
On the Role of Glutamate in Presynaptic Development: Possible Contributions of Presynaptic NMDA Receptors

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. General Properties of NMDA Receptors
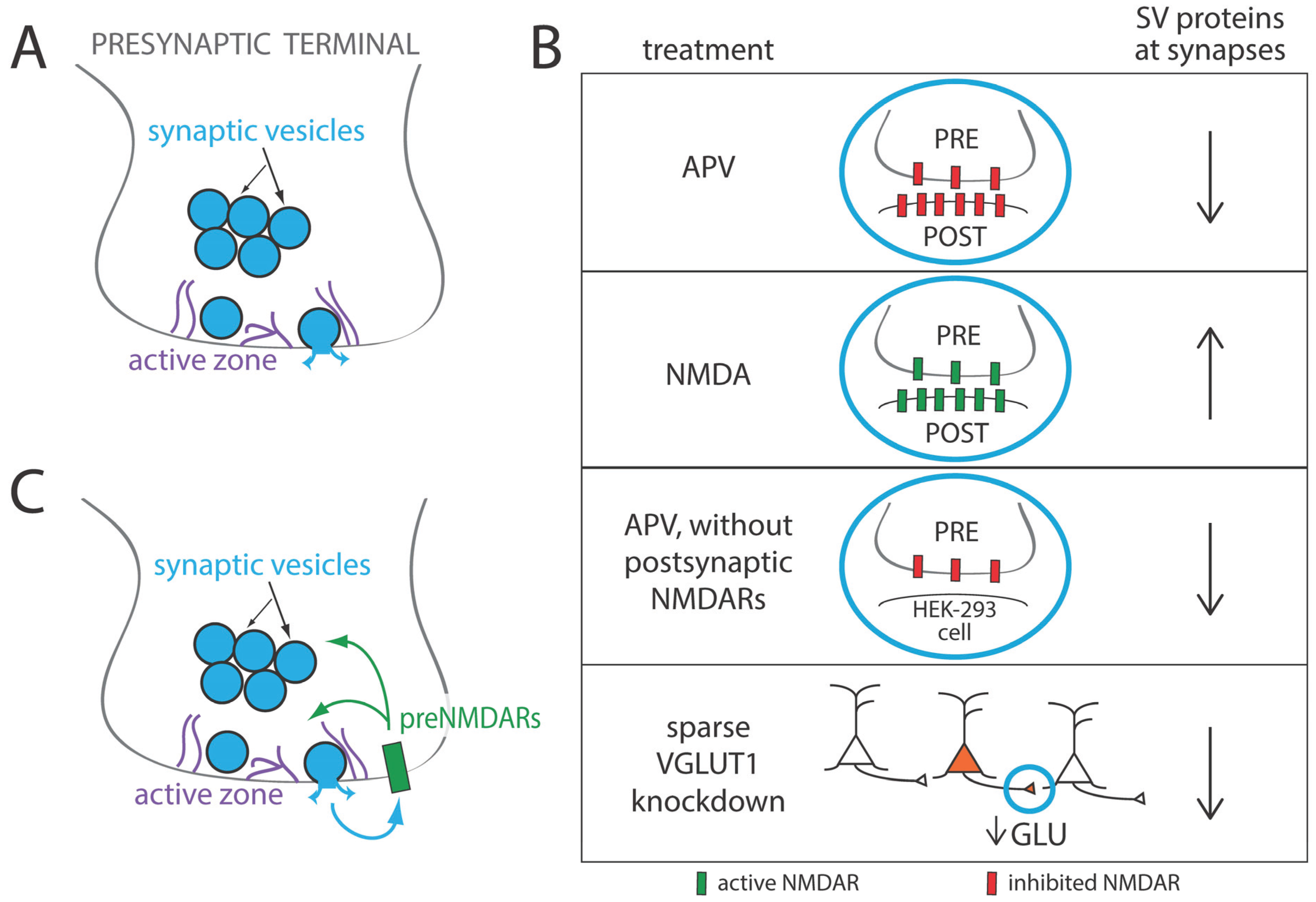
3. Regulation of Presynaptic Development by Glutamate and Synaptic Activity
4. Regulation of Synapse Formation by NMDA Receptors
5. Presynaptic NMDA Receptors
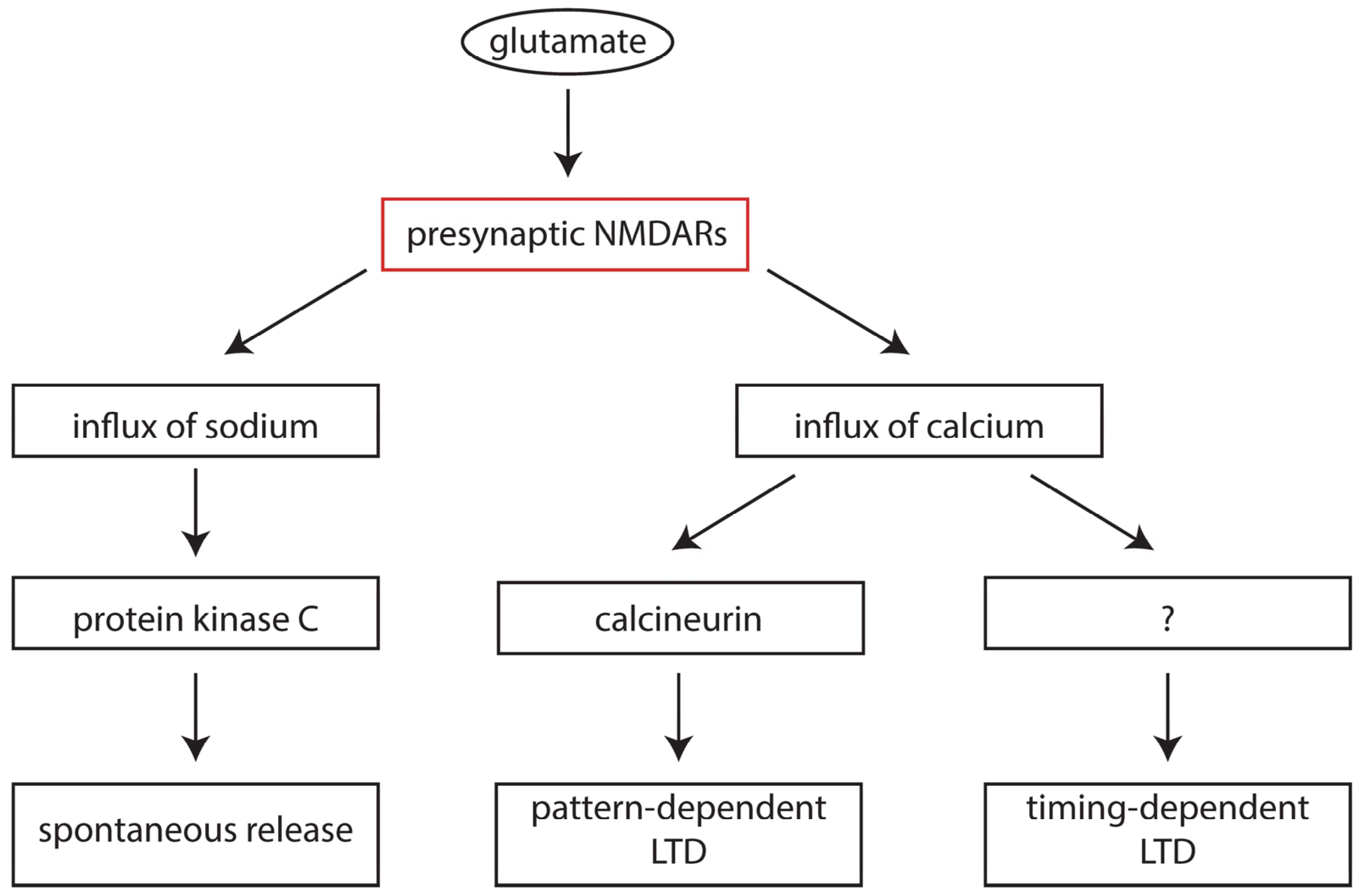
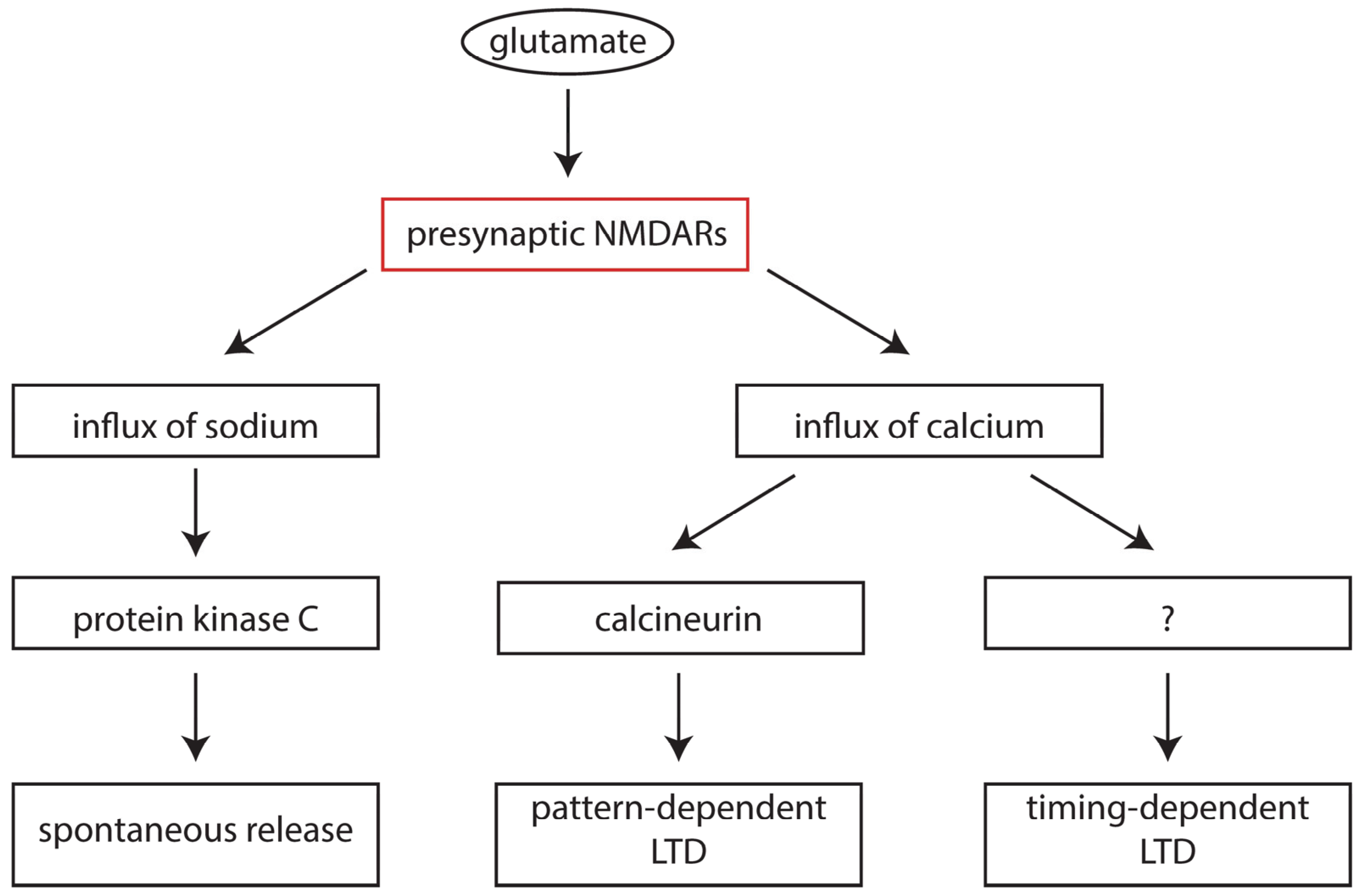
6. Presynaptic NMDA Receptors in Development
7. Relevance to Neurodevelopmental Disease
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Saito, Y.; Murakami, F.; Song, W.J.; Okawa, K.; Shimono, K.; Katsumaru, H. Developing corticorubral axons of the cat form synapses on filopodial dendritic protrusions. Neurosci. Lett. 1992, 147, 81–84. [Google Scholar] [CrossRef]
- Saito, Y.; Song, W.J.; Murakami, F. Preferential termination of corticorubral axons on spine-like dendritic protrusions in developing cat. J. Neurosci. 1997, 17, 8792–8803. [Google Scholar] [PubMed]
- Ziv, N.E.; Smith, S.J. Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron 1996, 17, 91–102. [Google Scholar] [CrossRef]
- Dailey, M.E.; Smith, S.J. The dynamics of dendritic structure in developing hippocampal slices. J. Neurosci. 1996, 16, 2983–2994. [Google Scholar] [PubMed]
- Fiala, J.C.; Feinberg, M.; Popov, V.; Harris, K.M. Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J. Neurosci. 1998, 18, 8900–8911. [Google Scholar] [PubMed]
- Jontes, J.D.; Buchanan, J.; Smith, S.J. Growth cone and dendrite dynamics in zebrafish embryos: Early events in synaptogenesis imaged in vivo. Nat. Neurosci. 2000, 3, 231–237. [Google Scholar] [PubMed]
- Ahmari, S.E.; Buchanan, J.; Smith, S.J. Assembly of presynaptic active zones from cytoplasmic transport packets. Nat. Neurosci. 2000, 3, 445–451. [Google Scholar] [PubMed]
- Washbourne, P.; Bennett, J.E.; McAllister, A.K. Rapid recruitment of NMDA receptor transport packets to nascent synapses. Nat. Neurosci. 2002, 5, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Shapira, M.; Zhai, R.G.; Dresbach, T.; Bresler, T.; Torres, V.I.; Gundelfinger, E.D.; Ziv, N.E.; Garner, C.C. Unitary assembly of presynaptic active zones from Piccolo-Bassoon transport vesicles. Neuron 2003, 38, 237–252. [Google Scholar] [CrossRef]
- Kraszewski, K.; Mundigl, O.; Daniell, L.; Verderio, C.; Matteoli, M.; de Camilli, P. Synaptic vesicle dynamics in living cultured hippocampal neurons visualized with CY3-conjugated antibodies directed against the lumenal domain of synaptotagmin. J. Neurosci. 1995, 15, 4328–4342. [Google Scholar] [PubMed]
- Sabo, S.L.; Gomes, R.A.; McAllister, A.K. Formation of presynaptic terminals at predefined sites along axons. J. Neurosci. 2006, 26, 10813–10825. [Google Scholar] [CrossRef] [PubMed]
- Cline, H.; Haas, K. The regulation of dendritic arbor development and plasticity by glutamatergic synaptic input: A review of the synaptotrophic hypothesis. J. Physiol. 2008, 586, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Mozhayeva, M.G.; Sara, Y.; Liu, X.; Kavalali, E.T. Development of vesicle pools during maturation of hippocampal synapses. J. Neurosci. 2002, 22, 654–665. [Google Scholar] [PubMed]
- Sceniak, M.P.; Berry, C.T.; Sabo, S.L. Facilitation of neocortical presynaptic terminal development by NMDA receptor activation. Neural Dev. 2012, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Berry, C.T.; Sceniak, M.P.; Zhou, L.; Sabo, S.L. Developmental up-regulation of vesicular glutamate transporter-1 promotes neocortical presynaptic terminal development. PLoS ONE 2012, 7, e50911. [Google Scholar] [CrossRef] [PubMed]
- Cull-Candy, S.G.; Leszkiewicz, D.N. Role of distinct NMDA receptor subtypes at central synapses. Sci. STKE 2004. [Google Scholar] [CrossRef] [PubMed]
- Pachernegg, S.; Strutz-Seebohm, N.; Hollmann, M. GluN3 subunit-containing NMDA receptors: Not just one-trick ponies. Trends Neurosci. 2012, 35, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Held, R.G.; Chang, S.C.; Yang, L.; Delpire, E.; Ghosh, A.; Hall, B.J. A critical role for GluN2B-containing NMDA receptors in cortical development and function. Neuron 2011, 72, 789–805. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Clemente, A.; Nicoll, R.A.; Roche, K.W. Diversity in NMDA receptor composition: Many regulators, many consequences. Neuroscientist 2013, 19, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Monyer, H.; Sprengel, R.; Schoepfer, R.; Herb, A.; Higuchi, M.; Lomeli, H.; Burnashev, N.; Sakmann, B.; Seeburg, P.H. Heteromeric NMDA receptors: Molecular and functional distinction of subtypes. Science 1992, 256, 1217–1221. [Google Scholar] [CrossRef] [PubMed]
- Larsen, R.S.; Corlew, R.J.; Henson, M.A.; Roberts, A.C.; Mishina, M.; Watanabe, M.; Lipton, S.A.; Nakanishi, N.; Perez-Otano, I.; Weinberg, R.J.; et al. NR3A-containing NMDARs promote neurotransmitter release and spike timing-dependent plasticity. Nat. Neurosci. 2011, 14, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Mothet, J.P.; le Bail, M.; Billard, J.M. Time and space profiling of NMDA receptor co-agonist functions. J. Neurochem. 2015, 135, 210–225. [Google Scholar] [CrossRef] [PubMed]
- Van Horn, M.R.; Sild, M.; Ruthazer, E.S. d-serine as a gliotransmitter and its roles in brain development and disease. Front. Cell. Neurosci. 2013. [Google Scholar] [CrossRef] [PubMed]
- Le Bail, M.; Martineau, M.; Sacchi, S.; Yatsenko, N.; Radzishevsky, I.; Conrod, S.; Ait Ouares, K.; Wolosker, H.; Pollegioni, L.; Billard, J.M.; et al. Identity of the NMDA receptor coagonist is synapse specific and developmentally regulated in the hippocampus. Proc. Natl. Acad. Sci. USA 2015, 112, E204–E213. [Google Scholar] [CrossRef] [PubMed]
- Papouin, T.; Ladepeche, L.; Ruel, J.; Sacchi, S.; Labasque, M.; Hanini, M.; Groc, L.; Pollegioni, L.; Mothet, J.P.; Oliet, S.H. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell 2012, 150, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, A.; Kumashiro, S.; Nishikawa, T.; Oka, T.; Takahashi, K.; Mito, T.; Takashima, S.; Doi, N.; Mizutani, Y.; Yamazaki, T.; et al. Embryonic development and postnatal changes in free D-aspartate and D-serine in the human prefrontal cortex. J. Neurochem. 1993, 61, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, A.; Nishikawa, T.; Oka, T.; Takahashi, K. Endogenous D-serine in rat brain: N-methyl-d-aspartate receptor-related distribution and aging. J. Neurochem. 1993, 60, 783–786. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, A.; Oka, T. Free D-aspartate and D-serine in the mammalian brain and periphery. Prog. Neurobiol. 1997, 52, 325–353. [Google Scholar] [CrossRef]
- Puyal, J.; Martineau, M.; Mothet, J.P.; Nicolas, M.T.; Raymond, J. Changes in D-serine levels and localization during postnatal development of the rat vestibular nuclei. J. Comp. Neurol. 2006, 497, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Verhage, M.; Maia, A.S.; Plomp, J.J.; Brussaard, A.B.; Heeroma, J.H.; Vermeer, H.; Toonen, R.F.; Hammer, R.E.; van den Berg, T.K.; Missler, M.; et al. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 2000, 287, 864–869. [Google Scholar] [CrossRef] [PubMed]
- Varoqueaux, F.; Sigler, A.; Rhee, J.S.; Brose, N.; Enk, C.; Reim, K.; Rosenmund, C. Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc. Natl. Acad. Sci. USA 2002, 99, 9037–9042. [Google Scholar] [CrossRef] [PubMed]
- Han, E.B.; Stevens, C.F. Development regulates a switch between post- and presynaptic strengthening in response to activity deprivation. Proc. Natl. Acad. Sci. USA 2009, 106, 10817–10822. [Google Scholar] [CrossRef] [PubMed]
- Wierenga, C.J.; Ibata, K.; Turrigiano, G.G. Postsynaptic expression of homeostatic plasticity at neocortical synapses. J. Neurosci. 2005, 25, 2895–2905. [Google Scholar] [CrossRef] [PubMed]
- Goold, C.P.; Nicoll, R.A. Single-cell optogenetic excitation drives homeostatic synaptic depression. Neuron 2010, 68, 512–528. [Google Scholar] [CrossRef] [PubMed]
- Murthy, V.N.; Schikorski, T.; Stevens, C.F.; Zhu, Y. Inactivity produces increases in neurotransmitter release and synapse size. Neuron 2001, 32, 673–682. [Google Scholar] [CrossRef]
- Ripley, B.; Otto, S.; Tiglio, K.; Williams, M.E.; Ghosh, A. Regulation of synaptic stability by AMPA receptor reverse signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.C.; Yasuda, R.; Ehlers, M.D. Metaplasticity at single glutamatergic synapses. Neuron 2010, 66, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Soler-Llavina, G.J.; Fuccillo, M.V.; Malenka, R.C.; Sudhof, T.C. Neuroligins/LRRTMs prevent activity- and Ca2+/calmodulin-dependent synapse elimination in cultured neurons. J. Cell Biol. 2011, 194, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Ultanir, S.K.; Kim, J.E.; Hall, B.J.; Deerinck, T.; Ellisman, M.; Ghosh, A. Regulation of spine morphology and spine density by NMDA receptor signaling in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 19553–19558. [Google Scholar] [CrossRef] [PubMed]
- Fremeau, R.T., Jr.; Kam, K.; Qureshi, T.; Johnson, J.; Copenhagen, D.R.; Storm-Mathisen, J.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science 2004, 304, 1815–1819. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, J.; Maia, A.S.; Camoletto, P.G.; Posthuma, G.; Roubos, E.W.; Oorschot, V.M.; Klumperman, J.; Verhage, M. Quantification of synapse formation and maintenance in vivo in the absence of synaptic release. Neuroscience 2004, 126, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Craig, A.M. Activity regulates the synaptic localization of the NMDA receptor in hippocampal neurons. Neuron 1997, 19, 801–812. [Google Scholar] [CrossRef]
- McKinney, R.A.; Luthi, A.; Bandtlow, C.E.; Gahwiler, B.H.; Thompson, S.M. Selective glutamate receptor antagonists can induce or prevent axonal sprouting in rat hippocampal slice cultures. Proc. Natl. Acad. Sci. USA 1999, 96, 11631–11636. [Google Scholar] [CrossRef] [PubMed]
- Harms, K.J.; Craig, A.M. Synapse composition and organization following chronic activity blockade in cultured hippocampal neurons. J. Comp. Neurol. 2005, 490, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Takada, N.; Yanagawa, Y.; Komatsu, Y. Activity-dependent maturation of excitatory synaptic connections in solitary neuron cultures of mouse neocortex. Eur. J. Neurosci. 2005, 21, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Lazarevic, V.; Schone, C.; Heine, M.; Gundelfinger, E.D.; Fejtova, A. Extensive remodeling of the presynaptic cytomatrix upon homeostatic adaptation to network activity silencing. J. Neurosci. 2011, 31, 10189–10200. [Google Scholar] [CrossRef] [PubMed]
- Burrone, J.; O’Byrne, M.; Murthy, V.N. Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature 2002, 420, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Sutton, M.A.; Ito, H.T.; Cressy, P.; Kempf, C.; Woo, J.C.; Schuman, E.M. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell 2006, 125, 785–799. [Google Scholar] [CrossRef] [PubMed]
- Frank, C.A.; Kennedy, M.J.; Goold, C.P.; Marek, K.W.; Davis, G.W. Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron 2006, 52, 663–677. [Google Scholar] [CrossRef] [PubMed]
- Branco, T.; Staras, K.; Darcy, K.J.; Goda, Y. Local dendritic activity sets release probability at hippocampal synapses. Neuron 2008, 59, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Hou, Q.; Zhang, D.; Jarzylo, L.; Huganir, R.L.; Man, H.Y. Homeostatic regulation of AMPA receptor expression at single hippocampal synapses. Proc. Natl. Acad. Sci. USA 2008, 105, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Thiagarajan, T.C.; Lindskog, M.; Tsien, R.W. Adaptation to synaptic inactivity in hippocampal neurons. Neuron 2005, 47, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Aoto, J.; Nam, C.I.; Poon, M.M.; Ting, P.; Chen, L. Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron 2008, 60, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Chubykin, A.A.; Atasoy, D.; Etherton, M.R.; Brose, N.; Kavalali, E.T.; Gibson, J.R.; Sudhof, T.C. Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron 2007, 54, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.; Hofer, S.B.; Pichler, B.; Buchanan, K.A.; Sjostrom, P.J.; Mrsic-Flogel, T.D. Functional specificity of local synaptic connections in neocortical networks. Nature 2011, 473, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Kerschensteiner, D.; Morgan, J.L.; Parker, E.D.; Lewis, R.M.; Wong, R.O. Neurotransmission selectively regulates synapse formation in parallel circuits in vivo. Nature 2009, 460, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Atasoy, D.; Ertunc, M.; Moulder, K.L.; Blackwell, J.; Chung, C.; Su, J.; Kavalali, E.T. Spontaneous and evoked glutamate release activates two populations of NMDA receptors with limited overlap. J. Neurosci. 2008, 28, 10151–10166. [Google Scholar] [CrossRef] [PubMed]
- Sara, Y.; Bal, M.; Adachi, M.; Monteggia, L.M.; Kavalali, E.T. Use-dependent AMPA receptor block reveals segregation of spontaneous and evoked glutamatergic neurotransmission. J. Neurosci. 2011, 31, 5378–5382. [Google Scholar] [CrossRef] [PubMed]
- Autry, A.E.; Adachi, M.; Nosyreva, E.; Na, E.S.; Los, M.F.; Cheng, P.F.; Kavalali, E.T.; Monteggia, L.M. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 2011, 475, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, V.A.; Ridenour, D.A.; Sabatini, B.L. Distinct structural and ionotropic roles of NMDA receptors in controlling spine and synapse stability. J. Neurosci. 2007, 27, 7365–7376. [Google Scholar] [CrossRef] [PubMed]
- Colonnese, M.T.; Constantine-Paton, M. Developmental period for N-methyl-d-aspartate (NMDA) receptor-dependent synapse elimination correlated with visuotopic map refinement. J. Comp. Neurol. 2006, 494, 738–751. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.; Kriegstein, A.R. GABA regulates excitatory synapse formation in the neocortex via NMDA receptor activation. J. Neurosci. 2008, 28, 5547–5558. [Google Scholar] [CrossRef] [PubMed]
- Ruthazer, E.S.; Li, J.; Cline, H.T. Stabilization of axon branch dynamics by synaptic maturation. J. Neurosci. 2006, 26, 3594–3603. [Google Scholar] [CrossRef] [PubMed]
- Luthi, A.; Schwyzer, L.; Mateos, J.M.; Gahwiler, B.H.; McKinney, R.A. NMDA receptor activation limits the number of synaptic connections during hippocampal development. Nat. Neurosci. 2001, 4, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Ohno, T.; Maeda, H.; Murabe, N.; Kamiyama, T.; Yoshioka, N.; Mishina, M.; Sakurai, M. Specific involvement of postsynaptic GluN2B-containing NMDA receptors in the developmental elimination of corticospinal synapses. Proc. Natl. Acad. Sci. USA 2010, 107, 15252–15257. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.; Zhang, X.; O’Brien, R.; Ehlers, M.D.; Huganir, R.L. Regulation of morphological postsynaptic silent synapses in developing hippocampal neurons. Nat. Neurosci. 1999, 2, 37–43. [Google Scholar] [PubMed]
- Li, J.; Erisir, A.; Cline, H. In vivo time-lapse imaging and serial section electron microscopy reveal developmental synaptic rearrangements. Neuron 2011, 69, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Nikonenko, I.; Jourdain, P.; Muller, D. Presynaptic remodeling contributes to activity-dependent synaptogenesis. J. Neurosci. 2003, 23, 8498–8505. [Google Scholar] [PubMed]
- Bacci, A.; Coco, S.; Pravettoni, E.; Schenk, U.; Armano, S.; Frassoni, C.; Verderio, C.; de Camilli, P.; Matteoli, M. Chronic blockade of glutamate receptors enhances presynaptic release and downregulates the interaction between synaptophysin-synaptobrevin-vesicle-associated membrane protein 2. J. Neurosci. 2001, 21, 6588–6596. [Google Scholar] [PubMed]
- Zhang, W.; Benson, D.L. Stages of synapse development defined by dependence on F-actin. J. Neurosci. 2001, 21, 5169–5181. [Google Scholar] [PubMed]
- Takeichi, M. The cadherin superfamily in neuronal connections and interactions. Nat. Rev. Neurosci. 2007, 8, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Sebeo, J.; Hsiao, K.; Bozdagi, O.; Dumitriu, D.; Ge, Y.; Zhou, Q.; Benson, D.L. Requirement for protein synthesis at developing synapses. J. Neurosci. 2009, 29, 9778–9793. [Google Scholar] [CrossRef] [PubMed]
- Aoki, C.; Venkatesan, C.; Go, C.G.; Mong, J.A.; Dawson, T.M. Cellular and subcellular localization of NMDA-R1 subunit immunoreactivity in the visual cortex of adult and neonatal rats. J. Neurosci. 1994, 14, 5202–5222. [Google Scholar] [PubMed]
- Berretta, N.; Jones, R.S. Tonic facilitation of glutamate release by presynaptic N-methyl-d-aspartate autoreceptors in the entorhinal cortex. Neuroscience 1996, 75, 339–344. [Google Scholar] [CrossRef]
- Fujisawa, S.; Aoki, C. In vivo blockade of N-methyl-d-aspartate receptors induces rapid trafficking of NR2B subunits away from synapses and out of spines and terminals in adult cortex. Neuroscience 2003, 121, 51–63. [Google Scholar] [CrossRef]
- Corlew, R.; Wang, Y.; Ghermazien, H.; Erisir, A.; Philpot, B.D. Developmental switch in the contribution of presynaptic and postsynaptic NMDA receptors to long-term depression. J. Neurosci. 2007, 27, 9835–9845. [Google Scholar] [CrossRef] [PubMed]
- DeBiasi, S.; Minelli, A.; Melone, M.; Conti, F. Presynaptic NMDA receptors in the neocortex are both auto- and heteroreceptors. Neuroreport 1996, 7, 2773–2776. [Google Scholar] [CrossRef] [PubMed]
- Charton, J.P.; Herkert, M.; Becker, C.M.; Schroder, H. Cellular and subcellular localization of the 2B-subunit of the NMDA receptor in the adult rat telencephalon. Brain Res. 1999, 816, 609–617. [Google Scholar] [CrossRef]
- McGuinness, L.; Taylor, C.; Taylor, R.D.; Yau, C.; Langenhan, T.; Hart, M.L.; Christian, H.; Tynan, P.W.; Donnelly, P.; Emptage, N.J. Presynaptic NMDARs in the hippocampus facilitate transmitter release at theta frequency. Neuron 2010, 68, 1109–1127. [Google Scholar] [CrossRef] [PubMed]
- Siegel, S.J.; Brose, N.; Janssen, W.G.; Gasic, G.P.; Jahn, R.; Heinemann, S.F.; Morrison, J.H. Regional, cellular, and ultrastructural distribution of N-methyl-d-aspartate receptor subunit 1 in monkey hippocampus. Proc. Natl. Acad. Sci. USA 1994, 91, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Jourdain, P.; Bergersen, L.H.; Bhaukaurally, K.; Bezzi, P.; Santello, M.; Domercq, M.; Matute, C.; Tonello, F.; Gundersen, V.; Volterra, A. Glutamate exocytosis from astrocytes controls synaptic strength. Nat. Neurosci. 2007, 10, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Aoki, C.; Fujisawa, S.; Mahadomrongkul, V.; Shah, P.J.; Nader, K.; Erisir, A. NMDA receptor blockade in intact adult cortex increases trafficking of NR2A subunits into spines, postsynaptic densities, and axon terminals. Brain Res. 2003, 963, 139–149. [Google Scholar] [CrossRef]
- Gill, I.; Droubi, S.; Giovedi, S.; Fedder, K.N.; Bury, L.A.; Bosco, F.; Sceniak, M.P.; Benfenati, F.; Sabo, S.L. Presynaptic NMDA receptors—Dynamics and distribution in developing axons in vitro and in vivo. J. Cell Sci. 2015, 128, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Herkert, M.; Rottger, S.; Becker, C.M. The NMDA receptor subunit NR2B of neonatal rat brain: Complex formation and enrichment in axonal growth cones. Eur. J. Neurosci. 1998, 10, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, M.D.; Fung, E.T.; O’Brien, R.J.; Huganir, R.L. Splice variant-specific interaction of the NMDA receptor subunit NR1 with neuronal intermediate filaments. J. Neurosci. 1998, 18, 720–730. [Google Scholar] [PubMed]
- Song, A.H.; Wang, D.; Chen, G.; Li, Y.; Luo, J.; Duan, S.; Poo, M.M. A selective filter for cytoplasmic transport at the axon initial segment. Cell 2009, 136, 1148–1160. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.Y.; Petralia, R.S.; Wang, Y.X.; Wenthold, R.J.; Brenowitz, S.D. Functional NMDA receptors at axonal growth cones of young hippocampal neurons. J. Neurosci. 2011, 31, 9289–9297. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Popescu, A.; Poo, M.M. Essential role of presynaptic NMDA receptors in activity-dependent BDNF secretion and corticostriatal LTP. Neuron 2014, 84, 1009–1022. [Google Scholar] [CrossRef] [PubMed]
- Sjostrom, P.J.; Turrigiano, G.G.; Nelson, S.B. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 2003, 39, 641–654. [Google Scholar] [CrossRef]
- Brasier, D.J.; Feldman, D.E. Synapse-specific expression of functional presynaptic NMDA receptors in rat somatosensory cortex. J. Neurosci. 2008, 28, 2199–2211. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, K.A.; Blackman, A.V.; Moreau, A.W.; Elgar, D.; Costa, R.P.; Lalanne, T.; Tudor Jones, A.A.; Oyrer, J.; Sjostrom, P.J. Target-specific expression of presynaptic NMDA receptors in neocortical microcircuits. Neuron 2012, 75, 451–466. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Moreno, A.; Paulsen, O. Spike timing-dependent long-term depression requires presynaptic NMDA receptors. Nat. Neurosci. 2008, 11, 744–745. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Moreno, A.; Kohl, M.M.; Reeve, J.E.; Eaton, T.R.; Collins, H.A.; Anderson, H.L.; Paulsen, O. Presynaptic induction and expression of timing-dependent long-term depression demonstrated by compartment-specific photorelease of a use-dependent NMDA receptor antagonist. J. Neurosci. 2011, 31, 8564–8569. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Gonzalez-Rueda, A.; Sampaio-Baptista, C.; Paulsen, O.; Rodriguez-Moreno, A. Distinct mechanisms of spike timing-dependent LTD at vertical and horizontal inputs onto L2/3 pyramidal neurons in mouse barrel cortex. Physiol. Rep. 2014. [Google Scholar] [CrossRef] [PubMed]
- Cochilla, A.J.; Alford, S. NMDA receptor-mediated control of presynaptic calcium and neurotransmitter release. J. Neurosci. 1999, 19, 193–205. [Google Scholar] [PubMed]
- Woodhall, G.; Evans, D.I.; Cunningham, M.O.; Jones, R.S. NR2B-containing NMDA autoreceptors at synapses on entorhinal cortical neurons. J. Neurophysiol. 2001, 86, 1644–1651. [Google Scholar] [PubMed]
- Mameli, M.; Carta, M.; Partridge, L.D.; Valenzuela, C.F. Neurosteroid-induced plasticity of immature synapses via retrograde modulation of presynaptic NMDA receptors. J. Neurosci. 2005, 25, 2285–2294. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Vicini, S.; Hsu, F.C.; Doshi, S.; Takano, H.; Coulter, D.A.; Lynch, D.R. Axonal alpha7 nicotinic ACh receptors modulate presynaptic NMDA receptor expression and structural plasticity of glutamatergic presynaptic boutons. Proc. Natl. Acad. Sci. USA 2010, 107, 16661–16666. [Google Scholar] [CrossRef] [PubMed]
- Kunz, P.A.; Roberts, A.C.; Philpot, B.D. Presynaptic NMDA receptor mechanisms for enhancing spontaneous neurotransmitter release. J. Neurosci. 2013, 33, 7762–7769. [Google Scholar] [CrossRef] [PubMed]
- Bender, V.A.; Bender, K.J.; Brasier, D.J.; Feldman, D.E. Two coincidence detectors for spike timing-dependent plasticity in somatosensory cortex. J. Neurosci. 2006, 26, 4166–4177. [Google Scholar] [CrossRef] [PubMed]
- Nevian, T.; Sakmann, B. Spine Ca2+ signaling in spike-timing-dependent plasticity. J. Neurosci. 2006, 26, 11001–11013. [Google Scholar] [CrossRef] [PubMed]
- Min, R.; Nevian, T. Astrocyte signaling controls spike timing-dependent depression at neocortical synapses. Nat. Neurosci. 2012, 15, 746–753. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Moreno, A.; Gonzalez-Rueda, A.; Banerjee, A.; Upton, A.L.; Craig, M.T.; Paulsen, O. Presynaptic self-depression at developing neocortical synapses. Neuron 2013, 77, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Corlew, R.; Brasier, D.J.; Feldman, D.E.; Philpot, B.D. Presynaptic NMDA receptors: Newly appreciated roles in cortical synaptic function and plasticity. Neuroscientist 2008, 14, 609–625. [Google Scholar] [CrossRef] [PubMed]
- Isaac, J.T.; Nicoll, R.A.; Malenka, R.C. Evidence for silent synapses: Implications for the expression of LTP. Neuron 1995, 15, 427–434. [Google Scholar] [CrossRef]
- Isaac, J.T.; Nicoll, R.A.; Malenka, R.C. Silent glutamatergic synapses in the mammalian brain. Can. J. Physiol. Pharmacol. 1999, 77, 735–737. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, P.S.; Mulle, C. Presynaptic glutamate receptors: Physiological functions and mechanisms of action. Nat. Rev. Neurosci. 2008, 9, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Urban-Ciecko, J.; Wen, J.A.; Parekh, P.K.; Barth, A.L. Experience-dependent regulation of presynaptic NMDARs enhances neurotransmitter release at neocortical synapses. Learn. Mem. 2015, 22, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Larsen, R.S.; Smith, I.T.; Miriyala, J.; Han, J.E.; Corlew, R.J.; Smith, S.L.; Philpot, B.D. Synapse-specific control of experience-dependent plasticity by presynaptic NMDA receptors. Neuron 2014, 83, 879–893. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Pallas, S.L. NMDA antagonists in the superior colliculus prevent developmental plasticity but not visual transmission or map compression. J. Neurophysiol. 2001, 86, 1179–1194. [Google Scholar] [PubMed]
- Simon, D.K.; Prusky, G.T.; O’Leary, D.D.; Constantine-Paton, M. N-methyl-d-aspartate receptor antagonists disrupt the formation of a mammalian neural map. Proc. Natl. Acad. Sci. USA 1992, 89, 10593–10597. [Google Scholar] [CrossRef] [PubMed]
- Ramoa, A.S.; Mower, A.F.; Liao, D.; Jafri, S.I. Suppression of cortical NMDA receptor function prevents development of orientation selectivity in the primary visual cortex. J. Neurosci. 2001, 21, 4299–4309. [Google Scholar] [PubMed]
- Bury, L.A.; Sabo, S.L. How it’s made: The synapse. Mol. Interv. 2010, 10, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Bourgeron, T. A synaptic trek to autism. Curr. Opin. Neurobiol. 2009, 19, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Huttenlocher, P.R.; Dabholkar, A.S. Regional differences in synaptogenesis in human cerebral cortex. J. Comp. Neurol. 1997, 387, 167–178. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiplex Targeted Sequencing Identifies Recurrently Mutated Genes in Autism Spectrum Disorders. Science 2012, 338, 1619–1622. [Google Scholar] [CrossRef] [PubMed]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Belmonte, M.K.; Allen, G.; Beckel-Mitchener, A.; Boulanger, L.M.; Carper, R.A.; Webb, S.J. Autism and abnormal development of brain connectivity. J. Neurosci. 2004, 24, 9228–9231. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Anderson, C.T.; Levitt, P.; Shepherd, G.M. Circuit-specific intracortical hyperconnectivity in mice with deletion of the autism-associated Met receptor tyrosine kinase. J. Neurosci. 2011, 31, 5855–5864. [Google Scholar] [CrossRef] [PubMed]
- Jamain, S.; Quach, H.; Betancur, C.; Rastam, M.; Colineaux, C.; Gillberg, I.C.; Soderstrom, H.; Giros, B.; Leboyer, M.; Gillberg, C.; et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003, 34, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Oliveira, G.; Coutinho, A.; Yang, C.; Feng, J.; Katz, C.; Sram, J.; Bockholt, A.; Jones, I.R.; Craddock, N.; et al. Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol. Psychiatry 2005, 10, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Talebizadeh, Z.; Lam, D.Y.; Theodoro, M.F.; Bittel, D.C.; Lushington, G.H.; Butler, M.G. Novel splice isoforms for NLGN3 and NLGN4 with possible implications in autism. J. Med. Genet. 2006. [Google Scholar] [CrossRef] [PubMed]
- Geschwind, D.H.; Levitt, P. Autism spectrum disorders: Developmental disconnection syndromes. Curr. Opin. Neurobiol. 2007, 17, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Helmeke, C.; Ovtscharoff, W., Jr.; Poeggel, G.; Braun, K. Juvenile emotional experience alters synaptic inputs on pyramidal neurons in the anterior cingulate cortex. Cereb. Cortex 2001, 11, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Poeggel, G.; Helmeke, C.; Abraham, A.; Schwabe, T.; Friedrich, P.; Braun, K. Juvenile emotional experience alters synaptic composition in the rodent cortex, hippocampus, and lateral amygdala. Proc. Natl. Acad. Sci. USA 2003, 100, 16137–16142. [Google Scholar] [CrossRef] [PubMed]
- Breslau, N.; Schultz, L.; Peterson, E. Sex differences in depression: A role for preexisting anxiety. Psychiatry Res. 1995, 58, 1–12. [Google Scholar] [CrossRef]
- Parker, G.; Wilhelm, K.; Mitchell, P.; Austin, M.P.; Roussos, J.; Gladstone, G. The influence of anxiety as a risk to early onset major depression. J. Affect. Disord. 1999, 52, 11–17. [Google Scholar] [CrossRef]
- Weissman, M.M.; Wickramaratne, P.; Nomura, Y.; Warner, V.; Verdeli, H.; Pilowsky, D.J.; Grillon, C.; Bruder, G. Families at high and low risk for depression: A 3-generation study. Arch. Gen. Psychiatry 2005, 62, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Leonardo, E.D.; Hen, R. Anxiety as a developmental disorder. Neuropsychopharmacology 2008, 33, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Endele, S.; Rosenberger, G.; Geider, K.; Popp, B.; Tamer, C.; Stefanova, I.; Milh, M.; Kortum, F.; Fritsch, A.; Pientka, F.K.; et al. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat. Genet. 2010, 42, 1021–1026. [Google Scholar] [CrossRef] [PubMed]
- Tarabeux, J.; Kebir, O.; Gauthier, J.; Hamdan, F.F.; Xiong, L.; Piton, A.; Spiegelman, D.; Henrion, E.; Millet, B.; Fathalli, F.; et al. Rare mutations in N-methyl-d-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl. Psychiatry 2011. [Google Scholar] [CrossRef] [PubMed]
- Autism Genome Project, C.; Szatmari, P.; Paterson, A.D.; Zwaigenbaum, L.; Roberts, W.; Brian, J.; Liu, X.Q.; Vincent, J.B.; Skaug, J.L.; Thompson, A.P.; et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 2007, 39, 319–328. [Google Scholar]
- Gai, X.; Xie, H.M.; Perin, J.C.; Takahashi, N.; Murphy, K.; Wenocur, A.S.; D’Arcy, M.; O’Hara, R.J.; Goldmuntz, E.; Grice, D.E.; et al. Rare structural variation of synapse and neurotransmission genes in autism. Mol. Psychiatry 2012, 17, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, F.F.; Gauthier, J.; Araki, Y.; Lin, D.T.; Yoshizawa, Y.; Higashi, K.; Park, A.R.; Spiegelman, D.; Dobrzeniecka, S.; Piton, A.; et al. Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am. J. Hum. Genet. 2011, 88, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Purcell, A.E.; Jeon, O.H.; Zimmerman, A.W.; Blue, M.E.; Pevsner, J. Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology 2001, 57, 1618–1628. [Google Scholar] [CrossRef] [PubMed]
- Blatt, G.J.; Fitzgerald, C.M.; Guptill, J.T.; Booker, A.B.; Kemper, T.L.; Bauman, M.L. Density and distribution of hippocampal neurotransmitter receptors in autism: An autoradiographic study. J. Autism Dev. Disord. 2001, 31, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Eadie, B.D.; Cushman, J.; Kannangara, T.S.; Fanselow, M.S.; Christie, B.R. NMDA receptor hypofunction in the dentate gyrus and impaired context discrimination in adult Fmr1 knockout mice. Hippocampus 2012, 22, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Maliszewska-Cyna, E.; Bawa, D.; Eubanks, J.H. Diminished prevalence but preserved synaptic distribution of N-methyl-d-aspartate receptor subunits in the methyl CpG binding protein 2(MeCP2)-null mouse brain. Neuroscience 2010, 168, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Bangash, M.A.; Park, J.M.; Melnikova, T.; Wang, D.; Jeon, S.K.; Lee, D.; Syeda, S.; Kim, J.; Kouser, M.; Schwartz, J.; et al. Enhanced polyubiquitination of Shank3 and NMDA receptor in a mouse model of autism. Cell 2011, 145, 758–772. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, T.; Kulangara, K.; Antoniello, K.; Markram, H. Elevated NMDA receptor levels and enhanced postsynaptic long-term potentiation induced by prenatal exposure to valproic acid. Proc. Natl. Acad. Sci. USA 2007, 104, 13501–13506. [Google Scholar] [CrossRef] [PubMed]
- Etherton, M.; Foldy, C.; Sharma, M.; Tabuchi, K.; Liu, X.; Shamloo, M.; Malenka, R.C.; Sudhof, T.C. Autism-linked neuroligin-3 R451C mutation differentially alters hippocampal and cortical synaptic function. Proc. Natl. Acad. Sci. USA 2011, 108, 13764–13769. [Google Scholar] [CrossRef] [PubMed]
- Sceniak, M.P.; Lang, M.; Enomoto, A.C.; James Howell, C.; Hermes, D.J.; Katz, D.M. Mechanisms of functional hypoconnectivity in the medial prefrontal cortex of Mecp2 null mice. Cereb. Cortex 2015. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, P.R.; Lahiri, S.; Rajamma, U. Glutamate mediated signaling in the pathophysiology of autism spectrum disorders. Pharmacol. Biochem. Behav. 2012, 100, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Chez, M.G.; Burton, Q.; Dowling, T.; Chang, M.; Khanna, P.; Kramer, C. Memantine as adjunctive therapy in children diagnosed with autistic spectrum disorders: An observation of initial clinical response and maintenance tolerability. J. Child Neurol. 2007, 22, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Kron, M.; Howell, C.J.; Adams, I.T.; Ransbottom, M.; Christian, D.; Ogier, M.; Katz, D.M. Brain activity mapping in Mecp2 mutant mice reveals functional deficits in forebrain circuits, including key nodes in the default mode network, that are reversed with ketamine treatment. J. Neurosci. 32, 13860–13872. [CrossRef] [PubMed]
- Alkondon, M.; Costa, A.C.; Radhakrishnan, V.; Aronstam, R.S.; Albuquerque, E.X. Selective blockade of NMDA-activated channel currents may be implicated in learning deficits caused by lead. FEBS Lett. 1990, 261, 124–130. [Google Scholar] [CrossRef]
- Gavazzo, P.; Zanardi, I.; Baranowska-Bosiacka, I.; Marchetti, C. Molecular determinants of Pb2+ interaction with NMDA receptor channels. Neurochem. Int. 2008, 52, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Neal, A.P.; Worley, P.F.; Guilarte, T.R. Lead exposure during synaptogenesis alters NMDA receptor targeting via NMDA receptor inhibition. Neurotoxicology 2011, 32, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Omelchenko, I.A.; Nelson, C.S.; Allen, C.N. Lead inhibition of N-methyl-d-aspartate receptors containing NR2A, NR2C and NR2D subunits. J. Pharmacol. Exp. Ther. 1997, 282, 1458–1464. [Google Scholar] [PubMed]
- Ujihara, H.; Albuquerque, E.X. Developmental change of the inhibition by lead of NMDA-activated currents in cultured hippocampal neurons. J. Pharmacol. Exp. Ther. 1992, 263, 868–875. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fedder, K.N.; Sabo, S.L. On the Role of Glutamate in Presynaptic Development: Possible Contributions of Presynaptic NMDA Receptors. Biomolecules 2015, 5, 3448-3466. https://doi.org/10.3390/biom5043448
Fedder KN, Sabo SL. On the Role of Glutamate in Presynaptic Development: Possible Contributions of Presynaptic NMDA Receptors. Biomolecules. 2015; 5(4):3448-3466. https://doi.org/10.3390/biom5043448
Chicago/Turabian StyleFedder, Karlie N., and Shasta L. Sabo. 2015. "On the Role of Glutamate in Presynaptic Development: Possible Contributions of Presynaptic NMDA Receptors" Biomolecules 5, no. 4: 3448-3466. https://doi.org/10.3390/biom5043448
APA StyleFedder, K. N., & Sabo, S. L. (2015). On the Role of Glutamate in Presynaptic Development: Possible Contributions of Presynaptic NMDA Receptors. Biomolecules, 5(4), 3448-3466. https://doi.org/10.3390/biom5043448