
High Intrinsic Aerobic Capacity Protects against Ethanol-Induced Hepatic Injury and Metabolic Dysfunction: Study Using High Capacity Runner Rat Model



Abstract
:1. Introduction
2. Results
2.1. Animal Characteristics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | HCR-C | HCR-E |
|---|---|---|
| Body Weight (grams) | 311.1 ± 11.3 | 326.7 ± 11.6 |
| Fat Pad Weight (grams) | 21.5 ± 2.2 | 19.7 ± 1.8 |
| Food consumption (g/week) | 53 ± 2 | 53.1 ± 2.1 |
| Serum Glucose (mg/mL) | 195.8 ± 8.7 | 183.7 ± 9.2 |
| Serum Insulin (ng/mL) | 4.3 ± 0.5 | 4.9 ± 0.5 |
| Serum Free Fatty Acids (umol/L) | 285.4 ± 28.7 | 298.1 ± 27.9 |
| Serum Triglycerides (mg/dL) | 127.7 ± 3.6 | 123.3 ± 3.9 |
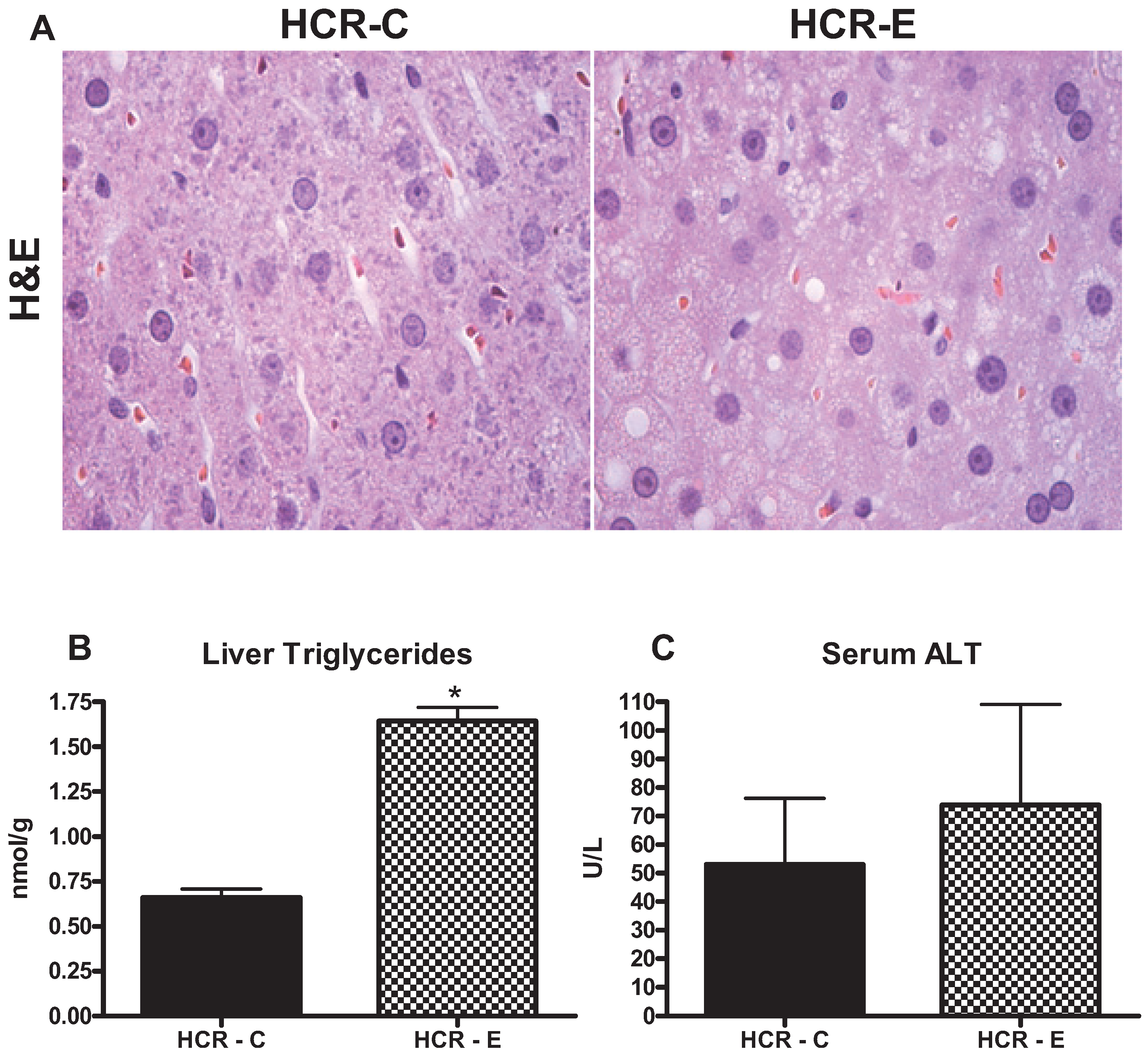
2.2. Hepatic Steatosis and Injury
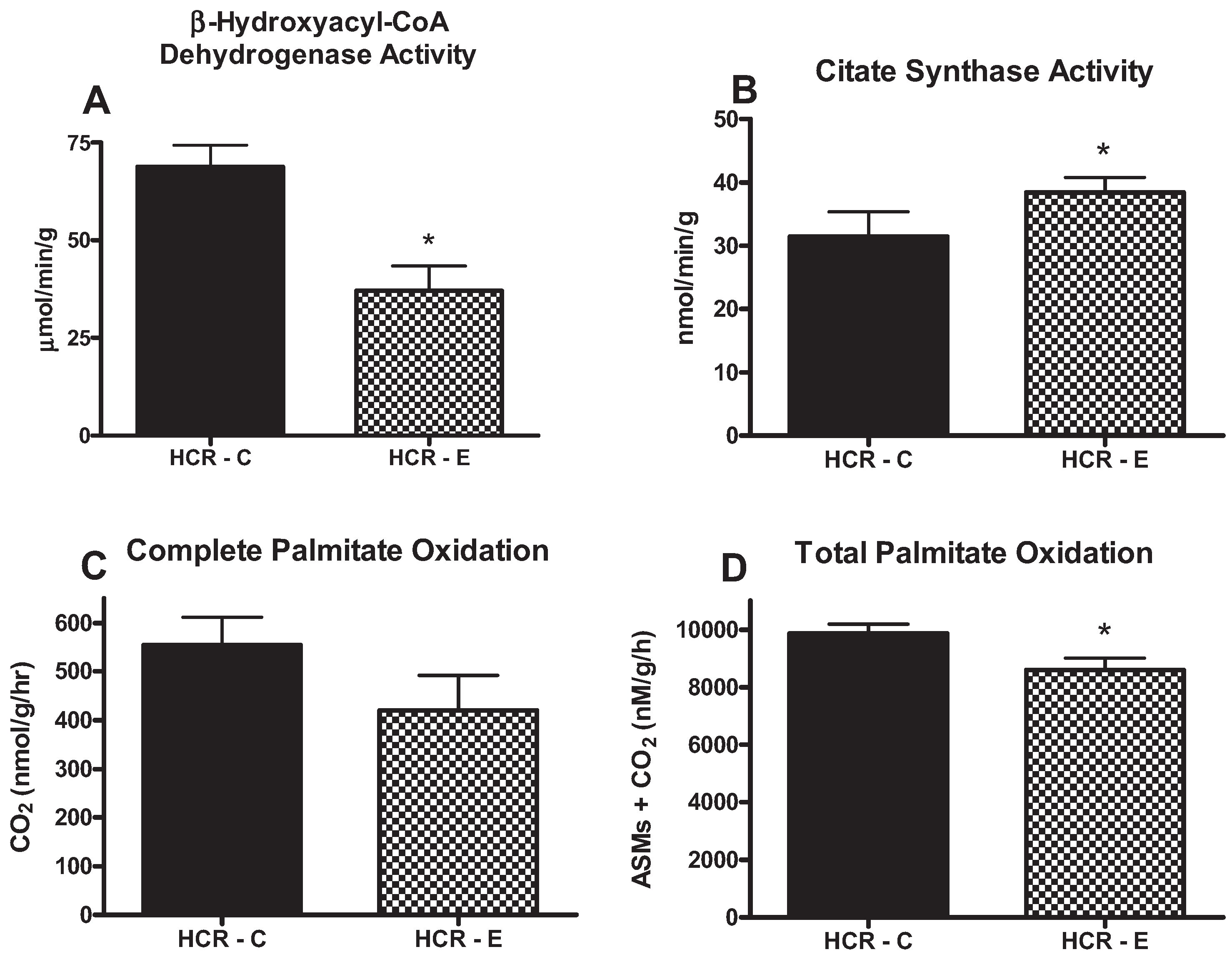
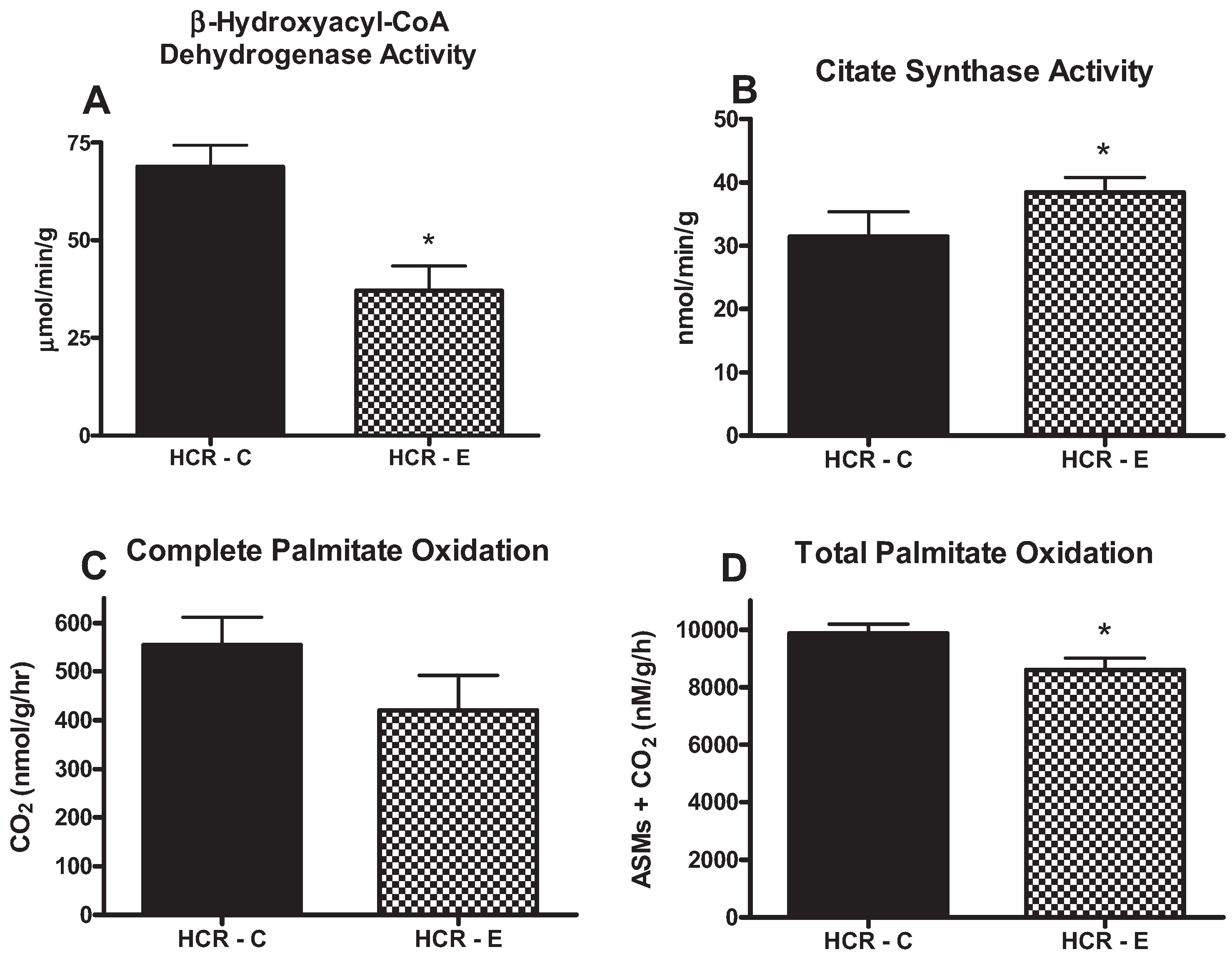
2.3. Fatty Acid Oxidation and Hepatic Mitochondrial Enzyme Activities
2.4. Markers of Oxidative Stress
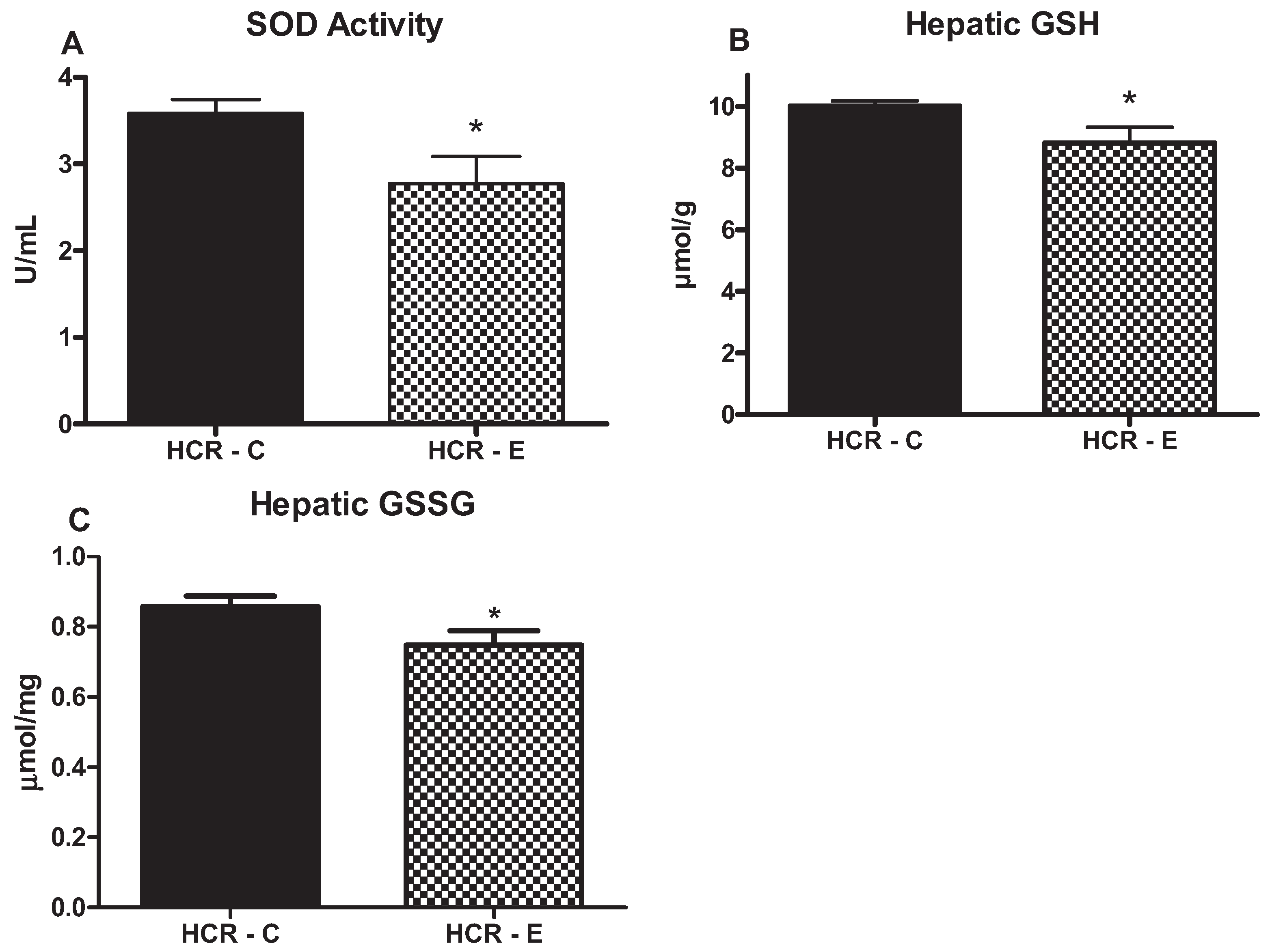
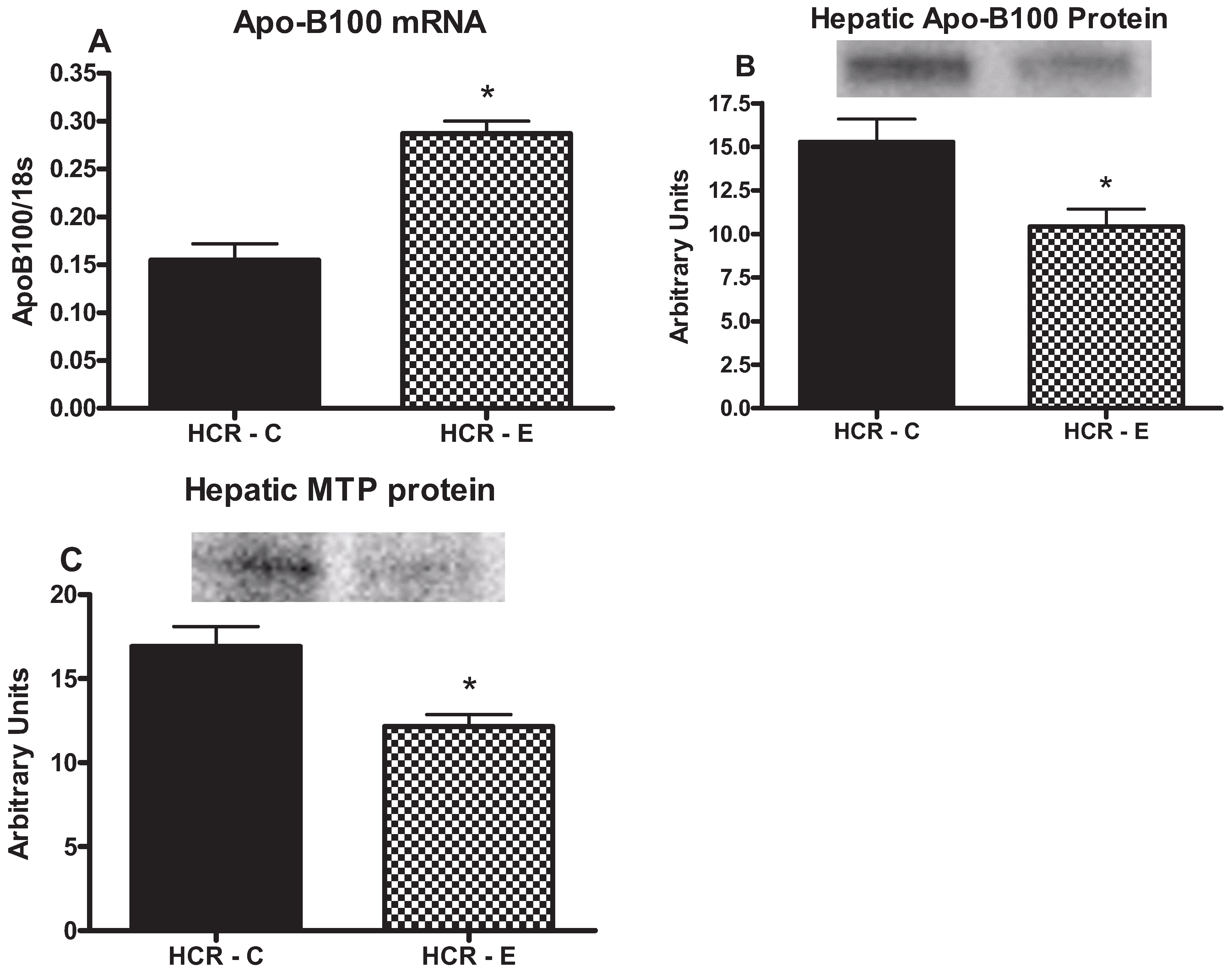
2.5. Markers of TG Export
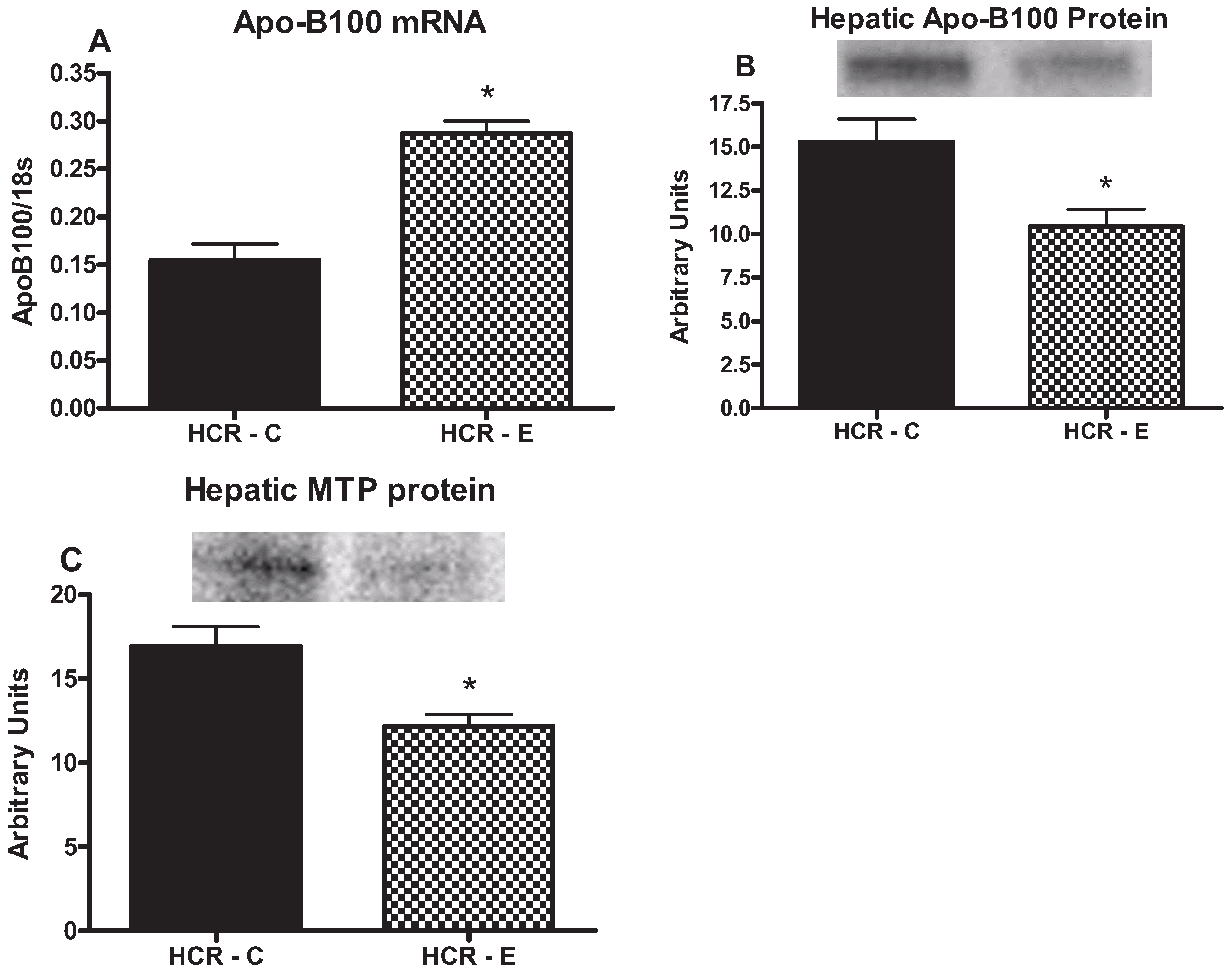
3. Discussion
4. Experimental Section
4.1. Animal Strains and Protocols
4.2. Tissue Homogenization
4.3. Fatty Acid Oxidation
4.4. Hepatic Lipid Content
4.5. Western Blotting
4.6. Serum Assays
4.7. H & E and Staining of Liver Sections, SOD Activity and Glutathione Assays
4.8. Real-Time PCR
4.9. Hepatic Mitchondrial Enzymes
4.10. Statistics
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- CDC. Alcohol-Attributable Deaths and Years of Potential Life Lost—United States, 2001. Morb. Mortal. Wkly. Rep. 2004, 53, 866–870. [Google Scholar]
- Brunt, E.M.; Tiniakos, D.G. Histopathology of nonalcoholic fatty liver disease. World J. Gastroenterol. 2010, 16, 5286–5296. [Google Scholar] [CrossRef] [PubMed]
- Coleman, W.B.; Cunningham, C.C. Effect of chronic ethanol consumption on hepatic mitochondrial transcription and translation. Biochim. Biophys. Acta 1991, 1058, 178–186. [Google Scholar] [CrossRef]
- Church, T.; Kuk, J.L.; Ross, R.; Priest, E.L.; Biltoft, E.; Blair, S.N. Association of cardiorespiratory fitness, body mass index, and waist circumference to nonalcoholic fatty liver disease. Gastroenterology 2006, 130, 2023–2030. [Google Scholar] [CrossRef] [PubMed]
- McMillan, K.P.; Kuk, J.L.; Church, T.S.; Blair, S.N.; Ross, R. Independent associations between liver fat, visceral adipose tissue, and metabolic risk factors in men. Appl. Physiol. Nutr. Metab. 2007, 32, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.; Prakash, M.; Froelicher, V.; Do, D.; Partington, S.; Atwood, J.E. Exercise capacity and mortality among men referred for exercise testing. N. Engl. J. Med. 2002, 346, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Fromenty, B.; Vadrot, N.; Massart, J.; Turlin, B.; Barri-Ova, N.; Lettéron, P.; Fautrel, A.; Robin, M.A. Chronic ethanol consumption lessens the gain of body weight, liver triglycerides, and diabetes in obese ob/ob mice. J. Pharmacol. Exp. Ther. 2009, 331, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ruiz, I.; Rodriguez-Juan, C.; Diaz-Sanjuan, T.; del Hoyo, P.; Colina, F.; Muñoz-Yagüe, T.; Solís-Herruzo, J.A. Uric acid and anti-TNF antibody improve mitochondrial dysfunction in ob/ob mice. Hepatology 2006, 44, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Wisloff, U.; Najjar, S.M.; Ellingsen, O.; Haram, P.M.; Swoap, S.; Al-Share, Q.; Fernström, M.; Rezaei, K.; Lee, S.J.; Koch, L.G.; et al. Cardiovascular risk factors emerge after artificial selection for low aerobic capacity. Science 2005, 307, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Noland, R.C.; Thyfault, J.P.; Henes, S.T.; Whitfield, B.R.; Woodlief, T.L.; Evans, J.R.; Lust, J.A.; Britton, S.L.; Koch, L.G.; Dudek, R.W.; et al. Artificial selection for high-capacity endurance running is protective against high-fat diet-induced insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E31–E41. [Google Scholar] [CrossRef] [PubMed]
- Thyfault, J.P.; Rector, R.S.; Uptergrove, G.M.; Borengasser, S.J.; Morris, E.M.; Wei, Y.; Laye, M.J.; Burant, C.F.; Qi, N.R.; Ridenhour, S.E.; et al. Rats selectively bred for low aerobic capacity have reduced hepatic mitochondrial oxidative capacity and susceptibility to hepatic steatosis and injury. J. Physiol. 2009, 587, 1805–1816. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.M.; Whaley-Connell, A.T.; Thyfault, J.P.; Britton, S.L.; Koch, L.G.; Wei, Y.; Ibdah, J.A.; Sowers, J.R. Low aerobic capacity and high-fat diet contribute to oxidative stress and IRS-1 degradation in the kidney. Am. J. Nephrol. 2009, 30, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Rector, R.S.; Thyfault, J.P.; Morris, R.T.; Laye, M.J.; Borengasser, S.J.; Booth, F.W.; Ibdah, J.A. Daily exercise increases hepatic fatty acid oxidation and prevents steatosis in Otsuka Long-Evans Tokushima Fatty rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G619–G626. [Google Scholar] [CrossRef] [PubMed]
- Rector, R.S.; Thyfault, J.P.; Laye, M.J.; Morris, R.T.; Borengasser, S.J.; Uptergrove, G.M.; Chakravarthy, M.V.; Booth, F.W.; Ibdah, J.A. Cessation of daily exercise dramatically alters precursors of hepatic steatosis in Otsuka Long-Evans Tokushima Fatty (OLETF) rats. J. Physiol. 2008, 586, 4241–4249. [Google Scholar] [CrossRef] [PubMed]
- Laye, M.J.; Rector, R.S.; Warner, S.O.; Naples, S.P.; Perretta, A.L.; Uptergrove, G.M.; Laughlin, M.H.; Thyfault, J.P.; Booth, F.W.; Ibdah, J.A. Changes in visceral adipose tissue mitochondrial content with type 2 diabetes and daily voluntary wheel running in OLETF rats. J. Physiol. 2009, 587, 3729–3739. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M.; Yeon, J.E.; Tong, M.; Longato, L.; Chaudhry, R.; Pang, M.Y.; Duan, K.; Wands, J.R. Insulin resistance in experimental alcohol-induced liver disease. J. Gastroenterol. Hepatol. 2008, 23, e477–e486. [Google Scholar] [CrossRef] [PubMed]
- Rector, R.S.; Thyfault, J.P.; Uptergrove, G.M.; Morris, E.M.; Naples, S.P.; Borengasser, S.J.; Mikus, C.R.; Laye, M.J.; Laughlin, M.H.; Booth, F.W.; et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J. Hepatol. 2010, 52, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Pang, M.; de la Monte, S.M.; Longato, L.; Tong, M.; He, J.; Chaudhry, R.; Duan, K.; Ouh, J.; Wands, J.R. PPARdelta agonist attenuates alcohol-induced hepatic insulin resistance and improves liver injury and repair. J. Hepatol. 2009, 50, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Tabouy, L.; Zamora, A.J.; Oliva, L.; Montet, A.M.; Beauge, F.; Montet, J.C. Ursodeoxycholate protects against ethanol-induced liver mitochondrial injury. Life Sci. 1998, 6, 2259–2270. [Google Scholar] [CrossRef]
- Cunningham, C.C.; Coleman, W.B.; Spach, P.I. The effects of chronic ethanol consumption on hepatic mitochondrial energy metabolism. Alcohol. Alcohol. 1990, 25, 127–136. [Google Scholar] [PubMed]
- Kukielka, E.; Dicker, E.; Cederbaum, A.I. Increased production of reactive oxygen species by rat liver mitochondria after chronic ethanol treatment. Arch. Biochem. Biophys. 1994, 309, 377–386. [Google Scholar] [CrossRef] [PubMed]
- McVicker, B.L.; Tuma, P.L.; Kharbanda, K.K.; Lee, S.M.; Tuma, D.J. Relationship between oxidative stress and hepatic glutathione levels in ethanol-mediated apoptosis of polarized hepatic cells. World J. Gastroenterol. 2009, 15, 2609–2616. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, M.D.; Nakagami, M.; Bradford, B.U.; Uesugi, T.; Mason, R.P.; Connor, H.D.; Dikalova, A.; Kadiiska, M. Thurman RG Overexpression of manganese superoxide dismutase prevents alcohol-induced liver injury in the rat. J. Biol. Chem. 2001, 276, 36664–36672. [Google Scholar] [CrossRef] [PubMed]
- Grunnet, N.; Kondrup, J. The effect of ethanol on the beta-oxidation of fatty acids. Alcohol. Clin. Exp. Res. 1986, 10, 64S–68S. [Google Scholar] [CrossRef] [PubMed]
- Hoek, J.B.; Cahill, A.; Pastorino, J.G. Alcohol and mitochondria: A dysfunctional relationship. Gastroenterology 2002, 122, 2049–2063. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Alcoholic fatty liver: Its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol 2004, 34, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Lakshman, M.R. Some novel insights into the pathogenesis of alcoholic steatosis. Alcohol 2004, 34, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Brinton, E.A. Effects of ethanol intake on lipoproteins and atherosclerosis. Curr. Opin. Lipidol. 2010, 21, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; Ibdah, J.A. Role of mitochondria in alcoholic liver disease. World J. Gastroenterol. 2014, 20, 2136–2142. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, G.; Scopacasa, F.; Colao, A.; Capone, D.; Tarantino, M.; Grimaldi, E.; Savastano, S. Serum Bcl-2 concentrations in overweight-obese subjects with nonalcoholic fatty liver disease. World J. Gastroenterol. 2011, 17, 5280–5288. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, G.; Colao, A.; Capone, D.; Conca, P.; Tarantino, M.; Grimaldi, E.; Chianese, D.; Finelli, C.; Contaldo, F.; Scopacasa, F.; et al. Circulating levels of cytochrome C, gamma-glutamyl transferase, triglycerides and unconjugated bilirubin in overweight/obese patients with non-alcoholic fatty liver disease. J. Biol. Regul. Homeost. Agents 2011, 25, 47–56. [Google Scholar] [PubMed]
- Bakillah, A.; Hussain, M.M. Binding of microsomal triglyceride transfer protein to lipids results in increased affinity for apolipoprotein B: Evidence for stable microsomal MTP-lipid complexes. J. Biol. Chem. 2001, 276, 31466–31473. [Google Scholar] [CrossRef] [PubMed]
- Cahill, A.; Sykora, P. Alcoholic liver disease and the mitochondrial ribosome: Methods of analysis. Methods Mol. Biol. 2008, 447, 381–394. [Google Scholar] [PubMed]
- Lin, M.C.; Li, J.J.; Wang, E.J.; Princler, G.L.; Kauffman, F.C.; Kung, H.F. Ethanol down-regulates the transcription of microsomal triglyceride transfer protein gene. FASEB J. 1997, 11, 1145–1152. [Google Scholar] [PubMed]
- Sugimoto, T.; Yamashita, S.; Ishigami, M.; Sakai, N.; Hirano, K.; Tahara, M.; Matsumoto, K.; Nakamura, T.; Matsuzawa, Y. Decreased microsomal triglyceride transfer protein activity contributes to initiation of alcoholic liver steatosis in rats. J. Hepatol. 2002, 36, 157–162. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szary, N.; Rector, R.S.; Uptergrove, G.M.; Ridenhour, S.E.; Shukla, S.D.; Thyfault, J.P.; Koch, L.G.; Britton, S.L.; Ibdah, J.A. High Intrinsic Aerobic Capacity Protects against Ethanol-Induced Hepatic Injury and Metabolic Dysfunction: Study Using High Capacity Runner Rat Model. Biomolecules 2015, 5, 3295-3308. https://doi.org/10.3390/biom5043295
Szary N, Rector RS, Uptergrove GM, Ridenhour SE, Shukla SD, Thyfault JP, Koch LG, Britton SL, Ibdah JA. High Intrinsic Aerobic Capacity Protects against Ethanol-Induced Hepatic Injury and Metabolic Dysfunction: Study Using High Capacity Runner Rat Model. Biomolecules. 2015; 5(4):3295-3308. https://doi.org/10.3390/biom5043295
Chicago/Turabian StyleSzary, Nicholas, R. Scott Rector, Grace M. Uptergrove, Suzanne E. Ridenhour, Shivendra D. Shukla, John P. Thyfault, Lauren G. Koch, Steven L. Britton, and Jamal A. Ibdah. 2015. "High Intrinsic Aerobic Capacity Protects against Ethanol-Induced Hepatic Injury and Metabolic Dysfunction: Study Using High Capacity Runner Rat Model" Biomolecules 5, no. 4: 3295-3308. https://doi.org/10.3390/biom5043295
APA StyleSzary, N., Rector, R. S., Uptergrove, G. M., Ridenhour, S. E., Shukla, S. D., Thyfault, J. P., Koch, L. G., Britton, S. L., & Ibdah, J. A. (2015). High Intrinsic Aerobic Capacity Protects against Ethanol-Induced Hepatic Injury and Metabolic Dysfunction: Study Using High Capacity Runner Rat Model. Biomolecules, 5(4), 3295-3308. https://doi.org/10.3390/biom5043295