil, C.L.; Medina, R.J.; Das, S.K.; Muddana, S.K.; Olson, P.R.; et al. Intravitreal AAV2.COMP-Ang1 Prevents Neurovascular Degeneration in a Murine Model of Diabetic Retinopathy. Diabetes 2015, 64, 4247–4259. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.H.; Park, S.W.; Kim, J.H.; Park, Y.J.; Cho, C.H.; Kim, J.H. Angiopoietin 2 induces astrocyte apoptosis via alphavbeta5-integrin signaling in diabetic retinopathy. Cell Death Dis 2016, 7, e2101. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, W.; Moon, S.O.; Sung, M.J.; Kim, D.H.; Kang, K.P.; Jang, K.Y.; Lee, S.Y.; Park, B.H.; Koh, G.Y.; et al. Renoprotective effect of COMP-angiopoietin-1 in db/db mice with type 2 diabetes. Nephrol. Dial. Transplant. 2007, 22, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.; Dei Cas, A.; Long, D.A.; White, K.E.; Hayward, A.; Ku, C.H.; Woolf, A.S.; Bilous, R.; Viberti, G.; Gnudi, L. Podocyte-specific expression of angiopoietin-2 causes proteinuria and apoptosis of glomerular endothelia. J. Am. Soc. Nephrol. 2007, 18, 2320–2329. [Google Scholar] [CrossRef] [PubMed]
- Dessapt-Baradez, C.; Woolf, A.S.; White, K.E.; Pan, J.; Huang, J.L.; Hayward, A.A.; Price, K.L.; Kolatsi-Joannou, M.; Locatelli, M.; Diennet, M.; et al. Targeted glomerular angiopoietin-1 therapy for early diabetic kidney disease. J. Am. Soc. Nephrol. 2014, 25, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Li, T.; Zhang, C.; Chen, Q.; Li, Z.; Liu, J.; Wang, Y. Therapeutic effect of alprostadil in diabetic nephropathy: Possible roles of angiopoietin-2 and IL-18. Cell. Physiol. Biochem. 2014, 34, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Khairoun, M.; van den Heuvel, M.; van den Berg, B.M.; Sorop, O.; de Boer, R.; van Ditzhuijzen, N.S.; Bajema, I.M.; Baelde, H.J.; Zandbergen, M.; Duncker, D.J.; et al. Early systemic microvascular damage in pigs with atherogenic diabetes mellitus coincides with renal angiopoietin dysbalance. PLoS ONE 2015, 10, e0121555. [Google Scholar] [CrossRef] [PubMed]
- David, S.; Kumpers, P.; Lukasz, A.; Fliser, D.; Martens-Lobenhoffer, J.; Bode-Boger, S.M.; Kliem, V.; Haller, H.; Kielstein, J.T. Circulating angiopoietin-2 levels increase with progress of chronic kidney disease. Nephrol. Dial. Transplant. 2010, 25, 2571–2576. [Google Scholar] [CrossRef] [PubMed]
- David, S.; John, S.G.; Jefferies, H.J.; Sigrist, M.K.; Kumpers, P.; Kielstein, J.T.; Haller, H.; McIntyre, C.W. Angiopoietin-2 levels predict mortality in CKD patients. Nephrol. Dial. Transplant. 2012, 27, 1867–1872. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.C.; Lai, T.S.; Chiang, C.K.; Chen, Y.M.; Wu, M.S.; Chu, T.S.; Wu, K.D.; Lin, S.L. Angiopoietin-2 is associated with albuminuria and microinflammation in chronic kidney disease. PLoS ONE 2013, 8, e54668. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.C.; Chiu, Y.W.; Tsai, J.C.; Kuo, H.T.; Lee, S.C.; Hung, C.C.; Lin, M.Y.; Hwang, S.J.; Kuo, M.C.; Chen, H.C. Association of angiopoietin-2 with renal outcome in chronic kidney disease. PLoS ONE 2014, 9, e108862. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Niu, J.; Ding, W.; Zhang, M.; Yang, M.; Gu, Y. Angiopoietin-1 attenuates angiotensin II-induced ER stress in glomerular endothelial cells via a Tie2 receptor/ERK1/2-p38 MAPK-dependent mechanism. Mol. Cell. Endocrinol. 2016, 428, 118–132. [Google Scholar] [CrossRef] [PubMed]
- Koutroubakis, I.E.; Xidakis, C.; Karmiris, K.; Sfiridaki, A.; Kandidaki, E.; Kouroumalis, E.A. Potential role of soluble angiopoietin-2 and Tie2 in patients with inflammatory bowel disease. Eur. J. Clin. Investig. 2006, 36, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Ganta, V.C.; Cromer, W.; Mills, G.L.; Traylor, J.; Jennings, M.; Daley, S.; Clark, B.; Mathis, J.M.; Bernas, M.; Boktor, M.; et al. Angiopoietin-2 in experimental colitis. Inflamm. Bowel Dis. 2010, 16, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, K.A.; Kapsoritakis, A.N.; Kapsoritaki, A.I.; Manolakis, A.C.; Tiaka, E.K.; Tsiopoulos, F.D.; Tsiompanidis, I.A.; Potamianos, S.P. Angiogenin, angiopoietin-1, angiopoietin-2, and endostatin serum levels in inflammatory bowel disease. Inflamm. Bowel Dis. 2011, 17, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Algaba, A.; Linares, P.M.; Encarnacion Fernandez-Contreras, M.; Figuerola, A.; Calvet, X.; Guerra, I.; de Pousa, I.; Chaparro, M.; Gisbert, J.P.; Bermejo, F. The effects of infliximab or adalimumab on vascular endothelial growth factor and angiopoietin 1 angiogenic factor levels in inflammatory bowel disease: Serial observations in 37 patients. Inflamm. Bowel Dis. 2014, 20, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.X.; Gu, S.Z.; Zhang, S.; Ren, Y.; Sang, L.X.; Dai, C. Angiopoietin and vascular endothelial growth factor expression in colorectal disease models. World J. Gastroenterol. 2015, 21, 2645–2650. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.D.; Campbell, A.I.; Robb, M.; Ng, D.; Stewart, D.J. Protective role of angiopoietin-1 in experimental pulmonary hypertension. Circ. Res. 2003, 92, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, C.C.; Du, L.; Chu, D.; Cho, A.J.; Kido, M.; Wolf, P.L.; Jamieson, S.W.; Thistlethwaite, P.A. Induction of pulmonary hypertension by an angiopoietin 1/TIE2/serotonin pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 12331–12336. [Google Scholar] [CrossRef] [PubMed]
- Dewachter, L.; Adnot, S.; Fadel, E.; Humbert, M.; Maitre, B.; Barlier-Mur, A.M.; Simonneau, G.; Hamon, M.; Naeije, R.; Eddahibi, S. Angiopoietin/Tie2 pathway influences smooth muscle hyperplasia in idiopathic pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2006, 174, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Kugathasan, L.; Ray, J.B.; Deng, Y.; Rezaei, E.; Dumont, D.J.; Stewart, D.J. The angiopietin-1-Tie2 pathway prevents rather than promotes pulmonary arterial hypertension in transgenic mice. J. Exp. Med. 2009, 206, 2221–2234. [Google Scholar] [CrossRef] [PubMed]
- Kumpers, P.; Nickel, N.; Lukasz, A.; Golpon, H.; Westerkamp, V.; Olsson, K.M.; Jonigk, D.; Maegel, L.; Bockmeyer, C.L.; David, S.; et al. Circulating angiopoietins in idiopathic pulmonary arterial hypertension. Eur. Heart J. 2010, 31, 2291–2300. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kim, H.S. Serial changes of serum endostatin and angiopoietin-1 levels in preterm infants with severe bronchopulmonary dysplasia and subsequent pulmonary artery hypertension. Neonatology 2014, 106, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, K.; Sapadin, A.; Shoji, T.; Fleischmajer, R.; Lebwohl, M. Altered expression of angiopoietins and Tie2 endothelium receptor in psoriasis. J. Investig. Dermatol. 2001, 116, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Markham, T.; Mullan, R.; Goldesn-Mason, L.; Rogers, S.; Bresnihan, B.; Fitzgerald, O.; Fearon, U.; Veale, D.J. Resolution of endothelial activation and down-regulation of Tie2 receptor in psoriatic skin after infliximab therapy. J. Am. Acad. Dermatol. 2006, 54, 1003–1012. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
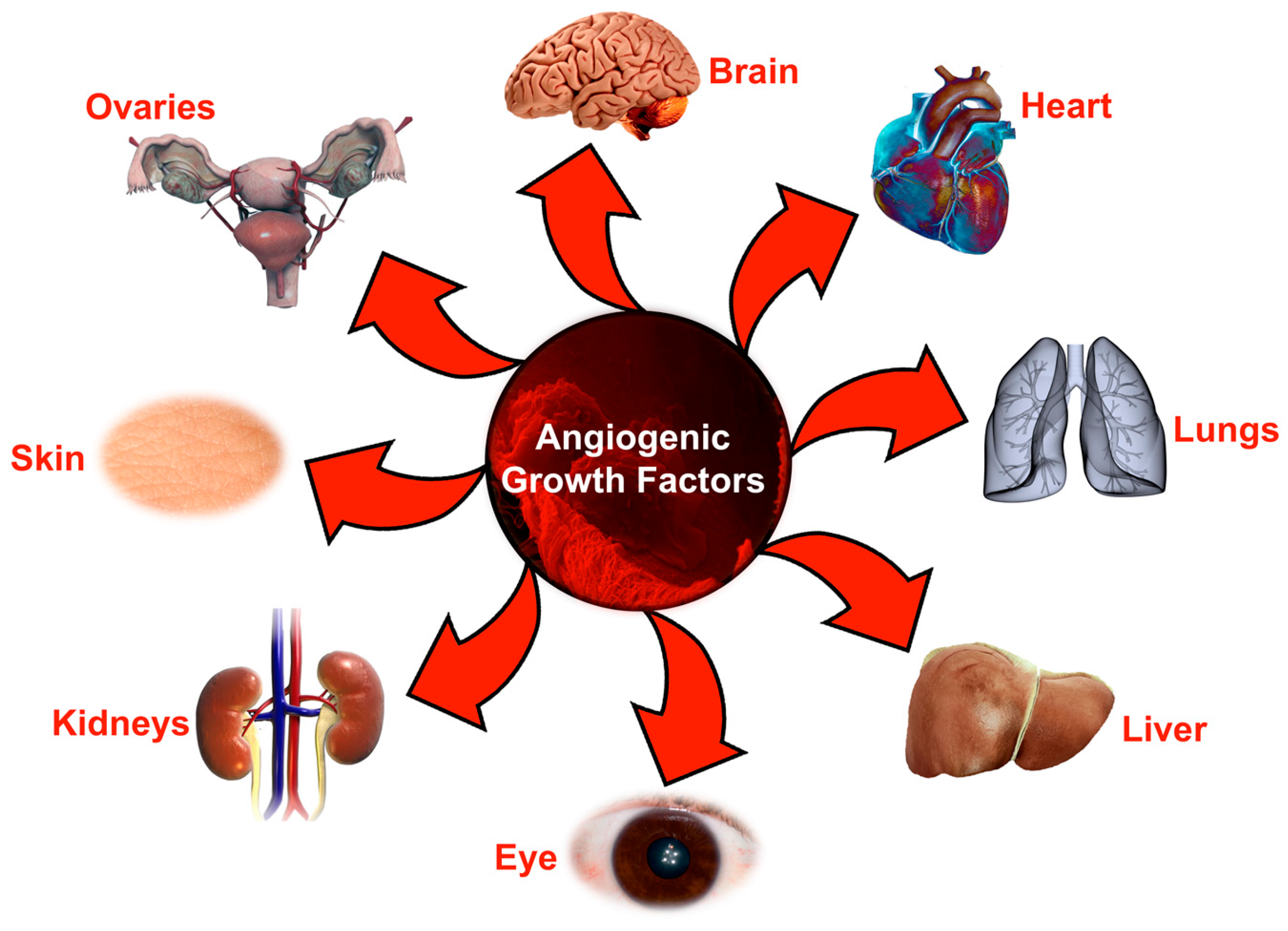{kind=link}
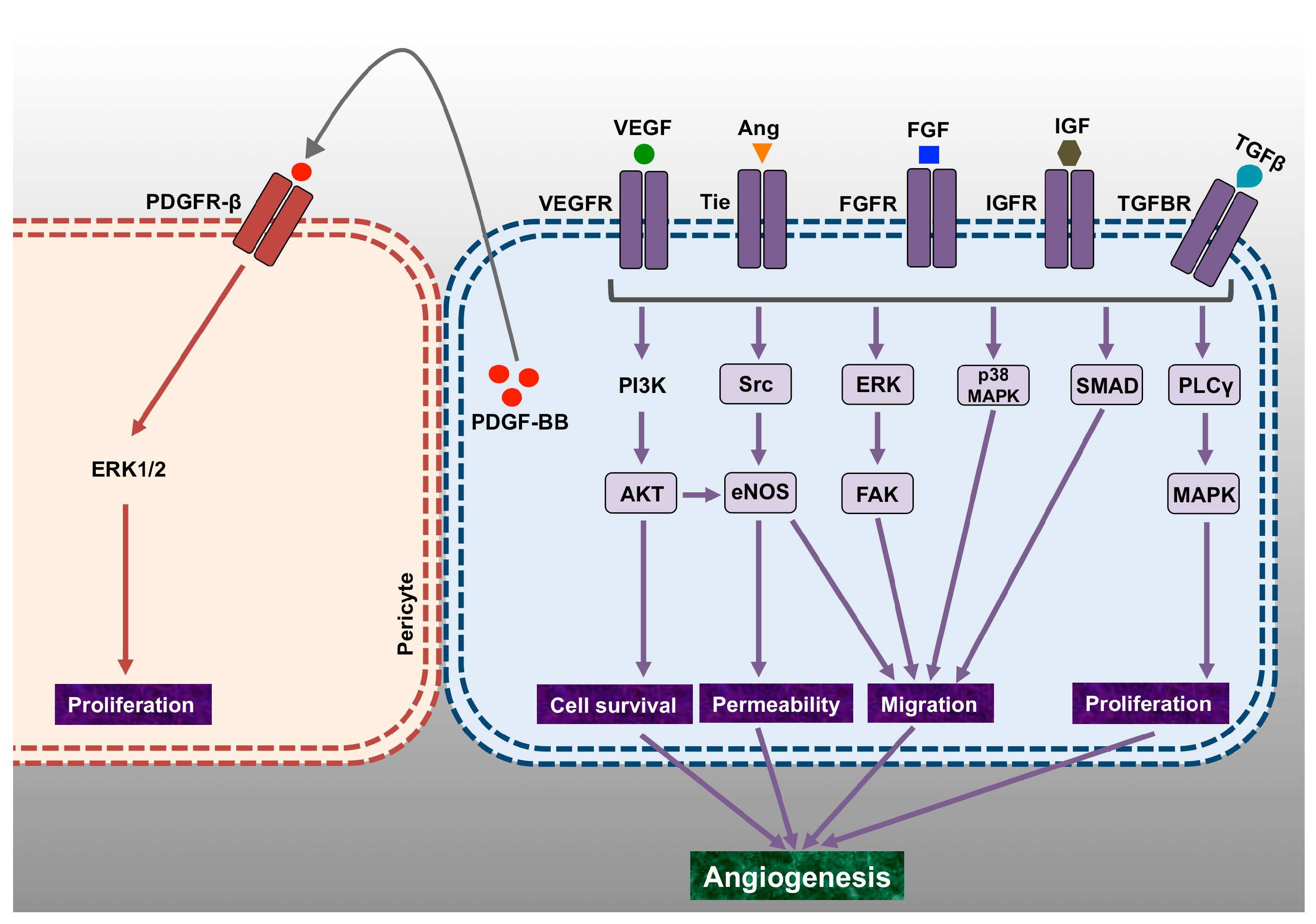{kind=link}
| Author and Year | Target Studied | Study Details | Main Findings and Conclusions |
|---|---|---|---|
| Urakawa, et al. (2006) [205] | FGF23 | In vitro: HEK293 cells, peak rapid cells (derived from HEK293 cells), CHO cells, and L6 myogenic cells | - Klotho was crucial for endogenous FGF23 function and klotho by itself was unable to mediate intracellular signaling - Membrane Klotho in concert with the FGF23 receptor increased the FGF23 receptor specificity and mediated the activity of Klotho-dependent FGF23 - The combined action of Klotho and FGFR1(IIIc) constituted the FGF23 receptor |
| In vivo: Klotho-deficient (kl/kl) mice, Klotho-insufficient (kl/+) mice, Fgf23(+/−) and Fgf23(−/−) and Fgf23(+/−)(kl+/−) mice | |||
| Fliser, et al. (2007) [206] | FGF23 | Human studies: 227 white patients who were between 18 and 65 years of age and had non-diabetic CKD and various degrees of renal impairment were recruited. The primary cause of kidney disease was glomerulonephritis in 97 (biopsy-confirmed in 90) patients, adult polycystic kidney disease in 37 patients, interstitial nephritis in 24 patients, other types of kidney disease in 43 patients, and unknown in 26 patients | - In the baseline cohort, a substantial inverse correlation was noted between glomerular filtration rate and levels of both c-terminal and intact FGF23 - About 65 patients recorded a doubling of serum creatinine and/or fatal kidney failure and had a considerably lesser glomerular filtration rate at baseline. Contrastingly, they had higher levels of intact parathormone, serum phosphate and c-terminal and intact FGF23 - Both c-terminal and intact FGF23 independently projected the advancement of CKD |
| Isakova et al. (2011) [207] | FGF23 | Human studies: 3879 participants with CKD stages 2 through 4 who were enrolled in the Chronic Renal Insufficiency Cohort between June 2003 and September 2008 | - Greater concentrations of FGF23 were independently linked to a higher risk of death - Increased FGF23 was an independent risk factor for end-stage renal disease in patients with somewhat well-preserved kidney function and for mortality within the range of CKD |
| Gutiérrez, et al. (2008) [208] | FGF23 | Human studies: FGF23 levels were assessed in 10,044 patients who were starting hemodialysis treatment Mortality was analyzed in a nested case-control sample of 200 subjects who died and 200 who survived during the first year of hemodialysis treatment | - Elevated FGF23 levels were related to an increased risk of mortality when studied either on a continuous scale or in quartiles - Elevated FGF23 levels independently correlated with mortality among patients who were commencing hemodialysis treatment |
| Isakova et al. (2011) [207] | FGF23 | Human studies: 67 adults undergoing peritoneal dialysis for treatment of ESRD were studied | - FGF23 levels were associated with serum phosphate, residual kidney function, phosphate clearance and dialysis vintage - Higher FGF23 was linked with loss of residual renal function and larger dialysis vintage independent of population distribution, laboratory values, peritoneal dialysis method and suitability, and treatment with vitamin D mimics and phosphate binding agents - FGF23 could be a steadier indicator of phosphate uptake in ESRD than PTH or serum phosphate |
| Isakova et al. (2011) [207] | FGF23 | Human studies: FGF23 was measured in baseline samples from 3879 patients in the Chronic Renal Insufficiency Cohort study, which is a distinct group of patients with CKD stage 2–4 | - Mean serum phosphate and median PTH quantities were in the usual range, but median FGF23 was distinctly amplified than in healthy subjects and boosted considerably with diminishing eGFR - Increased FGF23 is a common manifestation of CKD that shows up earlier than elevated phosphate or PTH |
| Pavik et al. (2013) [209] | FGF23 | Human studies: 87 patients at various stages of CKD unaffected by polycystic kidney disease nor having undergone kidney transplantation, were registered | - Although soluble klotho and 1,25-dihydroxy vitamin D(3) levels decreased and FGF23 levels increased at early CKD stages, PTH levels were elevated only at more progressive stages of CKD |
| Gutiérrez, et al. (2005) [208] | FGF23 | Human studies: 80 patients from across the spectrum of CKD were enrolled in the study | - There was a negative correlation between FGF23 and PTH with eGFR, while calcitriol levels were linearly correlated with eGFR - Elevated Fe(PO4) levels correlated with reduced eGFR, and both increased FGF23 and PTH were independently linked to increased Fe(PO4) - Although higher FGF23 and lower 25(OH)D3 levels were independent predictors of reduced calcitriol, the effects of calcitriol levels on kidney function and hyperphosphatemia were totally abolished when corrected for FGF23 - FGF23 levels rose early in CKD prior to the development of serum mineral anomalies and were independently associated with serum phosphate, Fe(PO4), and calcitriol insufficiency |
| Floege et al. (1995) [210] | FGF2 | In vivo: PHN was induced in male Sprague Dawley rats | - After treatment with FGF2, podocytes of PHN rats exhibited substantial escalations in mitoses, pseudocyst development, foot process retraction, focal detachment from the glomerular basement membrane, and desmin appearance - FGF2-injected PHN rats had increased glomerulosclerosis in comparison to control animals. Also, FGF2 provoked proteinuria and podocyte injury in rats infused with 10% of the routine PHN-serum dose - FGF2 augmented podocyte damage, resulting in amplified glomerular protein permeability and faster glomerulosclerosis |
| Guan et al. (2014) [211] | Klotho and FGF2 | HK-2 cells, a proximal tubular cell line, and the normal rat kidney blast cell line NRK-49 F HK-2 cells, a proximal tubular cell line, and the normal rat kidney F | - In vitro, FGF2 produced tubulo-epithelial plasticity and reduced klotho expression. Recombinant klotho protein could constrain FGF2 activity - The FGF2 effects were mediated via ERK1/2 that were repressed by klotho. Klotho also inhibited FGF2-mediated fibroblast proliferation - Increased FGF2 and reduced klotho was associated with UUO-induced renal fibrosis in WT mice. FGF2−/− mice mostly preserved klotho expression and exhibited only slight renal fibrosis after UUO injury - Intake of klotho protein in UUO mice notably abridged renal fibrosis, accompanied with a striking reduction in FGF2 production and activity |
| In vitro: HK-2 cells, a proximal tubular cell line, and the normal rat kidney fibroblast cell line NRK-49 F | |||
| In vivo: Male C57BL/6 mice, WT mice, and FGF2-knockout (FGF2−/−) mice. UUO was performed to induce renal fibrosis | |||
| Rossaint et al. (2016) [212] | FGF23 | In vitro: Hematopoietic stem cells were isolated from WT mice | - Although leukocyte recruitment into inflamed areas and host defense is deterred by CKD, FGF23 nullification during CKD in mice reinstated leukocyte recruitment and host defense - FGF23 inhibited chemokine-activated leukocyte capture on the endothelium, and reduction in FGFR2 on PMNs salvaged host defense in these mice - In vitro, FGF23 inhibited PMN adhesion, arrest under flow, and transendothelial movement. Additionally, FGF23 binding to FGFR2 counteracted selectin- and chemokine-stimulated β2 integrin activation on PMNs via activation of PKA and impeding the activation of the small GTPase Rap1 |
| In vivo: Chronic kidney failure in mice was induced by 5/6-nephrectomy in male C57BL/6 mice E. coli-induced pneumonia was established | |||
| Rossini et al. (2005) [213] | FGF1 | Human studies: Formalin-fixed, paraffin-embedded kidney tissues from normal control kidneys, PLN, NPLN, AIN, and from transplant nephrectomies with acute rejection and CAN were included in this study | - FGF1 was detected in mesangial cells, glomerular endothelial, visceral, and parietal epithelial cells in normal kidney tissues. FGFR1 staining displayed a comparable pattern but was also detected in tubular epithelium, arterial endothelium, and smooth muscle - FGF1 expression was augmented over normal in glomerular parenchymal cells in podocytes and parietal epithelial cells only in CAN. Intruding glomerular and interstitial inflammatory cells in affected glomeruli also showed FGF1 and FGFR1 expression - Increased FGFR1 but not FGF1 was observed in tubular cells in diseased kidneys vs. normal kidneys |
| Silswal et al. (2014) [214] | FGF23 | In vitro: Primary mouse endothelial cells were isolated from aorta by enzymatic digestion | - All four subtypes of FGF receptors were found in male mouse aortae - Exogenous FGF23 neither stimulated contraction of aortic rings nor relaxed the rings precontracted with prostaglandin F2α - Pretreatment with FGF23 led to a ~36% prevention of endothelium-relied relaxation stimulated by acetylcholine (ACh) in precontracted aortic rings. This was inhibited by the FGFR antagonist - Col4a3−/− CKD mice exhibited exceedingly raised serum FGF23 levels and had compromised endothelium-dependent relaxation. They also showed abridged nitrate production as compared to WT - Exogenous FGF23 caused increased superoxide levels in endothelial cells and aortic rings |
| In vivo: Male C57BL/6J mice were used to study the severe effects of FGF23. Male Col4a3−/− mice (background 129 Sv/J), a model of human autosomal-recessive Alport syndrome and litter-matched WT mice were also used in this study | |||
| Hindricks et al. (2014) [215] | FGF21 | Human studies: Study cohort 1: Out of all 532 patients, those on PPARα- and PPARγ-agonists were excluded, and 499 patients stayed in the study that were categorized into CKD stages 1–5 according to the National Kidney Foundation—KDOQI guidelines Study cohort 2: 32 patients undergoing elective partial or total unilateral nephrectomy (model for acute kidney dysfunction) were enrolled | - In study cohort 1, circulating FGF21 was considerably dissimilar between CKD stages with maximum values detected in stage 5 when corrected for age, gender and body mass index - eGFR was a robust independent and negative predictor of FGF21 - In study cohort 2, FGF21 augmented appreciably post-surgery when matched to presurgical values - Moreover, relative changes in FGF21 levels were independently and positively associated with comparative changes in creatinine levels - Overall, FGF21 was elevated in both CKD and acute kidney disease |
| Pathological Condition | Author and Year | Angiopoietin (Ang) Isoform | Main Findings and Conclusions |
|---|---|---|---|
| Vascular calcification | Chang et al. (2014) [295] | Ang-2 | - The Ang-2 serum levels correlated independently with the severity of arterial stiffness in 416 CKD patients when measured by pulse wave velocity - Plasma levels of Ang-2 also augmented in mice subjected to 5/6 subtotal nephrectomy or UUO. Although Ang-2 was distinctly elevated in tubular epithelial cells of fibrotic kidneys, it was decreased in other tissues like aorta and lung, post 5/6 subtotal nephrectomy - Collagen and profibrotic genes in aortic vascular smooth muscle cells were up-regulated in mice with 5/6 subtotal nephrectomy and in mice generating human Ang-2 - Ang-2 induced expression of endothelial cytokines and adhesion molecules for monocytes, elevated aortic Ly6C (low) macrophages, and stimulated the expression of the profibrotic TGFβ1 in aortic endothelial cells and Ly6C (low) macrophages - Ang-2 blockade with recombinant protein L1-10 decreased the expression of monocyte chemokines, profibrotic cytokines, and collagen in the aorta of mice after 5/6 subtotal nephrectomy. Thus Ang-2 could be the missing link between kidney fibrosis and arterial rigidity |
| Diabetic retinopathy | (1) Joussen et al. (2002) [296] | Ang-1 | - Intravitreal administration of Ang-1 to newly diabetic rats, stabilized retinal VEGF and intercellular adhesion molecule-1 mRNA and protein levels, resulting in reduced leukocyte adhesion, endothelial cell damage, and blood-retinal barrier collapse - Similarly, Adenovirus-Ang-1 when administered systemically to mice with established diabetes, repressed leukocyte adhesion and endothelial cell damage and blood-retinal barrier collapse - These alterations were accompanied by a decline in recognized facilitators of VEGF bioactivity and leukocyte adhesion, namely, retinal eNOS, nitric oxide, Akt (protein kinase B), and MAP kinase - Overall, these results reveal novel vascular and anti-inflammatory bioactivities for Ang-1 and identify its role in directly guarding the retinal vasculature in diabetes |
| (2) Hammes et al. (2004) [297] | Ang-2 | - The expression of Ang-2 and -1 in relation to the evolution of pericyte loss in diabetic rat retinae was studied, using quantitative retinal morphometry, and in retinae from mice with heterozygous Ang deficiency (Ang-2 LacZ knock-in mice). Recombinant Ang-2 was injected into eyes of nondiabetic rats, and pericyte numbers were quantitated in retinal capillaries - Ang-1 was present in the normal maturing retina and was upregulated 2.5-fold in diabetic retinae over 3 months of diabetes - In contrast, Ang-2 was consistently upregulated more than 30-fold in the retinae of diabetic rats, preceding the onset of pericyte loss - Heterozygous Ang-2 deficiency completely prevented diabetes-induced pericyte loss and reduced the number of acellular capillary segments - Injection of Ang-2 into the eyes of normal rats induced a dose-dependent pericyte loss. These data show that upregulation of Ang-2 plays a critical role in the loss of pericytes in the diabetic retina | |
| (3) Pfister (2010) [298] | Ang-2 | - The effects of retinal overexpression of human Ang-2 (mOpsinhAng2 mouse) on vascular morphology in non-diabetic and streptozotocin-induced diabetic animals were investigated. Pericyte (PC) coverage and acellular capillary (AC) formation were quantitated in retinal digest preparations after 3 and 6 months of diabetes duration - The degree of retinopathy in non-diabetic mOpsinhAng2 mice at 3 months (−21% PC, +49% AC) was comparable to age-matched diabetic wild type mice - Diabetic mOpsinhAng2 mice exhibited significantly worse vascular pathology than wild type counterparts at 6 months. Quantitative PCR revealed that human Ang-2 mRNA was highly overexpressed in retinas of transgenic mice - Overexpression of Ang-2 in the retina enhances vascular pathology, indicating that Ang-2 plays an essential role in diabetic vasoregression via destabilization of pericytes | |
| (4) Rangasamy (2011) [299] | Ang-2 | - Ang-2 mRNA and protein increased in the retinal tissues after 8 weeks of diabetes and in high-glucose-treated cells. Intravitreal injection of Ang-2 in rats produced a significant increase in retinal vascular permeability - Ang-2 increased human retinal endothelial cells monolayer permeability that was associated with a decrease in VE-cadherin and a change in monolayer morphology - High glucose and Ang-2 produced a significant increase in VE-cadherin phosphorylation - Increased Ang-2 alters VE-cadherin function, leading to increased vascular permeability. Thus, Ang-2 may play an important role in increased vasopermeability in diabetic retinopathy | |
| (5) Park (2014) [300] | Ang-2 | - Pericyte loss occurred with an Ang-2 increase in the diabetic mouse retina and that the source of Ang-2 could be the endothelial cell. Ang-2 induced pericyte apoptosis via the p53 pathway under high glucose, whereas Ang-2 alone did not induce apoptosis - Integrin, not Tie2 receptor, was involved for Ang-2-induced pericyte apoptosis under high glucose as an Ang-2 receptor. High glucose changed the integrin expression pattern, which increased integrin α3 and β1 in the pericyte - Furthermore, Ang-2-induced pericyte apoptosis in vitro was effectively attenuated via p53 suppression by blocking integrin α3 and β1 - Although intravitreal injection of Ang-2 induced pericyte loss in C57BL/6J mice retina in vivo, intravitreal injection of anti-integrin α3 and β1 antibodies attenuated Ang-2-induced pericyte loss. In conclusion, Ang-2 induced pericyte apoptosis under high glucose via α3β1 integrin | |
| (6) Cahoon (2015) [301] | Ang-1 | - In early diabetic retinopathy, adeno-associated virus serotype 2 encoding a more stable, soluble, and potent form of Ang-1 (AAV2.COMP-Ang-1) restored leukocyte-endothelial interaction, retinal oxygenation, vascular density, vascular marker expression, vessel permeability, retinal thickness, inner retinal cellularity, and retinal neurophysiological response to levels comparable with nondiabetic controls - In late diabetic retinopathy, AAV2.COMP-Ang-1 enhanced the therapeutic benefit of intravitreally delivered endothelial colony-forming cells by promoting their integration into the vasculature and thereby stemming further visual decline - AAV2.COMP-Ang-1 single-dose gene therapy can prevent neurovascular pathology, support vascular regeneration, and stabilize vision | |
| (7) Yun (2016) [302] | Ang-2 | - Vascular leakage occurred with astrocyte loss in early diabetic mice (streptozotocin-induced diabetic retinopathy) retina as Ang-2 increased. The astrocyte loss and vascular leakage were inhibited by intravitreal injection of Ang-2-neutralizing antibody - In vitro, Ang-2 aggravated high glucose-induced astrocyte apoptosis via GSK-3β activation. Ang-2 directly bound to αvβ5 integrin, which was abundant in astrocyte, and the blockade of αvβ5 integrin, in vitro, effectively attenuated Ang-2-induced astrocyte apoptosis - In vivo, intravitreal injection of anti-αvβ5-integrin antibody inhibited astrocyte loss in early diabetic retinopathy. Taken together, Ang-2 induced astrocyte apoptosis under high glucose via αvβ5-integrin/GSK-3β/β-catenin pathway, and hence Ang-2/integrin signaling could be a potential therapeutic target to prevent the vascular leakage by astrocyte loss in early diabetic retinopathy | |
| Diabetic nephropathy | (1) Lee (2007) [303] | Ang-1 | - COMP-Ang-1 reduced albuminuria and decreased mesangial expansion, thickening of the glomerular basement membrane and podocyte foot process broadening and effacement - COMP-Ang-1 suppressed both renal expression of intercellular adhesion molecule-1 and monocyte chemoattractant protein-1 and monocyte/macrophage infiltration in diabetic db/db mice - COMP-Ang-1 also reduced renal tissue levels of TGFβ1, alpha-smooth muscle actin, fibronectin, as well as Smad 2/3 expression, but increased Smad 7 - In HUVECs grown in high glucose concentrations of glucose, recombinant COMP-Ang-1 protein decreased nuclear factor-kappaB (NF-kappaB) expression. COMP-Ang-1-mediated inhibition of increased NF-kappaB-DNA binding in nuclear extracts from HUVECs grown in high glucose was significantly blocked by soluble Tie2 receptor-Fc - In addition, COMP-Ang-1 significantly decreased fasting blood glucose level, epididymal fat weight to body weight ratio, and epididymal adipocyte size in diabetic db/db mice. After intraperitoneal glucose challenge, COMP-Ang-1 significantly lowered plasma glucose levels and thus, in conclusion, delayed the fibrotic changes in the kidney of diabetic db/db mice through its anti-inflammatory or metabolic effects |
| (2) Davis (2007) [304] | Ang-2 | - When the transgene was induced in mice with inducible podocyte-specific Ang-2 overexpression for up to 10 weeks, mice had significant increases in both albuminuria and glomerular endothelial apoptosis, with significant decreases of both vascular endothelial growth factor-A and nephrin proteins, critical for maintenance of glomerular endothelia and filtration barrier functional integrity, respectively - There was, however, no significant change of systemic BP, creatinine clearance, or markers of renal fibrosis, and podocytes appeared structurally intact. In kidneys of young animals in which Ang-2 had been upregulated during organogenesis, increased apoptosis occurred in just-formed glomeruli - In vitro, short-term exposure of isolated wild-type murine glomeruli to exogenous Ang-2 led to decreased levels of vascular endothelial growth factor-A protein. These novel results provide insight into molecular mechanisms underlying proteinuric disorders involving Ang-2 | |
| (3) Dessapt-Baradez (2014) [305] | Ang-1 | - Decreased Ang-1, VEGF-A upregulation, decreased soluble VEGFR-1, and increased VEGFR-2 phosphorylation (pVEGFR-2) was observed in streptozotocin-induced type 1 diabetic mice, accompanied by marked albuminuria, nephromegaly, hyperfiltration, glomerular ultrastructural alterations, and aberrant angiogenesis - Podocyte-specific inducible repletion of Ang-1 in diabetic mice caused a 70% reduction of albuminuria and prevented diabetes-induced glomerular endothelial cell proliferation; hyperfiltration and renal morphology were unchanged - Ang-1 repletion in diabetic mice increased Tie2 phosphorylation, elevated soluble VEGFR-1, and was paralleled by a decrease in pVEGFR-2 and increased endothelial nitric oxide synthase Serine (1177) phosphorylation - Diabetes-induced nephrin phosphorylation was also reduced in mice with Ang-1 repletion. Ang-1 therapy could be a renoprotective tool in diabetic nephropathy | |
| (4) Luo (2014) [306] | Ang-2 | - Alprostadil treatment caused a significant decrease in the renal damage parameters in streptozotocin-induced diabetic nephropathy - Both Ang-2 and IL-18 were significantly increased in these mice and in glomerular endothelial cells cultured in high glucose; however, their expression was greatly reduced by alprostadil treatment - Ang-2 could also increase IL-18 expression in cultured endothelial cells under high glucose, and this response was partially blocked by Ang-2 siRNA - Ang-2 and IL-18 may be associated with the development and progression of diabetic nephropathy in mice | |
| (5) Khairoun (2015) [307] | Ang-1 + Ang-2 | - An increase in the capillary tortuosity index in streptozotocin-induced + atherogenic diet (DM + ATH) pigs was reported as compared to the control groups. Kidney biopsies showed marked glomerular lesions consisting of mesangial expansion and podocyte lesions - A disturbed Ang-2/Ang-1 balance was observed in the cortex of the kidney, as evidenced by increased expression of Ang-2 in DM + ATH pigs as compared to control pigs - In the setting of diabetes mellitus, atherogenesis leads to the augmentation of mucosal capillary tortuosity, indicative of systemic microvascular damage, while, an imbalance in renal angiopoietins was correlated with the development of diabetic nephropathy | |
| Chronic kidney disease (CKD) | (1) David et al. (2010) [308] | Ang-2 | - 44 untreated non-smokers with varying stages of CKD 1–4 and 19 patients on dialysis (CKD stage 5) were recruited for measuring Ang-2 levels. Ang-2 measurements were also recorded in 15 healthy subjects prior to and 72 h after kidney donation - The median Ang-2 levels gradually augmented within the following groups: healthy controls: 0.77 ng/mL; CKD 1: 0.83 ng/mL; CKD 2: 0.93 ng/mL; CKD 3: 1.13 ng/mL; CKD 4: 1.75 ng/mL; and CKD 5: 4.87 ng/mL - Ang-2 was linked to the extent of CKD as supported by an inverse association with the GFR and positive association with homocysteine and phosphate - Also, Ang-2 positively correlated with the nitric oxide synthase inhibitor; asymmetric dimethylarginine levels and had inverse association with the mean alterations in GFR at 72 h post kidney donation |
| (2) David et al. (2012) [309] | Ang-2 | - In 128 CKD patients (43 CKD Stage 4, 85 CKD Stage 5 (57 hemodialysis, 28 peritoneal dialysis)), Ang-2 levels were considerably greater than in controls - Ang-2 was considerably greater in dialysis than in Stage 4 CKD patients and was associated with indicators of vascular disease (cholesterol, hsCRP, osteoprotegerin (OPG)) - Increased Ang-2 did not correlate with the extent of vascular calcification or with arterial stiffness - Cox-regression analysis concluded Ang-2 as an independent predictor of mortality in CKD patients | |
| (3) Chang et al. (2013) [310] | Ang-2 | - 416 CKD patients were classified into stages 3 to 5 by urine albumin-creatinine ratio as normoalbuminuria (<30 mg/g), microalbuminuria (30–300 mg/g), or macroalbuminuria (>300 mg/g). Ang-2 and VEGF were increased, and soluble Tie2 levels in the plasma were reduced in the subgroups of albuminuria; with Ang-1 levels remaining unchanged - Linear regression analysis revealed a positive association between urine ACR and plasma Ang-2 only and not VEGF or soluble Tie2. Multivariate linear regression studies exhibited that plasma Ang-2 levels independently correlated with ACR and extremely sensitive C-reactive protein - In conclusion, plasma Ang-2 was related to albuminuria and markers of systemic microinflammation in CKD patients | |
| (4) Tsai et al. (2014) [311] | Ang-2 | - In 621 patients with stages 3–5 CKD, 224 patients (36.1%) proceeded to begin dialysis and 165 (26.6%) reached doubling creatinine. 85 subjects (13.9%) had a quick decay in renal function. Ang-2 quartile was divided at 1494.1, 1948.8, and 2593.1 pg/mL - The linear mixed-effects model demonstrated a further swift decline in estimated glomerular filtration rate over time in patients with quartile 3 or more of Ang-2 than those with the lowest quartile of Ang-2 - Ang-2 could be an independent predictor of severe renal outcome in CKD | |
| (5) Bi et al. (2016) [312] | Ang-1 | - Ang-1 appreciably reduced the angiotensin II-stimulated expression of the ER stress response proteins GRP78, GRP94, p-PERK, and CHOP, suggesting that Ang-1-facilitated cellular protection happens after the ER stress response - Tie2 inhibition using inhibitors and siRNA overturned these observations, inferring the prominence of Tie2 activation in preventing ER stress - The shielding effects of Ang-1 were attributed to the activation of ERK1/2 and p38 MAPK, which were significantly reduced when inhibited with specific inhibitors of these pathways (PD98059 and SB203580 respectively), along with moderate increase in the expression of chaperones involved in folding proteins - In conclusion, Ang-1 reduced ER stress-mediated cellular dysfunction and death via the Tie2 receptor/ERK1/2-p38 MAPK pathways in glomerular endothelial cells, which are principally associated with CKD |
| Pathological Condition | Author and Year | Angiopoietin (Ang) Isoform | Main Findings and Conclusions |
|---|---|---|---|
| IBD | (1) Koutroubakis et al. (2006) [313] | Ang-2 | - In 160 IBD patients (79 UC and 81 CD) and in 80 corresponding healthy controls, median serum Ang-2 and Tie2 levels were notably greater in both the UC and CD patients in comparison to the healthy controls - The IBD patients detected early (diagnosis < 2 years) had considerably greater median serum Ang-2 levels but lower median serum Tie2 levels in comparison to patients with late IBD (diagnosis > 2 years) - The CD patients with dynamic disease showed appreciably greater levels of Ang-2 than in non-active disease patients. Interestingly, levels of both Ang-2 and Tie2 in the serum did not associate with laboratory markers such as ESR, CRP, white blood cell number, platelet count and albumin levels |
| (2) Ganta et al. (2010) [314] | Ang-2 | - Numerous main alterations were observed in the development of IBD in Ang-2(−/−) mice. Weight variations and disease activity differences were insignificant in WT and Ang-2(−/−) + DSS treated mice, while leukocyte intrusion, inflammation and blood and lymphatic vessel density was substantially diminished compared to WT + DSS mice - Gut capillary friability and water export appeared considerably earlier in Ang-2(−/−) + DSS mice vs. WT. Also, colon sizes were appreciably condensed in Ang-2(−/−) and gut histopathology was less impaired in Ang-2(−/−) in comparison to WT + DSS treated mice - The reduction in serum protein concentration in WT + DSS was less severe in Ang-2(−/−) + DSS, consequently PLE a characteristic trait of IBD was relieved by Ang-2(−/−) - In conclusion, in DSS colitis, Ang-2 facilitated inflammatory hemangiogenesis, lymphangiogenesis, and neutrophil intrusion to alleviate some clinical characteristics of IBD | |
| (3) Oikonomou et al. (2011) [315] | Ang-1 + Ang-2 | - In 52 patients with UC, 59 with CD, and 55 healthy controls (HC), Ang-1 concentrations were considerably smaller in IBD patients compared to HC and were increased in smokers compared to non-smoker UC patients - IBD patients showed elevated Ang-2 levels compared to HC, whereas CD patients (disease only in colon) had notably lower Ang-2 levels when compared to other disease sites | |
| (4) Algaba et al. (2014) [316] | Ang-1 | - In 37 patients with IBD treated with infliximab (16 with Crohn’s disease and 6 with ulcerative colitis) or adalimumab (15 with Crohn’s disease) and 40 healthy control subjects, Ang-1 levels diminished prior to each treatment dose in patients who achieved retardation of the disease - Elevated baseline VEGF levels anticipated for a dismal response to anti-TNF-alpha therapy, while elevated Ang-1 levels were linked with disease reduction - Serum VEGF and Ang-1 levels decreased post anti-TNF-alpha therapy and thus could be good predictors of response to therapy against IBD | |
| (5) Liu et al. (2015) [317] | Ang-1 + Ang-2 | - Dysplasia and cancer were investigated in rats that received three rounds of 3.5% DSS with intraperitoneal pretreatment of DMH (CRC group). Colitis was investigated in rats that received three rounds of 3.5% DSS and intraperitoneal pretreatment with saline in UC group - CRC and UC groups exhibited the symptoms of serious colitis with diarrhea, rectal bleeding, wasting, and weight loss compared to controls - The mean length of the colon of was suggestively shorter in the CRC and UC group than in control rats. Only the CRC group had multiple tumors in the colorectal area (absent in UC and controls) - CRC and UC groups had strikingly augmented levels of Ang-1, Ang-2, Tie2, and VEGF protein in the colorectum compared to control group - In conclusion, aberrant expression of Ang-1, Ang-2, Tie2, and VEGF in UC-originated colorectal cancer may culminate in chronic colitis and pathologic angiogenesis | |
| PH | (1) Zhao et al. (2003) [318] | Ang-1 | - Ang-1 cDNA or null (pFLAG-CMV-1) vector was transfected into rat pulmonary artery smooth muscle cells. Fisher 344 rats were treated with monocrotaline (MCT) with or without transfer of 5 × 10(5) Ang-1 or null-transfected cells through the right jugular vein - 28 days post gene delivery, plasmid-derived Ang-1 mRNA was steadily and strongly expressed as assessed by RT-PCR in lungs from Ang-1 gene therapy group. Although Tie2 levels were noticeably reduced in rats treated with MCT, this effect was partly nullified by Ang-1 gene therapy - MCT-treated animals demonstrated 77% mortality by 28 days, while in pAng-1-treated animals, mortality was just 14% by 28 days. Moreover, measurement of the right ventricular systolic pressure and the right to left ventricular plus septal weight ratio was decreased in the Ang-1 group, as compared to MCT-treated group - Ang-1 gene transfer prevented the increased endothelial apoptosis and reduced endothelial NO synthase mRNA levels observed in the MCT-treated animals - Thus, cell-based gene therapy of Ang-1 enhanced survival and pulmonary hemodynamics in animal model of PH |
| (2) Sullivan et al. (2003) [319] | Ang-1 | - Constitutive Ang-1 expression (Adeno-Ang-1) in the lung of genetically engineered animals resulted in severe PH. Aberrant proliferation of smooth muscle cells (hyperplasia) causes diffuse medial thickening in small pulmonary vessels in these animals, a manifestation commonly observed in human PH - Ang-1/Tie2 signaling stimulated pulmonary arteriolar endothelial cells to manufacture and secrete serotonin (5-hydroxytryptamine), a strong smooth muscle mitogen. Also, elevated levels of serotonin were detected in human and rodent pulmonary hypertensive lung tissue as well - In conclusion, Ang-1/Tie2/serotonin paracrine pathway mediated pulmonary hypertensive vasculopathy, making them potential therapeutic targets to treat PH | |
| (3) Dewachter (2006) [320] | Ang-1 | - PA-SMCs and P-ECs were obtained and grown from PH patients. Tie2 expression was 4-fold elevated in lungs and P-ECs from these patients compared to controls, accompanied with an equivalent escalation in phosphorylated lung Tie2 - However, Ang-1 and Ang-2 levels in lungs, P-ECs, and PA-SMCs were similar. Cultivation of PA-SMCs with medium collected from P-EC cultures provoked distinct proliferation. This effect was enhanced in P-ECs from PH patients rather than from control population - When P-ECs from either PH patients or control subjects were pre-incubated with Ang-1, they produced an additional increase in PA-SMC proliferation Fluoxetine, an inhibitor of the mitogenic activity of serotonin, decreased the proliferative effect of P-EC medium - Although Ang-1 improved the production of ET-1, serotonin, the amount of tryptophan hydroxylase-1 (the rate-limiting enzyme of serotonin synthesis) mRNA, preproET-1, and ET-1-converting enzyme when added to P-ECs from PH patients, platelet-derived growth factor-BB or epidermal growth factor levels were particularly unaffected - Ang-1/Tie2 signaling is involved in PH and contributes to PA-SMC hyperplasia through enhanced stimulation of endothelium-resulting growth factors synthesized by P-ECs | |
| (4) Kugathasan et al. (2009) [321] | Ang-1 | - Right ventricular systolic pressure was moderately elevated in Tie2-deficient mice [Tie2(+/−)] when compared with WT littermate controls. The pressure was worsened when chronically stimulated with clinically significant PAH that triggered 5-HT or IL-6 - Although excess Ang-1 expression in transgenic mice had no adverse effect on pulmonary hemodynamics, it diminished the reaction to 5-HT. Incubation with 5-HT or IL-6 also reduced lung Ang-1 expression and subsequent Tie2 activation, thus promoting apoptosis in the Tie2(+/−) group lungs - Tie2 knockdown led to enhanced sensitivity to apoptosis after 5-HT treatment in cultured pulmonary artery endothelial cells - Z-VAD (pan-caspase inhibitor) treatment of Tie2-deficient mice, prohibited the pulmonary hypertensive reaction to 5-HT. Together these results decisively ascertain that the Ang-1-Tie2 pathway mediates the protective endothelial survival signaling in PH | |
| (5) Kümpers et al. (2010) [322] | Ang-2 | - Plasma levels of Ang-1, Ang-2, soluble Tie2 (sTie2), and VEGF were increased in IPAH patients compared with controls. Among the angiogenic growth factors, Ang-2, but not Ang-1, sTie2, and VEGF were associated with cardiac index, PVR, and SvO(2) - Greater Ang-2 expression was an independent risk factor for mortality. 3 months after initiation of therapy, changes in Ang-2 levels paralleled with the variations in mean right atrial pressure, PVR and were inversely related to the variations in SvO(2) - Plexiform lesions from IPAH lung tissue showed increased expression of Ang-2 mRNA and protein. These findings suggest that Ang-2 may take part in the pathogenesis of IPAH, and circulating Ang-2 might serve as a potential diagnostic biomarker for determining disease gravity and treatment efficacy in IPAH patients | |
| (6) Kim D and Kim H (2014) [323] | Ang-1 | - Circulating endostatin and Ang-1 in early life were linked to the development of PH in preterm infants with extreme BPD - The PH group regularly undertook more assertive respiratory management than the non-PH group, over 1 month after birth - The PH group demonstrated remarkably greater endostatin level and the ratio of endostatin to Ang-1 on day 7 of life when compared to the non-PH group or no/mild BPD groups - Additionally, the ratio of endostatin to Ang-1 on day 1 was considerably greater in the PH group in comparison to the no/mild BPD group - In conclusion, serum endostatin to Ang-1 ratio may represent compromised angiogenesis, which may play a role in the development of PH | |
| Psoriasis | (1) Kuroda et al. (2001) [324] | Ang-1 + Ang-2 | - Involved psoriasis skin demonstrated increased Ang-1, Ang-2, and Tie2 expression as compared to uninvolved psoriasis skin, healthy skin, and persistent spongiotic dermatitis skin. Extremely vascularized papillary dermis of involved psoriasis skin showed Ang-1 expression in the stromal cells, while Ang-2 was observed in endothelial cells around the multiplying epidermis that richly expressed VEGF - Aberrant expression of VEGF and FGF2 in involved psoriasis skin led to increased Ang-2 and Tie2 expression in dermal microvascular endothelial cell cultures - Overexpression of Ang-1, Ang-2, and Tie2 was related to the development of microvascular spread in psoriasis, suggesting that the Ang-Tie2 pathway may coordinate with VEGF and FGF2 together to foster neoangiogenesis in psoriasis - 5 psoriatic patients treated with PUVA (psoralen and ultraviolet A radiation) and two patients treated with tazarotene showed evident decrease in Ang-2 expression in skin lesions, indicating that modulation of Ang-2 may be crucial in regulating vascular proliferation in anti-psoriatic treatments |
| (2) Markham et al. (2006) [325] | Ang-1 + Ang-2 | - 16 patients with moderate to severe psoriasis and associated psoriatic arthritis (n = 13) were given infliximab infusions at baseline and at 2 and 6 weeks. The baseline levels of Ang-1/2, VEGF, Tie2, and TNF-alpha mRNA and protein were greater in pre-involved skin in comparison to uninvolved skin - Infliximab significantly reduced Ang-2, VEGF and Tie2 protein expression along with a reduction in Ang-1 and Tie2 mRNA expression. This was accompanied by a substantial decrease in the inflammatory infiltrate scores and CD31 expression, signifying inactivation of endothelial cell machinery - 12 weeks after treatment, there was a 93% mean decrease in the Psoriasis Area and Severity Index, and a noteworthy reduction in Disease Activity Score 28 and mean Health Assessment Questionnaire scores. These results decisively indicate that TNF-alpha is an important watchdog of the Ang/Tie2 pathway |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matkar, P.N.; Ariyagunarajah, R.; Leong-Poi, H.; Singh, K.K. Friends Turned Foes: Angiogenic Growth Factors beyond Angiogenesis. Biomolecules 2017, 7, 74. https://doi.org/10.3390/biom7040074
Matkar PN, Ariyagunarajah R, Leong-Poi H, Singh KK. Friends Turned Foes: Angiogenic Growth Factors beyond Angiogenesis. Biomolecules. 2017; 7(4):74. https://doi.org/10.3390/biom7040074
Chicago/Turabian StyleMatkar, Pratiek N., Ramya Ariyagunarajah, Howard Leong-Poi, and Krishna K. Singh. 2017. "Friends Turned Foes: Angiogenic Growth Factors beyond Angiogenesis" Biomolecules 7, no. 4: 74. https://doi.org/10.3390/biom7040074
APA StyleMatkar, P. N., Ariyagunarajah, R., Leong-Poi, H., & Singh, K. K. (2017). Friends Turned Foes: Angiogenic Growth Factors beyond Angiogenesis. Biomolecules, 7(4), 74. https://doi.org/10.3390/biom7040074