
Alcohol and Cancer: Mechanisms and Therapies

{kind=link}
{kind=link}
Abstract
1. Introduction
2. Epidemiologic Evidence for Alcohol-Associated Cancers
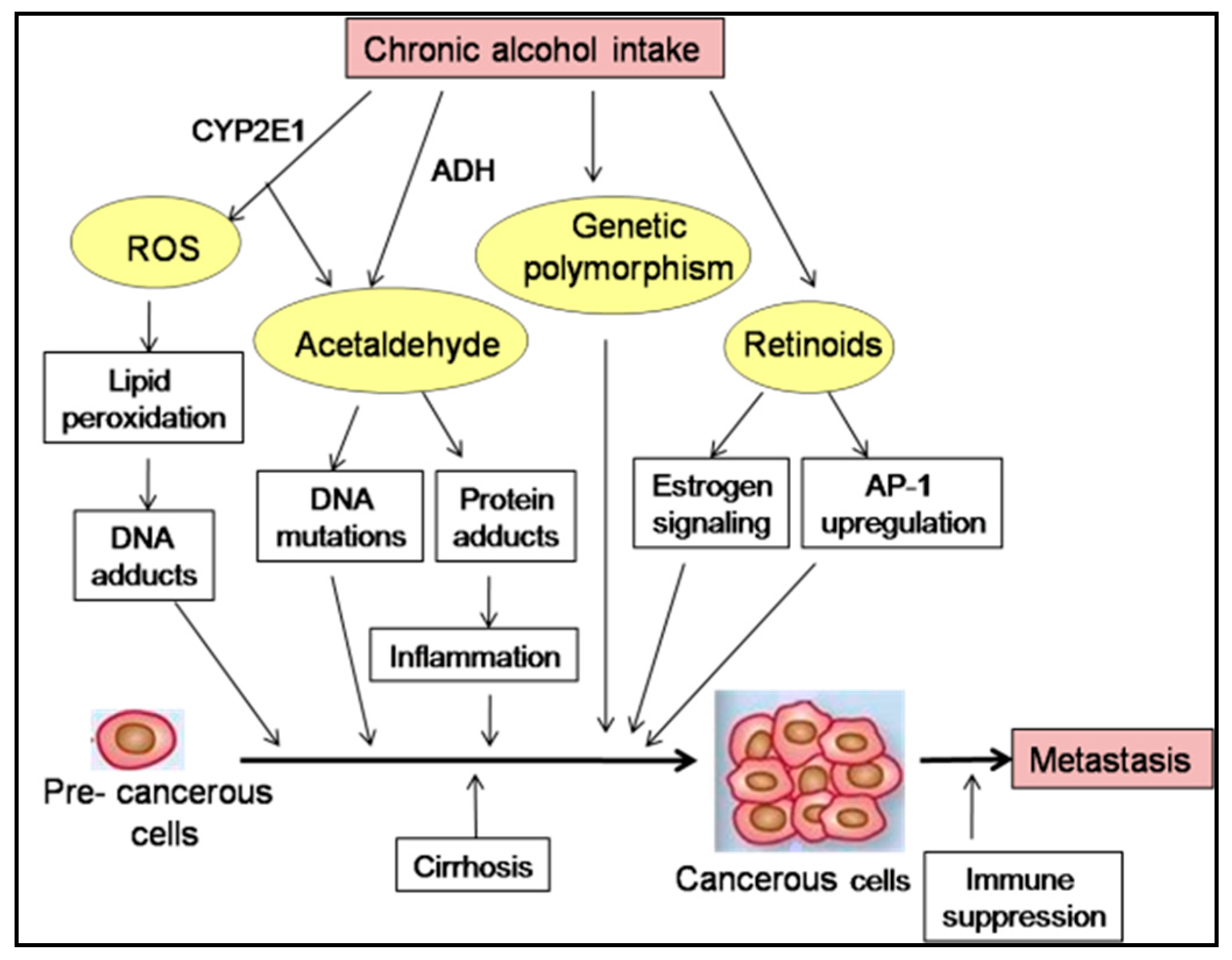
3. Alcohol: Tumor Initiator or Promoter?
4. Alcohol and Different Types of Cancer
4.1. Liver Cancer
4.2. Breast Cancer
4.3. Esophageal Cancer
4.4. Pancreatic Cancer
5. Potential Molecular Mechanisms
5.1. Genetic Polymorphism
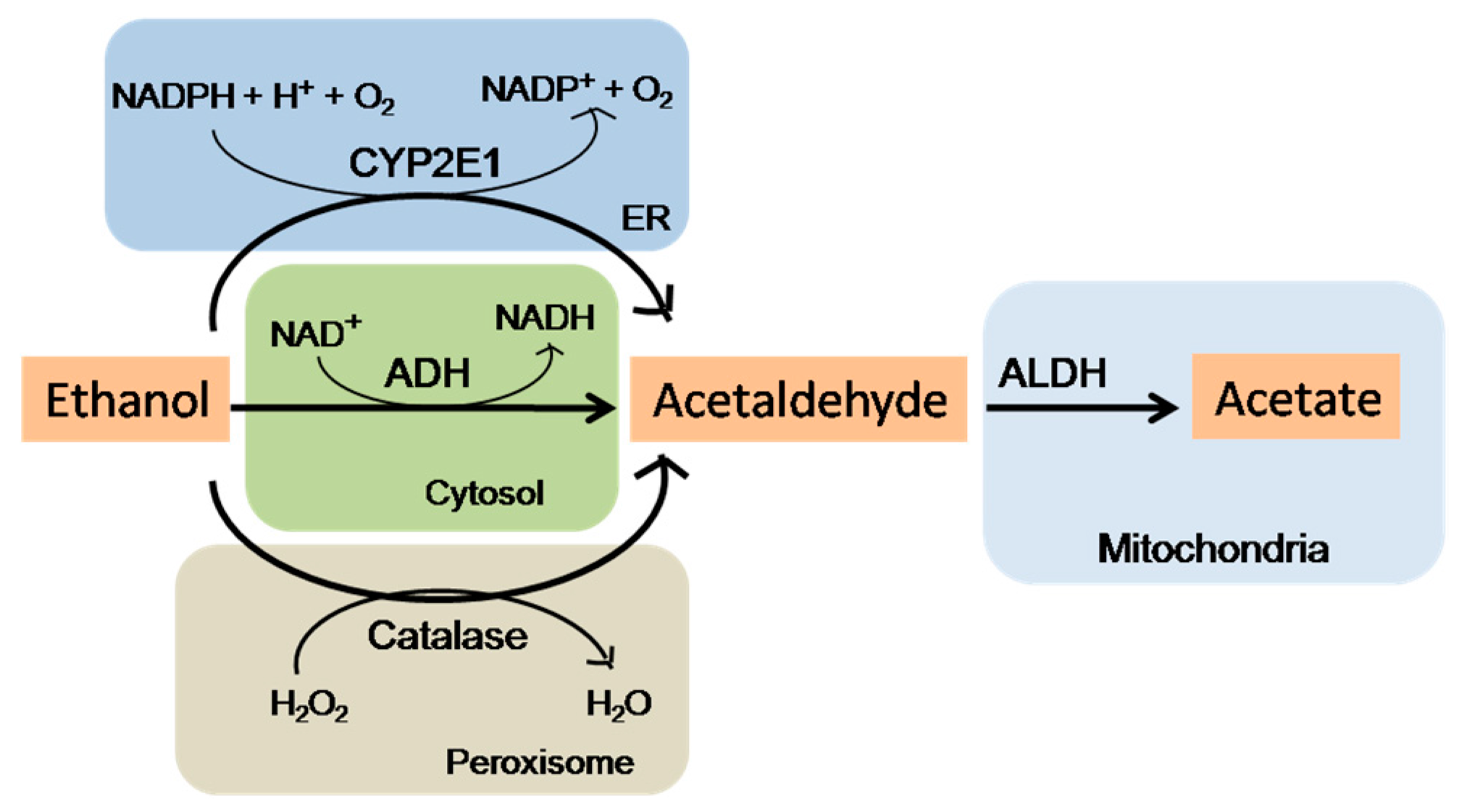
5.2. Oxidative Stress
5.3. Retinoic Acid Metabolism
6. Animal Models
6.1. Chemically Induced Models
6.2. Genetically Modified Models (GMM)
6.3. Xenograft Models
7. Alcohol-Induced Immune Modulation in Cancer
7.1. Innate Immune Surveillance
7.2. Adaptive Immune Surveillance
8. Prospective Strategies for Tumor Immunotherapy in Alcoholics
9. Conclusions and Future Approaches
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Centers for Disease Control and Prevention. Alcohol-Related Disease Impact (ARDI) Application, 2013. Available online: www.cdc.gov/ARDI (accessed on 10 August 2017).
- World Health Organization. Global Status Report on Alcohol and Health, 2014; World Health Organization: Luxembourg, 2014; ISBN 978-924-156-475-5. Available online: http://www.who.int/substance_abuse/publications/global_alcohol_report/msb_gsr_2014_1.pdf?ua=1 (accessed on 22 June 2017).
- Center for Behavioral Health Statistics and Quality. 2014 National Survey on Drug Use and Health: Detailed Tables; Substance Abuse and Mental Health Services Administration: Rockville, MD, USA, 2015. Available online: https://www.samhsa.gov/data/sites/default/files/NSDUH-DetTabs2014/NSDUH-DetTabs2014.pdf (accessed on 22 June 2017).
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Alcohol consumption and ethyl carbamate. In IARC Monographs on the Evaluation of Carcinogenic Risks in Humans; International Agency for Research on Cancer: Lyon, France, 2010; Volume 96, pp. 1281–1383. [Google Scholar]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Personal habits and indoor combustions. In IARC Monographs on the Evaluation of Carcinogenic Risks in Humans; International Agency for Research on Cancer: Lyon, France, 2012; Volume 100E, pp. 373–472. [Google Scholar]
- Testino, G. The burden of cancer attributable to alcohol consumption. Maedica 2011, 6, 313–320. [Google Scholar] [PubMed]
- Meadows, G.G.; Zhang, H. Effects of alcohol on tumor growth, metastasis, immune response, and host survival. Alcohol Res. 2015, 37, 311–322. [Google Scholar] [PubMed]
- Seitz, H.K.; Stickel, F.; Homann, N. Pathogenetic mechanisms of upper aerodigestive tract cancer in alcoholics. Int. J. Cancer 2004, 108, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Stickel, F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat. Rev. Cancer 2007, 7, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Gyamfi, M.A.; Wan, Y.J. Pathogenesis of alcoholic liver disease: The role of nuclear receptors. Exp. Biol. Med. 2010, 235, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Li, X.; Ma, Q.; Xu, Q.; Duan, W.; Lei, J.; Zhang, L.; Wu, Z. Chronic alcohol exposure exacerbates inflammation and triggers pancreatic acinar-to-ductal metaplasia through pi3k/akt/ikk. Int. J. Mol. Med. 2015, 35, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Druesne-Pecollo, N.; Tehard, B.; Mallet, Y.; Gerber, M.; Norat, T.; Hercberg, S.; Latino-Martel, P. Alcohol and genetic polymorphisms: Effect on risk of alcohol-related cancer. Lancet Oncol. 2009, 10, 173–180. [Google Scholar] [CrossRef]
- Boffetta, P.; Hashibe, M. Alcohol and cancer. Lancet Oncol. 2006, 7, 149–156. [Google Scholar] [CrossRef]
- Boffetta, P.; Hashibe, M.; La Vecchia, C.; Zatonski, W.; Rehm, J. The burden of cancer attributable to alcohol drinking. Int. J. Cancer 2006, 119, 884–887. [Google Scholar] [CrossRef] [PubMed]
- Poschl, G.; Seitz, H.K. Alcohol and cancer. Alcohol Alcohol. 2004, 39, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Corrao, G.; Bagnardi, V.; Zambon, A.; La Vecchia, C. A meta-analysis of alcohol consumption and the risk of 15 diseases. Prev. Med. 2004, 38, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Morgan, T.R.; Mandayam, S.; Jamal, M.M. Alcohol and hepatocellular carcinoma. Gastroenterology 2004, 127, S87–S96. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, M.; Mostafa, M.; Sabry, M.; el-Farrash, M.; Yousef, T. Aflatoxins as a risk factor for hepatocellular carcinoma in Egypt, mansoura gastroenterology center study. Hepatogastroenterology 2008, 55, 1754–1759. [Google Scholar] [PubMed]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Bouvard, V.; Altieri, A.; Cogliano, V.; WHO International Agency for Research on Cancer Monograph Working Group. Carcinogenicity of alcoholic beverages. Lancet Oncol. 2007, 8, 292–293. [Google Scholar] [CrossRef]
- Schwab, M. Encyclopedia of Cancer, 3rd ed.; Springer: Berlin, Germany, 2011; ISBN 978-364-216-483-5. [Google Scholar]
- Longnecker, M.P. Alcoholic beverage consumption in relation to risk of breast cancer: Meta-analysis and review. Cancer Causes Control 1994, 5, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Kopp, T.I.; Jensen, D.M.; Ravn-Haren, G.; Cohen, A.; Sommer, H.M.; Dragsted, L.O.; Tjonneland, A.; Hougaard, D.M.; Vogel, U. Alcohol-related breast cancer in postmenopausal women—Effect of cyp19a1, pparg and ppargc1a polymorphisms on female sex-hormone levels and interaction with alcohol consumption and nsaid usage in a nested case-control study and a randomised controlled trial. BMC Cancer 2016, 16, 283. [Google Scholar] [CrossRef] [PubMed]
- Dallal, C.M.; Tice, J.A.; Buist, D.S.; Bauer, D.C.; Lacey, J.V., Jr.; Cauley, J.A.; Hue, T.F.; Lacroix, A.; Falk, R.T.; Pfeiffer, R.M.; et al. Estrogen metabolism and breast cancer risk among postmenopausal women: A case-cohort study within B~FIT. Carcinogenesis 2014, 35, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Welsch, T.; Kleeff, J.; Seitz, H.K.; Buchler, P.; Friess, H.; Buchler, M.W. Update on pancreatic cancer and alcohol-associated risk. J. Gastroenterol. Hepatol. 2006, 21 (Suppl. 3), S69–S75. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Pöschl, G.; Salaspuro, M.P. Alcohol, Tobacco and Cancer; Karger: Basel, Switzerland, 2006. [Google Scholar]
- Cho, E.; Smith-Warner, S.A.; Ritz, J.; van den Brandt, P.A.; Colditz, G.A.; Folsom, A.R.; Freudenheim, J.L.; Giovannucci, E.; Goldbohm, R.A.; Graham, S.; et al. Alcohol intake and colorectal cancer: A pooled analysis of 8 cohort studies. Ann. Intern. Med. 2004, 140, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Corrao, G.; Bagnardi, V.; Zambon, A.; Arico, S. Exploring the dose-response relationship between alcohol consumption and the risk of several alcohol-related conditions: A meta-analysis. Addiction 1999, 94, 1551–1573. [Google Scholar] [CrossRef] [PubMed]
- McKinnell, R.; Parchment, R.; Perantoni, A.; Pierce, G.; Damjanov, I. The Biological Basis of Cancer, 2nd ed.; Cambridge University Press: Cambridge, UK, 2006; ISBN 978-051-181-664-2. [Google Scholar]
- Tsutsumi, M.; George, J.; Ishizawa, K.; Fukumura, A.; Takase, S. Effect of chronic dietary ethanol in the promotion of n-nitrosomethylbenzylamine-induced esophageal carcinogenesis in rats. J. Gastroenterol. Hepatol. 2006, 21, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Hilakivi-Clarke, L.; Cabanes, A.; de Assis, S.; Wang, M.; Khan, G.; Shoemaker, W.J.; Stevens, R.G. In utero alcohol exposure increases mammary tumorigenesis in rats. Br. J. Cancer 2004, 90, 2225–2231. [Google Scholar] [CrossRef] [PubMed]
- Brandon-Warner, E.; Walling, T.L.; Schrum, L.W.; McKillop, I.H. Chronic ethanol feeding accelerates hepatocellular carcinoma progression in a sex-dependent manner in a mouse model of hepatocarcinogenesis. Alcohol. Clin. Exp. Res. 2012, 36, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Simanowski, U.A.; Garzon, F.T.; Rideout, J.M.; Peters, T.J.; Koch, A.; Berger, M.R.; Einecke, H.; Maiwald, M. Possible role of acetaldehyde in ethanol-related rectal cocarcinogenesis in the rat. Gastroenterology 1990, 98, 406–413. [Google Scholar] [CrossRef]
- Beland, F.A.; Benson, R.W.; Mellick, P.W.; Kovatch, R.M.; Roberts, D.W.; Fang, J.L.; Doerge, D.R. Effect of ethanol on the tumorigenicity of urethane (ethyl carbamate) in b6c3f1 mice. Food Chem. Toxicol. 2005, 43, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Watabiki, T.; Okii, Y.; Tokiyasu, T.; Yoshimura, S.; Yoshida, M.; Akane, A.; Shikata, N.; Tsubura, A. Long-term ethanol consumption in icr mice causes mammary tumor in females and liver fibrosis in males. Alcohol. Clin. Exp. Res. 2000, 24, 117S–122S. [Google Scholar] [PubMed]
- Jackson, C.L.; Hu, F.B.; Kawachi, I.; Williams, D.R.; Mukamal, K.J.; Rimm, E.B. Black-white differences in the relationship between alcohol drinking patterns and mortality among us men and women. Am. J. Public Health 2015, 105 (Suppl. 3), S534–S543. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Oneta, C.M. Gastrointestinal alcohol dehydrogenase. Nutr. Rev. 1998, 56, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wang, S.; Ren, Z.; Frank, J.A.; Yang, X.H.; Zhang, Z.; Ke, Z.J.; Shi, X.; Luo, J. Chronic ethanol exposure enhances the aggressiveness of breast cancer: The role of p38gamma. Oncotarget 2016, 7, 3489–3505. [Google Scholar] [CrossRef] [PubMed]
- Masso-Welch, P.A.; Tobias, M.E.; Vasantha Kumar, S.C.; Bodziak, M.; Mashtare, T., Jr.; Tamburlin, J.; Koury, S.T. Folate exacerbates the effects of ethanol on peripubertal mouse mammary gland development. Alcohol 2012, 46, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Scoccianti, C.; Cecchini, M.; Anderson, A.S.; Berrino, F.; Boutron-Ruault, M.C.; Espina, C.; Key, T.J.; Leitzmann, M.; Norat, T.; Powers, H.; et al. European code against cancer 4th edition: Alcohol drinking and cancer. Cancer Epidemiol. 2016, 45, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Petri, A.L.; Tjonneland, A.; Gamborg, M.; Johansen, D.; Hoidrup, S.; Sorensen, T.I.; Gronbaek, M. Alcohol intake, type of beverage, and risk of breast cancer in pre- and postmenopausal women. Alcohol. Clin. Exp. Res. 2004, 28, 1084–1090. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Orsini, N.; Mignone, L.; Saji, S.; Wolk, A. Alcohol intake and risk of breast cancer defined by estrogen and progesterone receptor status—A meta-analysis of epidemiological studies. Int. J. Cancer 2008, 122, 1832–1841. [Google Scholar] [CrossRef] [PubMed]
- Li, C.I.; Chlebowski, R.T.; Freiberg, M.; Johnson, K.C.; Kuller, L.; Lane, D.; Lessin, L.; O’Sullivan, M.J.; Wactawski-Wende, J.; Yasmeen, S.; et al. Alcohol consumption and risk of postmenopausal breast cancer by subtype: The women’s health initiative observational study. J. Natl. Cancer Inst. 2010, 102, 1422–1431. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Baer, D.; Friedman, G.D.; Udaltsova, N.; Shim, V.; Klatsky, A.L. Wine, liquor, beer and risk of breast cancer in a large population. Eur. J. Cancer 2009, 45, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Lew, J.Q.; Freedman, N.D.; Leitzmann, M.F.; Brinton, L.A.; Hoover, R.N.; Hollenbeck, A.R.; Schatzkin, A.; Park, Y. Alcohol and risk of breast cancer by histologic type and hormone receptor status in postmenopausal women: The nih-aarp diet and health study. Am. J. Epidemiol. 2009, 170, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Gago-Dominguez, M.; Castelao, J.E.; Gude, F.; Fernandez, M.P.; Aguado-Barrera, M.E.; Ponte, S.M.; Redondo, C.M.; Castelo, M.E.; Dominguez, A.N.; Garzon, V.M.; et al. Alcohol and breast cancer tumor subtypes in a Spanish cohort. Springerplus 2016, 5, 39. [Google Scholar] [CrossRef] [PubMed]
- Kwan, M.L.; Kushi, L.H.; Weltzien, E.; Maring, B.; Kutner, S.E.; Fulton, R.S.; Lee, M.M.; Ambrosone, C.B.; Caan, B.J. Epidemiology of breast cancer subtypes in two prospective cohort studies of breast cancer survivors. Breast Cancer Res. 2009, 11, R31. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Tang, Y.; Tan, X.; Li, Q.; Zhong, W.; Sun, X.; Jia, W.; McClain, C.J.; Zhou, Z. Activation of peroxisome proliferator-activated receptor-gamma by rosiglitazone improves lipid homeostasis at the adipose tissue-liver axis in ethanol-fed mice. Am. J. Phys. Gastrointest. Liver Phys. 2012, 302, G548–G557. [Google Scholar]
- Kang, L.; Sebastian, B.M.; Pritchard, M.T.; Pratt, B.T.; Previs, S.F.; Nagy, L.E. Chronic ethanol-induced insulin resistance is associated with macrophage infiltration into adipose tissue and altered expression of adipocytokines. Alcohol. Clin. Exp. Res. 2007, 31, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sebastian, B.M.; Tang, H.; McMullen, M.M.; Axhemi, A.; Jacobsen, D.W.; Nagy, L.E. Taurine supplementation prevents ethanol-induced decrease in serum adiponectin and reduces hepatic steatosis in rats. Hepatology 2009, 49, 1554–1562. [Google Scholar] [CrossRef] [PubMed]
- Fulham, M.A.; Mandrekar, P. Sexual dimorphism in alcohol induced adipose inflammation relates to liver injury. PLoS ONE 2016, 11, e0164225. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.S.; Foster, C.S. Concepts of epigenetics in prostate cancer development. Br. J. Cancer 2009, 100, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.H.; Wan, P.; Bernstein, L. A multiethnic population-based study of smoking, alcohol and body size and risk of adenocarcinomas of the stomach and esophagus (United States). Cancer Causes Control 2001, 12, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Lagergren, J.; Bergstrom, R.; Lindgren, A.; Nyren, O. The role of tobacco, snuff and alcohol use in the aetiology of cancer of the oesophagus and gastric cardia. Int. J. Cancer 2000, 85, 340–346. [Google Scholar] [CrossRef]
- Go, V.L.; Gukovskaya, A.; Pandol, S.J. Alcohol and pancreatic cancer. Alcohol 2005, 35, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Wang, F.; Holly, E.A.; Bracci, P.M. Risk of pancreatic cancer by alcohol dose, duration, and pattern of consumption, including binge drinking: A population-based study. Cancer Causes Control 2010, 21, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Testino, G. Alcoholic diseases in hepato-gastroenterology: A point of view. Hepatogastroenterology 2008, 55, 371–377. [Google Scholar] [PubMed]
- Orywal, K.; Szmitkowski, M. Alcohol dehydrogenase and aldehyde dehydrogenase in malignant neoplasms. Clin. Exp. Med. 2017, 17, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Jelski, W.; Kozlowski, M.; Laudanski, J.; Niklinski, J.; Szmitkowski, M. Alcohol dehydrogenase isoenzymes and aldehyde dehydrogenase activity in the sera of patients with esophageal cancer. Clin. Exp. Med. 2009, 9, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Jelski, W.; Zalewski, B.; Szmitkowski, M. The activity of class i, ii, iii, and iv alcohol dehydrogenase (ADH) isoenzymes and aldehyde dehydrogenase (ALDH) in liver cancer. Dig. Dis. Sci. 2008, 53, 2550–2555. [Google Scholar] [CrossRef] [PubMed]
- Orywal, K.; Jelski, W.; Zdrodowski, M.; Szmitkowski, M. The activity of class i, ii, iii and iv alcohol dehydrogenase isoenzymes and aldehyde dehydrogenase in cervical cancer. Clin. Biochem. 2011, 44, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
- Asakage, T.; Yokoyama, A.; Haneda, T.; Yamazaki, M.; Muto, M.; Yokoyama, T.; Kato, H.; Igaki, H.; Tsujinaka, T.; Kumagai, Y.; et al. Genetic polymorphisms of alcohol and aldehyde dehydrogenases, and drinking, smoking and diet in japanese men with oral and pharyngeal squamous cell carcinoma. Carcinogenesis 2007, 28, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Hiraki, A.; Matsuo, K.; Wakai, K.; Suzuki, T.; Hasegawa, Y.; Tajima, K. Gene-gene and gene-environment interactions between alcohol drinking habit and polymorphisms in alcohol-metabolizing enzyme genes and the risk of head and neck cancer in Japan. Cancer Sci. 2007, 98, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Coutelle, C.; Hohn, B.; Benesova, M.; Oneta, C.M.; Quattrochi, P.; Roth, H.J.; Schmidt-Gayk, H.; Schneeweiss, A.; Bastert, G.; Seitz, H.K. Risk factors in alcohol associated breast cancer: Alcohol dehydrogenase polymorphism and estrogens. Int. J. Oncol. 2004, 25, 1127–1132. [Google Scholar] [PubMed]
- Visvanathan, K.; Crum, R.M.; Strickland, P.T.; You, X.; Ruczinski, I.; Berndt, S.I.; Alberg, A.J.; Hoffman, S.C.; Comstock, G.W.; Bell, D.A.; et al. Alcohol dehydrogenase genetic polymorphisms, low-to-moderate alcohol consumption, and risk of breast cancer. Alcohol. Clin. Exp. Res. 2007, 31, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Covolo, L.; Gelatti, U.; Talamini, R.; Garte, S.; Trevisi, P.; Franceschi, S.; Franceschini, M.; Barbone, F.; Tagger, A.; Ribero, M.L.; et al. Alcohol dehydrogenase 3, glutathione s-transferase m1 and t1 polymorphisms, alcohol consumption and hepatocellular carcinoma (Italy). Cancer Causes Control 2005, 16, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Wu, J.; Cai, Q.; Chen, E.Z.; Jiang, Z.Y. Association between glu504lys polymorphism of ALDH2 gene and cancer risk: A meta-analysis. PLoS ONE 2015, 10, e0117173. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.J.; Wang, H.Y.; Li, X.Q.; Du, H.Z.; Zheng, C.J.; Chen, H.G.; Mu, X.Y.; Yang, C.X. Genetic polymorphisms of ADH and ALDH2association with esophageal cancer risk in southwest China. World J. Gastroenterol. 2007, 13, 5760–5764. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Chen, C.; Wu, D.C.; Lee, C.H.; Wu, C.I.; Lee, J.M.; Goan, Y.G.; Huang, S.P.; Lin, C.C.; Li, T.C.; et al. Interactive effects of lifetime alcohol consumption and alcohol and aldehyde dehydrogenase polymorphisms on esophageal cancer risks. Int. J. Cancer 2006, 119, 2827–2831. [Google Scholar] [CrossRef] [PubMed]
- Munaka, M.; Kohshi, K.; Kawamoto, T.; Takasawa, S.; Nagata, N.; Itoh, H.; Oda, S.; Katoh, T. Genetic polymorphisms of tobacco- and alcohol-related metabolizing enzymes and the risk of hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2003, 129, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Hara, M.; Higaki, Y.; Ichiba, M.; Horita, M.; Mizuta, T.; Eguchi, Y.; Yasutake, T.; Ozaki, I.; Yamamoto, K.; et al. Influence of alcohol consumption and gene polymorphisms of ADH2 and ALDH2 on hepatocellular carcinoma in a Japanese population. Int. J. Cancer 2006, 118, 1501–1507. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Tian, F.; Dai, L.; Chai, Y. Cytochrome p450 2e1 gene polymorphism and alcohol drinking on the risk of hepatocellular carcinoma: A meta-analysis. Mol. Biol. Rep. 2014, 41, 7645–7650. [Google Scholar] [CrossRef] [PubMed]
- Rossini, A.; Rapozo, D.C.; Soares Lima, S.C.; Guimaraes, D.P.; Ferreira, M.A.; Teixeira, R.; Kruel, C.D.; Barros, S.G.; Andreollo, N.A.; Acatauassu, R.; et al. Polymorphisms of gstp1 and gstt1, but not of cyp2a6, cyp2e1 or gstm1, modify the risk for esophageal cancer in a western population. Carcinogenesis 2007, 28, 2537–2542. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.X.; Matsuo, K.; Ito, H.; Hirose, K.; Wakai, K.; Saito, T.; Shinoda, M.; Hatooka, S.; Mizutani, K.; Tajima, K. Esophageal cancer risk by ALDH2 and ADH2 polymorphisms and alcohol consumption: Exploration of gene-environment and gene-gene interactions. Asian Pac. J. Cancer Prev. 2005, 6, 256–262. [Google Scholar] [PubMed]
- Tsutsumi, M.; Wang, J.S.; Takase, S.; Takada, A. Hepatic messenger RNA contents of cytochrome p4502e1 in patients with different p4502e1 genotypes. Alcohol Alcohol. Suppl. 1994, 29, 29–32. [Google Scholar] [CrossRef]
- Suzuki, T.; Matsuo, K.; Hasegawa, Y.; Hiraki, A.; Wakai, K.; Hirose, K.; Saito, T.; Sato, S.; Ueda, R.; Tajima, K. One-carbon metabolism-related gene polymorphisms and risk of head and neck squamous cell carcinoma: Case-control study. Cancer Sci. 2007, 98, 1439–1446. [Google Scholar] [CrossRef]
- Yang, C.X.; Matsuo, K.; Ito, H.; Shinoda, M.; Hatooka, S.; Hirose, K.; Wakai, K.; Saito, T.; Suzuki, T.; Maeda, T.; et al. Gene-environment interactions between alcohol drinking and the mthfr c677t polymorphism impact on esophageal cancer risk: Results of a case-control study in Japan. Carcinogenesis 2005, 26, 1285–1290. [Google Scholar] [CrossRef]
- Wang, J.; Gajalakshmi, V.; Jiang, J.; Kuriki, K.; Suzuki, S.; Nagaya, T.; Nakamura, S.; Akasaka, S.; Ishikawa, H.; Tokudome, S. Associations between 5,10-methylenetetrahydrofolate reductase codon 677 and 1298 genetic polymorphisms and environmental factors with reference to susceptibility to colorectal cancer: A case-control study in an Indian population. Int. J. Cancer 2006, 118, 991–997. [Google Scholar] [CrossRef]
- Frosst, P.; Blom, H.J.; Milos, R.; Goyette, P.; Sheppard, C.A.; Matthews, R.G.; Boers, G.J.; den Heijer, M.; Kluijtmans, L.A.; van den Heuvel, L.P.; et al. A candidate genetic risk factor for vascular disease: A common mutation in methylenetetrahydrofolate reductase. Nat. Genet. 1995, 10, 111–113. [Google Scholar] [CrossRef]
- Platek, M.E.; Shields, P.G.; Marian, C.; McCann, S.E.; Bonner, M.R.; Nie, J.; Ambrosone, C.B.; Millen, A.E.; Ochs-Balcom, H.M.; Quick, S.K.; et al. Alcohol consumption and genetic variation in methylenetetrahydrofolate reductase and 5-methyltetrahydrofolate-homocysteine methyltransferase in relation to breast cancer risk. Cancer Epidemiol. Prev. Biomark. 2009, 18, 2453–2459. [Google Scholar] [CrossRef]
- Saffroy, R.; Pham, P.; Chiappini, F.; Gross-Goupil, M.; Castera, L.; Azoulay, D.; Barrier, A.; Samuel, D.; Debuire, B.; Lemoine, A. The mthfr 677c >T polymorphism is associated with an increased risk of hepatocellular carcinoma in patients with alcoholic cirrhosis. Carcinogenesis 2004, 25, 1443–1448. [Google Scholar] [CrossRef]
- Haorah, J.; Ramirez, S.H.; Floreani, N.; Gorantla, S.; Morsey, B.; Persidsky, Y. Mechanism of alcohol-induced oxidative stress and neuronal injury. Free Radic. Biol. Med. 2008, 45, 1542–1550. [Google Scholar] [CrossRef]
- Wu, W.S. The signaling mechanism of ros in tumor progression. Cancer Metastasis Rev. 2006, 25, 695–705. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and nf-kappab signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef]
- Wang, F.; Yang, J.L.; Yu, K.K.; Xu, M.; Xu, Y.Z.; Chen, L.; Lu, Y.M.; Fang, H.S.; Wang, X.Y.; Hu, Z.Q.; et al. Activation of the nf-kappab pathway as a mechanism of alcohol enhanced progression and metastasis of human hepatocellular carcinoma. Mol. Cancer 2015, 14, 10. [Google Scholar] [CrossRef]
- Shinohara, M.; Adachi, Y.; Mitsushita, J.; Kuwabara, M.; Nagasawa, A.; Harada, S.; Furuta, S.; Zhang, Y.; Seheli, K.; Miyazaki, H.; et al. Reactive oxygen generated by nadph oxidase 1 (nox1) contributes to cell invasion by regulating matrix metalloprotease-9 production and cell migration. J. Biol. Chem. 2010, 285, 4481–4488. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Lin, Z.; Lamb, R.; Hulit, J.; Howell, A.; Sotgia, F.; Rubin, E.; Lisanti, M.P. Ethanol exposure induces the cancer-associated fibroblast phenotype and lethal tumor metabolism: Implications for breast cancer prevention. Cell Cycle 2013, 12, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Filaire, E.; Dupuis, C.; Galvaing, G.; Aubreton, S.; Laurent, H.; Richard, R.; Filaire, M. Lung cancer: What are the links with oxidative stress, physical activity and nutrition. Lung Cancer 2013, 82, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Paschos, A.; Pandya, R.; Duivenvoorden, W.C.; Pinthus, J.H. Oxidative stress in prostate cancer: Changing research concepts towards a novel paradigm for prevention and therapeutics. Prostate Cancer Prostatic Dis. 2013, 16, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.D. Alcohol, vitamin a, and cancer. Alcohol 2005, 35, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.H.; Gudas, L.J. Retinoids, retinoic acid receptors, and cancer. Annu. Rev. Pathol. 2011, 6, 345–364. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.C.; Hsueh, W.T.; Ou, C.Y.; Huang, C.C.; Lee, W.T.; Fang, S.Y.; Tsai, S.T.; Huang, J.S.; Wong, T.Y.; Wu, J.L.; et al. Alcohol drinking obliterates the inverse association between serum retinol and risk of head and neck cancer. Medicine 2015, 94, e1064. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Liu, C.; Smith, D.E.; Seitz, H.K.; Russell, R.M.; Wang, X.D. Restoration of retinoic acid concentration suppresses ethanol-enhanced c-jun expression and hepatocyte proliferation in rat liver. Carcinogenesis 2001, 22, 1213–1219. [Google Scholar] [CrossRef] [PubMed]
- Bakiri, L.; Wagner, E.F. Mouse models for liver cancer. Mol. Oncol. 2013, 7, 206–223. [Google Scholar] [CrossRef] [PubMed]
- Uehara, T.; Ainslie, G.R.; Kutanzi, K.; Pogribny, I.P.; Muskhelishvili, L.; Izawa, T.; Yamate, J.; Kosyk, O.; Shymonyak, S.; Bradford, B.U.; et al. Molecular mechanisms of fibrosis-associated promotion of liver carcinogenesis. Toxicol. Sci. 2013, 132, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Ambade, A.; Satishchandran, A.; Gyongyosi, B.; Lowe, P.; Szabo, G. Adult mouse model of early hepatocellular carcinoma promoted by alcoholic liver disease. World J. Gastroenterol. 2016, 22, 4091–4108. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Yang, F.; Huang, S.X.; Kuang, Z.P.; Luo, X.L.; Li, Y.D.; Wu, J.N.; Xie, Y.A. Two-stage model of chemically induced hepatocellular carcinoma in mouse. Oncol. Res. 2013, 20, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Vargo-Gogola, T.; Rosen, J.M. Modelling breast cancer: One size does not fit all. Nat. Rev. Cancer 2007, 7, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, F.; Nishikawa, A.; Lee, I.S.; Son, H.Y.; Nakamura, H.; Miyauchi, M.; Takahashi, M.; Hirose, M. Inhibition by methionine of pancreatic carcinogenesis in hamsters after initiation with N-nitrosobis(2-oxopropyl) amine. Cancer Lett. 2000, 152, 163–167. [Google Scholar] [CrossRef]
- Z’Graggen, K.; Warshaw, A.L.; Werner, J.; Graeme-Cook, F.; Jimenez, R.E.; Fernandez-Del Castillo, C. Promoting effect of a high-fat/high-protein diet in dmba-induced ductal pancreatic cancer in rats. Ann. Surg. 2001, 233, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Schuller, H.M.; Jorquera, R.; Reichert, A.; Castonguay, A. Transplacental induction of pancreas tumors in hamsters by ethanol and the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Cancer Res. 1993, 53, 2498–2501. [Google Scholar] [PubMed]
- Wendt, L.R.; Osvaldt, A.B.; Bersch, V.P.; Schumacher Rde, C.; Edelweiss, M.I.; Rohde, L. Pancreatic intraepithelial neoplasia and ductal adenocarcinoma induced by dmba in mice: Effects of alcohol and caffeine. Acta Cir. Bras. 2007, 22, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T. Colorectal carcinogenesis: Review of human and experimental animal studies. J. Carcinog. 2009, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Niwa, K.; Tanaka, T.; Sugie, S.; Shinoda, T.; Kato, K.; Tamaya, T.; Mori, H. Enhancing effect of ethanol or sake on methylazoxymethanol acetate-initiated large bowel carcinogenesis in aci/n rats. Nutr. Cancer 1991, 15, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, N.; Tsutsumi, M.; Fukura, M.; Yano, H.; Tsuchishima, M.; Takase, S. Effect of chronic dietary ethanol consumption on colonic cancer in rats induced by 1,1-dimethylhydrazine. Alcohol. Clin. Exp. Res. 2007, 31, S72–S76. [Google Scholar] [CrossRef] [PubMed]
- Frese, K.K.; Tuveson, D.A. Maximizing mouse cancer models. Nat. Rev. Cancer 2007, 7, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Valentine, J.L.; Lee, S.S.; Seaton, M.J.; Asgharian, B.; Farris, G.; Corton, J.C.; Gonzalez, F.J.; Medinsky, M.A. Reduction of benzene metabolism and toxicity in mice that lack cyp2e1 expression. Toxicol. Appl. Pharmacol. 1996, 141, 205–213. [Google Scholar] [CrossRef]
- Wong, F.W.; Chan, W.Y.; Lee, S.S. Resistance to carbon tetrachloride-induced hepatotoxicity in mice which lack cyp2e1 expression. Toxicol. Appl. Pharmacol. 1998, 153, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Skarnes, W.C.; Rosen, B.; West, A.P.; Koutsourakis, M.; Bushell, W.; Iyer, V.; Mujica, A.O.; Thomas, M.; Harrow, J.; Cox, T.; et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature 2011, 474, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Stagos, D.; Chen, Y.; Brocker, C.; Donald, E.; Jackson, B.C.; Orlicky, D.J.; Thompson, D.C.; Vasiliou, V. Aldehyde dehydrogenase 1b1: Molecular cloning and characterization of a novel mitochondrial acetaldehyde-metabolizing enzyme. Drug Metab. Dispos. 2010, 38, 1679–1687. [Google Scholar] [CrossRef] [PubMed]
- Lassen, N.; Bateman, J.B.; Estey, T.; Kuszak, J.R.; Nees, D.W.; Piatigorsky, J.; Duester, G.; Day, B.J.; Huang, J.; Hines, L.M.; et al. Multiple and additive functions of aldh3a1 and aldh1a1: Cataract phenotype and ocular oxidative damage in aldh3a1(-/-)/aldh1a1(-/-) knock-out mice. J. Biol. Chem. 2007, 282, 25668–25676. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Krishan, M.; Nebert, D.W.; Shertzer, H.G. Glutathione-deficient mice are susceptible to tcdd-induced hepatocellular toxicity but resistant to steatosis. Chem. Res. Toxicol. 2012, 25, 94–100. [Google Scholar] [CrossRef] [PubMed]
- McConnachie, L.A.; Mohar, I.; Hudson, F.N.; Ware, C.B.; Ladiges, W.C.; Fernandez, C.; Chatterton-Kirchmeier, S.; White, C.C.; Pierce, R.H.; Kavanagh, T.J. Glutamate cysteine ligase modifier subunit deficiency and gender as determinants of acetaminophen-induced hepatotoxicity in mice. Toxicol. Sci. 2007, 99, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Johansson, E.; Yang, Y.; Miller, M.L.; Shen, D.; Orlicky, D.J.; Shertzer, H.G.; Vasiliou, V.; Nebert, D.W.; Dalton, T.P. Oral n-acetylcysteine rescues lethality of hepatocyte-specific gclc-knockout mice, providing a model for hepatic cirrhosis. J. Hepatol. 2010, 53, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.J.; Bardeesy, N.; Sinha, M.; Lopez, L.; Tuveson, D.A.; Horner, J.; Redston, M.S.; DePinho, R.A. Activated kras and ink4a/arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes. Dev. 2003, 17, 3112–3126. [Google Scholar] [CrossRef] [PubMed]
- Tuveson, D.A.; Zhu, L.; Gopinathan, A.; Willis, N.A.; Kachatrian, L.; Grochow, R.; Pin, C.L.; Mitin, N.Y.; Taparowsky, E.J.; Gimotty, P.A.; et al. Mist1-krasg12d knock-in mice develop mixed differentiation metastatic exocrine pancreatic carcinoma and hepatocellular carcinoma. Cancer Res. 2006, 66, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.; Vickers, S.M.; Adsay, N.V.; Jhala, N.C.; Kim, H.G.; Schoeb, T.R.; Grizzle, W.E.; Klug, C.A. Inactivation of smad4 accelerates kras(g12d)-mediated pancreatic neoplasia. Cancer Res. 2007, 67, 8121–8130. [Google Scholar] [CrossRef] [PubMed]
- De Minicis, S.; Marzioni, M.; Benedetti, A.; Svegliati-Baroni, G. New insights in hepatocellular carcinoma: From bench to bedside. Ann. Transl. Med. 2013, 1, 15. [Google Scholar] [PubMed]
- Heindryckx, F.; Colle, I.; Van Vlierberghe, H. Experimental mouse models for hepatocellular carcinoma research. Int. J. Exp. Pathol. 2009, 90, 367–386. [Google Scholar] [CrossRef] [PubMed]
- Urnov, F.D.; Miller, J.C.; Lee, Y.L.; Beausejour, C.M.; Rock, J.M.; Augustus, S.; Jamieson, A.C.; Porteus, M.H.; Gregory, P.D.; Holmes, M.C. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 2005, 435, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, L.; Kendrick, S.L.; Gerard, R.D.; Zhu, H. Talen-mediated somatic mutagenesis in murine models of cancer. Cancer Res. 2014, 74, 5311–5321. [Google Scholar] [CrossRef] [PubMed]
- Mou, H.; Kennedy, Z.; Anderson, D.G.; Yin, H.; Xue, W. Precision cancer mouse models through genome editing with crispr-cas9. Genome Med. 2015, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Hutmacher, D.W.; Horch, R.E.; Loessner, D.; Rizzi, S.; Sieh, S.; Reichert, J.C.; Clements, J.A.; Beier, J.P.; Arkudas, A.; Bleiziffer, O.; et al. Translating tissue engineering technology platforms into cancer research. J. Cell. Mol. Med. 2009, 13, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.X.; Tang, Z.Y.; Lui, K.D.; Ye, S.L.; Xue, Q.; Gao, D.M.; Ma, Z.C. Establishment of a metastatic model of human hepatocellular carcinoma in nude mice via orthotopic implantation of histologically intact tissues. Int. J. Cancer 1996, 66, 239–243. [Google Scholar] [CrossRef]
- Busby, B.; Tan, W.; Covington, J.; Young, E.; Gu, J.-W. Chronic alcohol consumption increases tumor growth and angiogenesis of breast cancer in female mice. FASEB J. 2007, 21, A527. [Google Scholar]
- Garrido-Laguna, I.; Uson, M.; Rajeshkumar, N.V.; Tan, A.C.; de Oliveira, E.; Karikari, C.; Villaroel, M.C.; Salomon, A.; Taylor, G.; Sharma, R.; et al. Tumor engraftment in nude mice and enrichment in stroma- related gene pathways predict poor survival and resistance to gemcitabine in patients with pancreatic cancer. Clin. Cancer Res. 2011, 17, 5793–5800. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.I.; Park, O.; Gao, B. Abrogation of the antifibrotic effects of natural killer cells/interferon-gamma contributes to alcohol acceleration of liver fibrosis. Gastroenterology 2008, 134, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.I.; Gao, B. Innate immunity and alcoholic liver fibrosis. J. Gastroenterol. Hepatol. 2008, 23 (Suppl. 1), S112–S118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Meadows, G.G. Exogenous il-15 in combination with il-15r alpha rescues natural killer cells from apoptosis induced by chronic alcohol consumption. Alcohol. Clin. Exp. Res. 2009, 33, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Gallucci, R.M.; Pfister, L.J.; Meadows, G.G. Effects of ethanol consumption on enriched natural killer cells from c57bl/6 mice. Alcohol. Clin. Exp. Res. 1994, 18, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.J.; Wolcott, R.M.; Pruett, S.B. Ethanol decreases the number and activity of splenic natural killer cells in a mouse model for binge drinking. J. Pharmacol. Exp. Ther. 1994, 271, 722–729. [Google Scholar] [PubMed]
- Ben-Eliyahu, S.; Page, G.G.; Yirmiya, R.; Taylor, A.N. Acute alcohol intoxication suppresses natural killer cell activity and promotes tumor metastasis. Nat. Med. 1996, 2, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Baskic, D.; Vujanovic, L.; Arsenijevic, N.; Whiteside, T.L.; Myers, E.N.; Vujanovic, N.L. Suppression of natural killer-cell and dendritic-cell apoptotic tumoricidal activity in patients with head and neck cancer. Head Neck 2013, 35, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.F.; Mannam, V.K.; Craft, B.S.; Puneky, L.V.; Sheehan, N.T.; Lewis, R.E.; Cruse, J.M. Comparative analysis of innate immune system function in metastatic breast, colorectal, and prostate cancer patients with circulating tumor cells. Exp. Mol. Pathol. 2014, 96, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H.W.; Seidler, S.; Gassler, N.; Nattermann, J.; Luedde, T.; Trautwein, C.; Tacke, F. Interleukin-8 is activated in patients with chronic liver diseases and associated with hepatic macrophage accumulation in human liver fibrosis. PLoS ONE 2011, 6, e21381. [Google Scholar] [CrossRef] [PubMed]
- Kiecolt-Glaser, J.K.; Preacher, K.J.; MacCallum, R.C.; Atkinson, C.; Malarkey, W.B.; Glaser, R. Chronic stress and age-related increases in the proinflammatory cytokine il-6. Proc. Natl. Acad. Sci. USA 2003, 100, 9090–9095. [Google Scholar] [CrossRef] [PubMed]
- Strobel, O.; Dor, Y.; Alsina, J.; Stirman, A.; Lauwers, G.; Trainor, A.; Castillo, C.F.; Warshaw, A.L.; Thayer, S.P. In vivo lineage tracing defines the role of acinar-to-ductal transdifferentiation in inflammatory ductal metaplasia. Gastroenterology 2007, 133, 1999–2009. [Google Scholar] [CrossRef] [PubMed]
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Eaden, J.A.; Abrams, K.R.; Mayberry, J.F. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut 2001, 48, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, J.A.; Harrison, R.F.; Perry, I.; Balkwill, F.; Tselepis, C. Barrett’s metaplasia. Lancet 2000, 356, 2079–2085. [Google Scholar] [CrossRef]
- Ninomiya, T.; Akbar, S.M.; Masumoto, T.; Horiike, N.; Onji, M. Dendritic cells with immature phenotype and defective function in the peripheral blood from patients with hepatocellular carcinoma. J. Hepatol. 1999, 31, 323–331. [Google Scholar] [CrossRef]
- Szabo, G.; Dolganiuc, A. Subversion of plasmacytoid and myeloid dendritic cell functions in chronic HCV infection. Immunobiology 2005, 210, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Dolganiuc, A.; Mandrekar, P.; White, B. Inhibition of antigen-presenting cell functions by alcohol: Implications for hepatitis c virus infection. Alcohol 2004, 33, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Dolganiuc, A.; Kodys, K.; Kopasz, A.; Marshall, C.; Mandrekar, P.; Szabo, G. Additive inhibition of dendritic cell allostimulatory capacity by alcohol and hepatitis c is not restored by dc maturation and involves abnormal il-10 and il-2 induction. Alcohol. Clin. Exp. Res. 2003, 27, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Dolganiuc, A.; Kodys, K.; Kopasz, A.; Marshall, C.; Do, T.; Romics, L., Jr.; Mandrekar, P.; Zapp, M.; Szabo, G. Hepatitis c virus core and nonstructural protein 3 proteins induce pro- and anti-inflammatory cytokines and inhibit dendritic cell differentiation. J. Immunol. 2003, 170, 5615–5624. [Google Scholar] [CrossRef] [PubMed]
- Erdag, G.; Schaefer, J.T.; Smolkin, M.E.; Deacon, D.H.; Shea, S.M.; Dengel, L.T.; Patterson, J.W.; Slingluff, C.L., Jr. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res. 2012, 72, 1070–1080. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Eyles, J.; Puaux, A.L.; Wang, X.; Toh, B.; Prakash, C.; Hong, M.; Tan, T.G.; Zheng, L.; Ong, L.C.; Jin, Y.; et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J. Clin. Investig. 2010, 120, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Wang, X.; Jia, J.; Yuan, Y.; Wan, F.; Zhou, X.; Yang, H.; Ren, J.; Gu, J.; Lyerly, H.K. Elevated level of peripheral cd8(+)cd28(-) t lymphocytes are an independent predictor of progression-free survival in patients with metastatic breast cancer during the course of chemotherapy. Cancer Immunol. Immunother. 2013, 62, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Peng, L.; Yu, P.; Zhao, Y.; Shi, Y.; Mao, X.; Chen, W.; Cheng, P.; Wang, T.; Chen, N.; et al. Increased circulating th22 and th17 cells are associated with tumor progression and patient survival in human gastric cancer. J. Clin. Immunol. 2012, 32, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Hunt, J.D.; Robert, E.G.; Zieske, A.W.; Bautista, A.P.; Bukara, M.; Lei, D.; Shellito, J.E.; Nelson, S.; Kolls, J.K.; Skrepnik, N. Orthotopic human lung carcinoma xenografts in balb/c mice immunosuppressed with anti-cd4 monoclonal antibodies and chronic alcohol consumption. Cancer 2000, 88, 468–479. [Google Scholar] [CrossRef]
- Zhang, H.; Meadows, G.G. Chronic alcohol consumption enhances myeloid-derived suppressor cells in b16bl6 melanoma-bearing mice. Cancer Immunol. Immunother. 2010, 59, 1151–1159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhu, Z.; Meadows, G.G. Chronic alcohol consumption impairs distribution and compromises circulation of b cells in b16bl6 melanoma-bearing mice. J. Immunol. 2012, 189, 1340–1348. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Bando, Y.; Xiao, S.; Yang, K.; Anderson, A.C.; Kuchroo, V.K.; Khoury, S.J. Cd11b+ly-6c(hi) suppressive monocytes in experimental autoimmune encephalomyelitis. J. Immunol. 2007, 179, 5228–5237. [Google Scholar] [CrossRef] [PubMed]
- DiLillo, D.J.; Yanaba, K.; Tedder, T.F. B cells are required for optimal cd4+ and cd8+ t cell tumor immunity: Therapeutic b cell depletion enhances b16 melanoma growth in mice. J. Immunol. 2010, 184, 4006–4016. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Leitner, W.W.; Golding, B.; Scott, D. Inhibitory effects of b cells on antitumor immunity. Cancer Res. 2006, 66, 7741–7747. [Google Scholar] [CrossRef] [PubMed]
- Ruddell, A.; Harrell, M.I.; Furuya, M.; Kirschbaum, S.B.; Iritani, B.M. B lymphocytes promote lymphogenous metastasis of lymphoma and melanoma. Neoplasia 2011, 13, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, M.; Minard-Colin, V.; Matsushita, T.; Tedder, T.F. Regulatory b cell production of il-10 inhibits lymphoma depletion during cd20 immunotherapy in mice. J. Clin. Investig. 2011, 121, 4268–4280. [Google Scholar] [CrossRef] [PubMed]
- Parekh, V.V.; Lalani, S.; Kim, S.; Halder, R.; Azuma, M.; Yagita, H.; Kumar, V.; Wu, L.; Kaer, L.V. Pd-1/pd-l blockade prevents anergy induction and enhances the anti-tumor activities of glycolipid-activated invariant nkt cells. J. Immunol. 2009, 182, 2816–2826. [Google Scholar] [CrossRef] [PubMed]
- Vasiliou, V.; Zakhari, S.; Seitz, H.K.; Hoek, J.B. Biological Basis of Alcohol-Induced Cancer, 1st ed.; Springer International Publishing: Basel, Switzerland, 2015; ISBN 978-331-909-614-8. [Google Scholar]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ratna, A.; Mandrekar, P. Alcohol and Cancer: Mechanisms and Therapies. Biomolecules 2017, 7, 61. https://doi.org/10.3390/biom7030061
Ratna A, Mandrekar P. Alcohol and Cancer: Mechanisms and Therapies. Biomolecules. 2017; 7(3):61. https://doi.org/10.3390/biom7030061
Chicago/Turabian StyleRatna, Anuradha, and Pranoti Mandrekar. 2017. "Alcohol and Cancer: Mechanisms and Therapies" Biomolecules 7, no. 3: 61. https://doi.org/10.3390/biom7030061
APA StyleRatna, A., & Mandrekar, P. (2017). Alcohol and Cancer: Mechanisms and Therapies. Biomolecules, 7(3), 61. https://doi.org/10.3390/biom7030061