
In Vivo Acute on Chronic Ethanol Effects in Liver: A Mouse Model Exhibiting Exacerbated Injury, Altered Metabolic and Epigenetic Responses


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
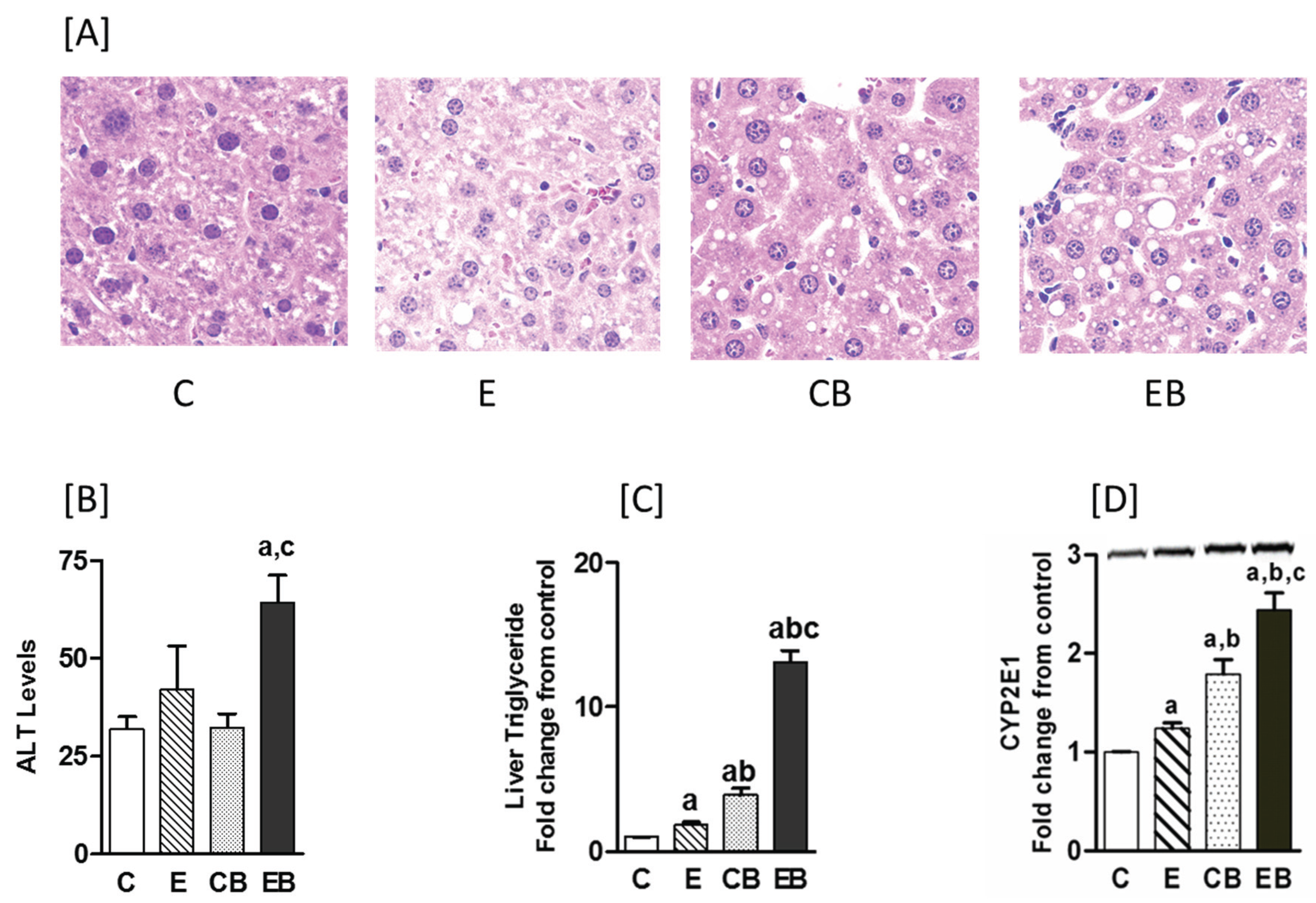
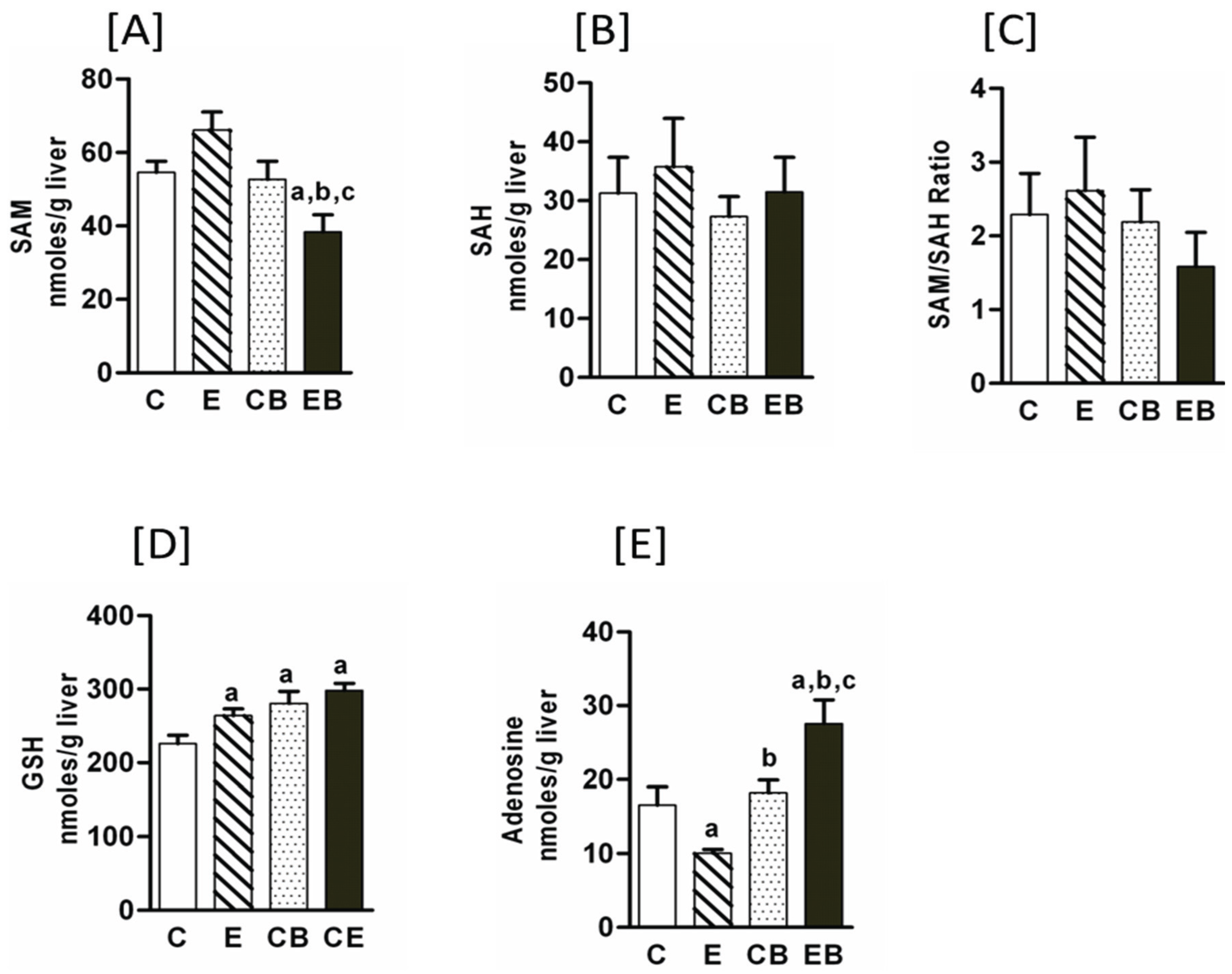
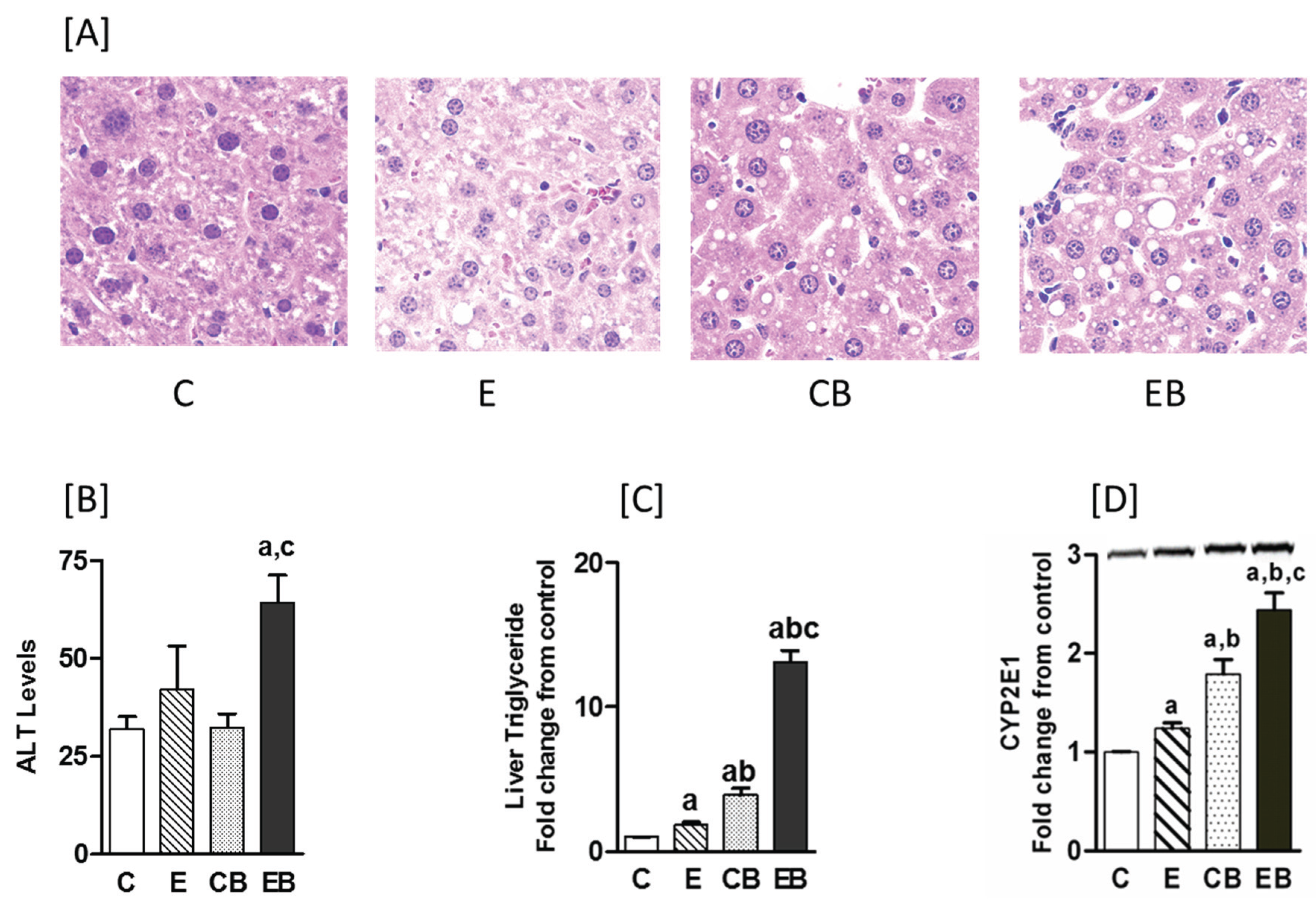
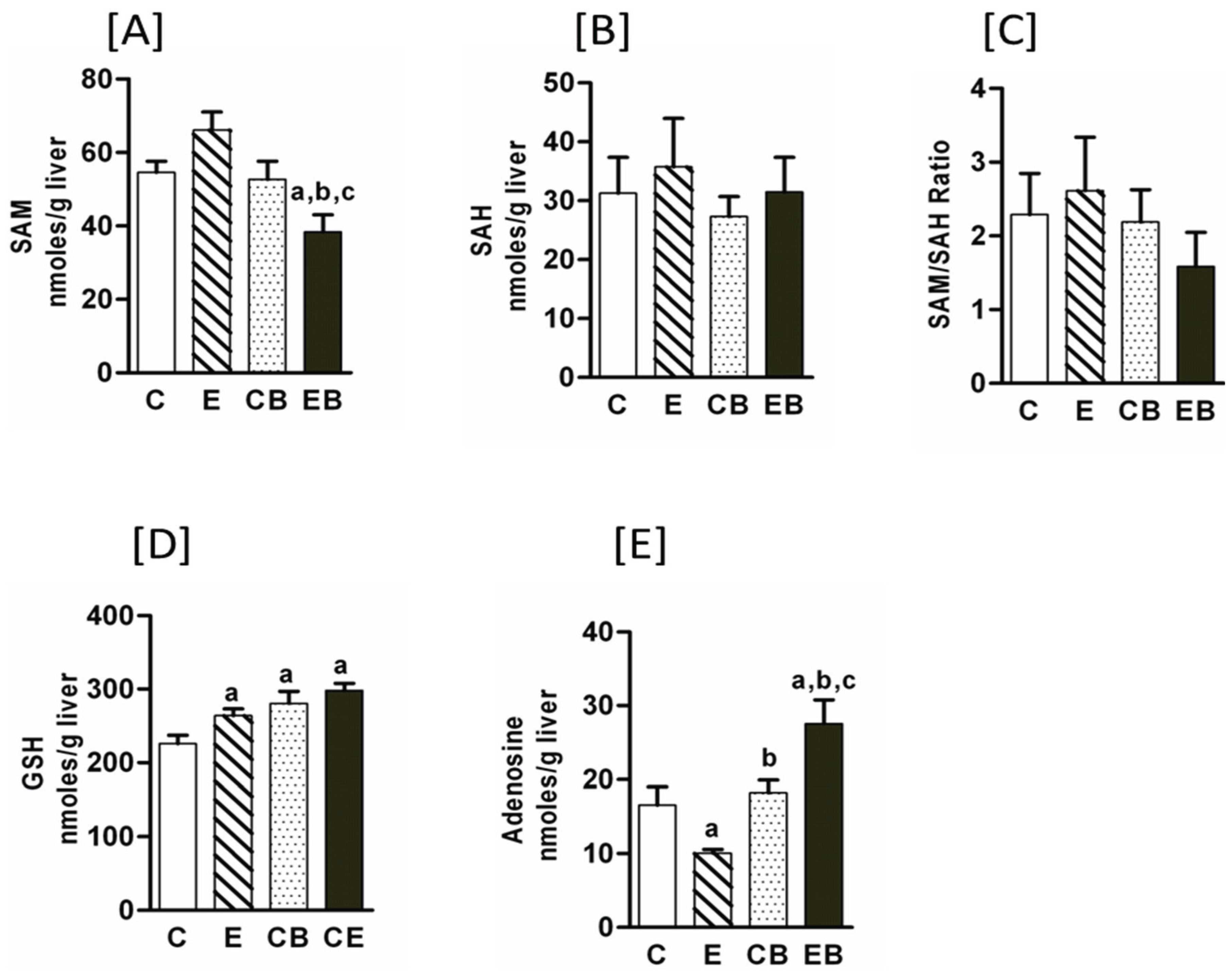
2.1. Increased Hepatic Steatosis, Necrosis, Adenosine, CYP2E Levels and Decreased S-Adenosylmethionine by Binge Administration in Chronic Ethanol Treated Mice
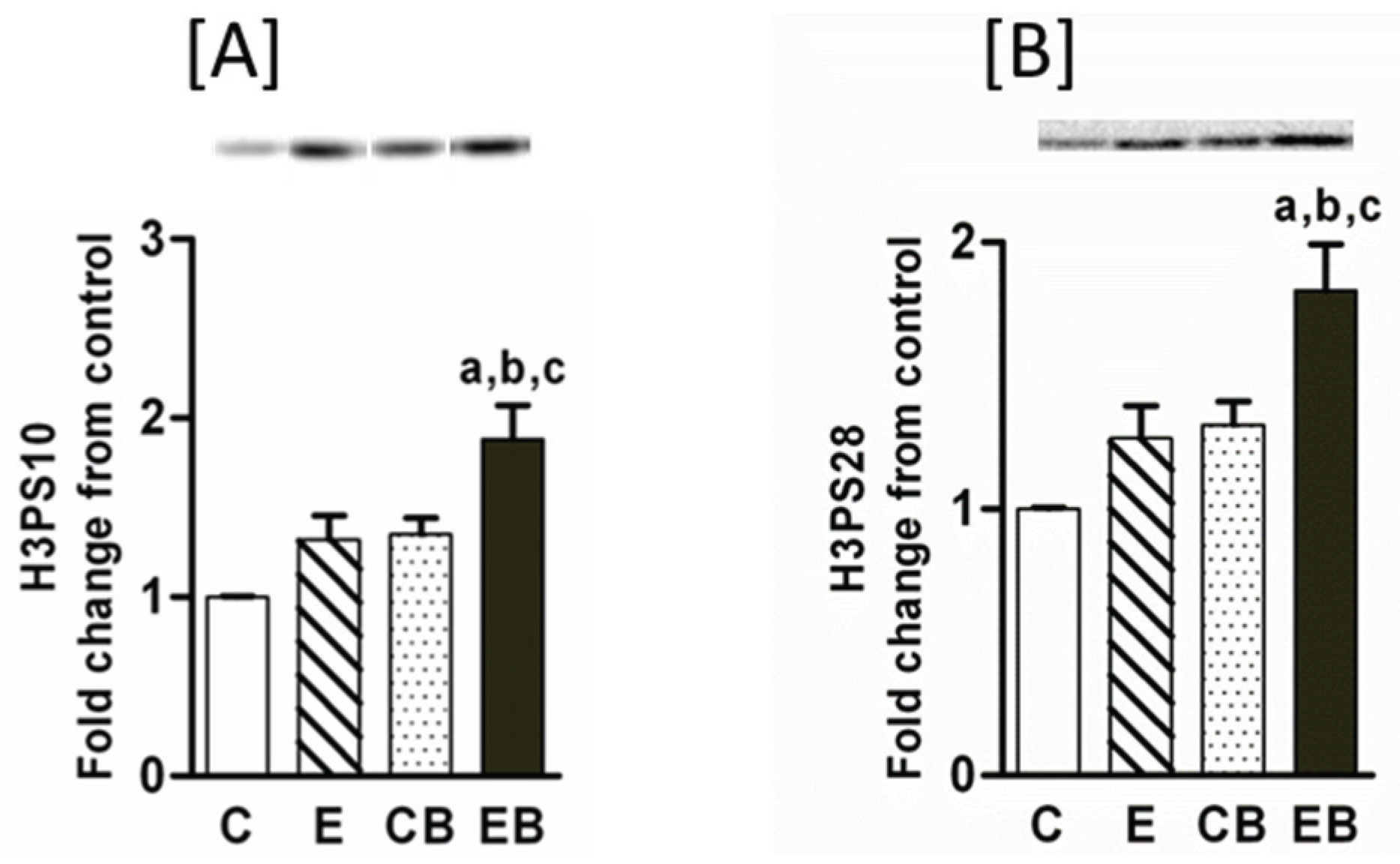
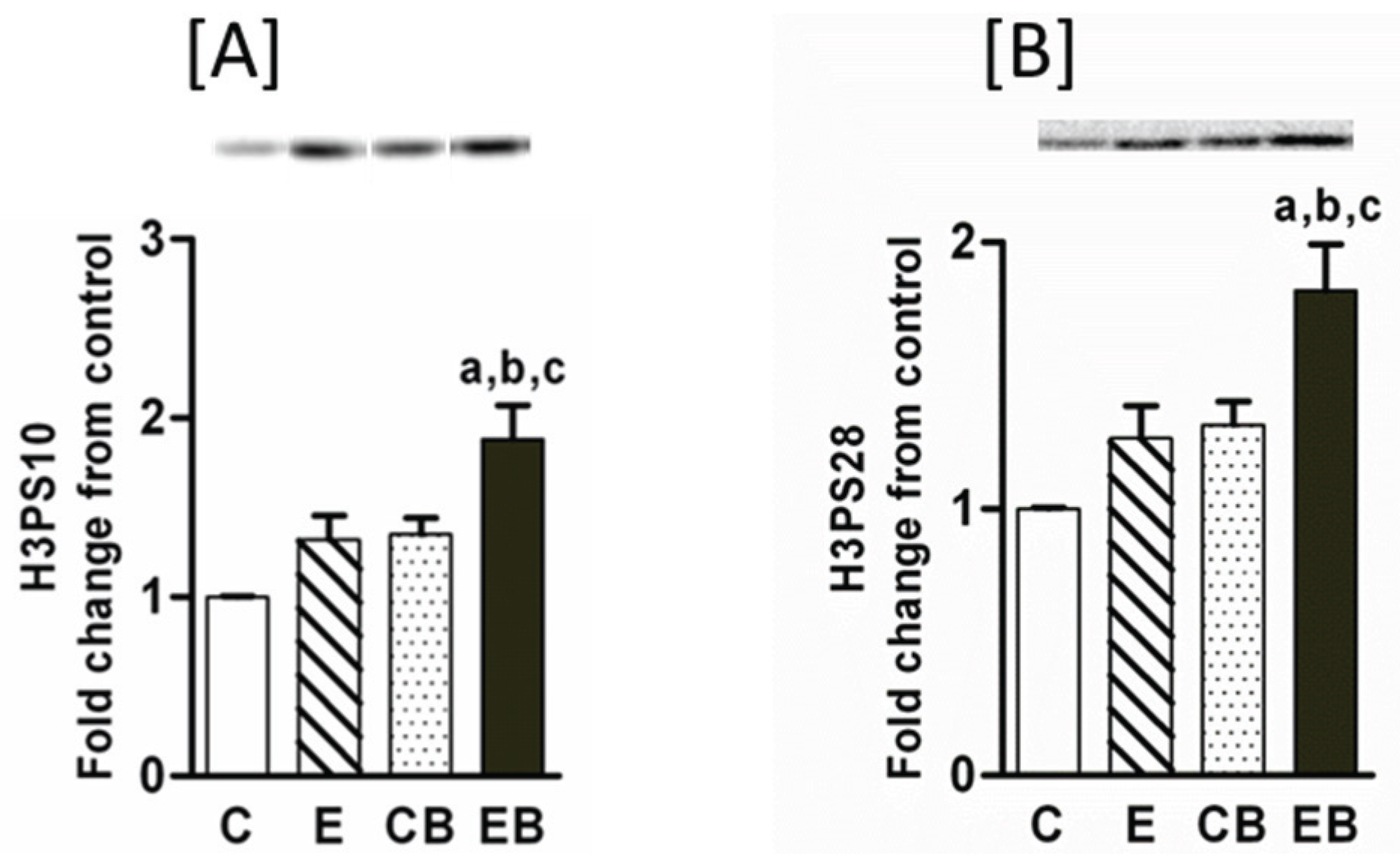
2.2. Increased Phosphorylation of Histone H3 after Chronic Ethanol-Binge
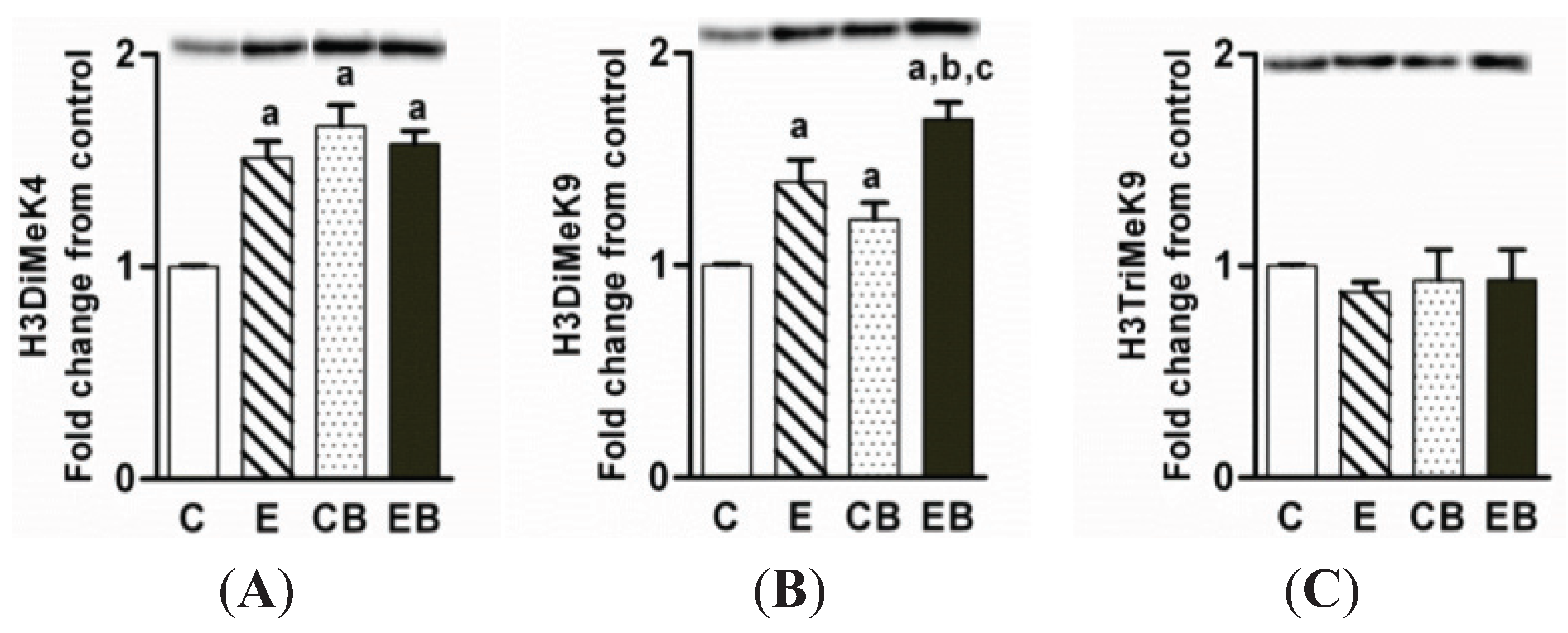
2.3. Levels of Dimethylated H3 K4, Dimethylated H3 K9, and Trimethylated H3K9
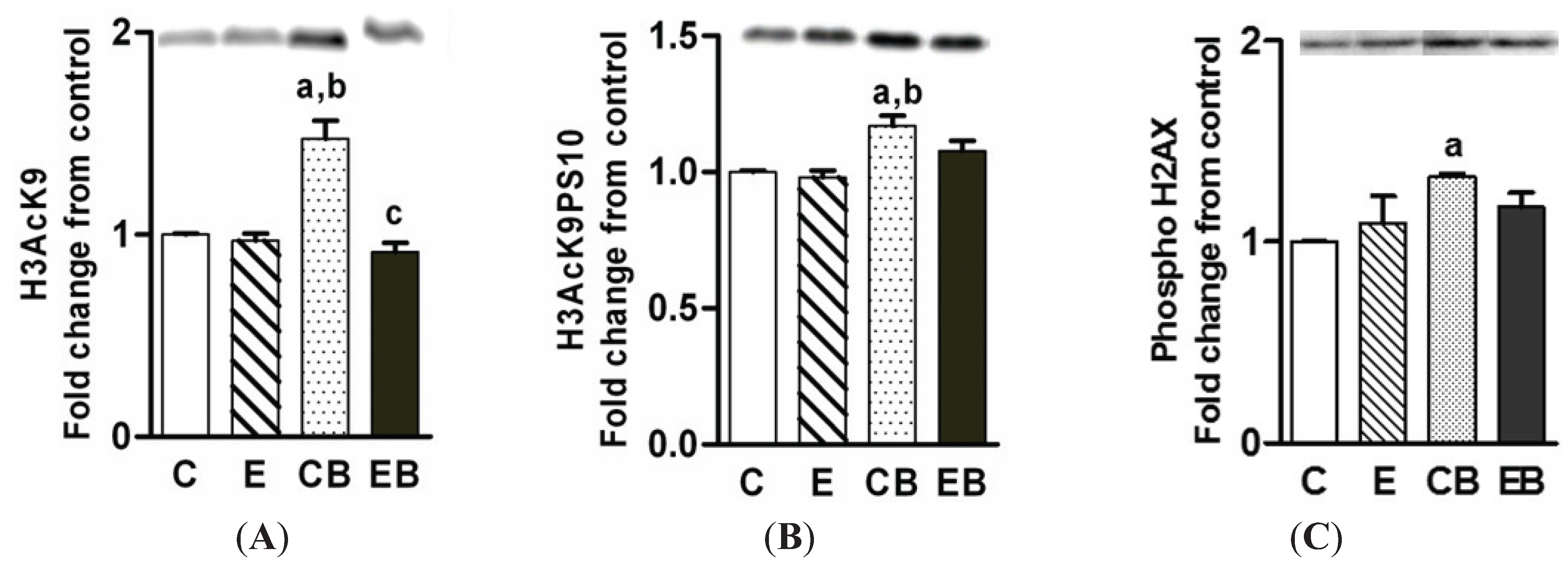
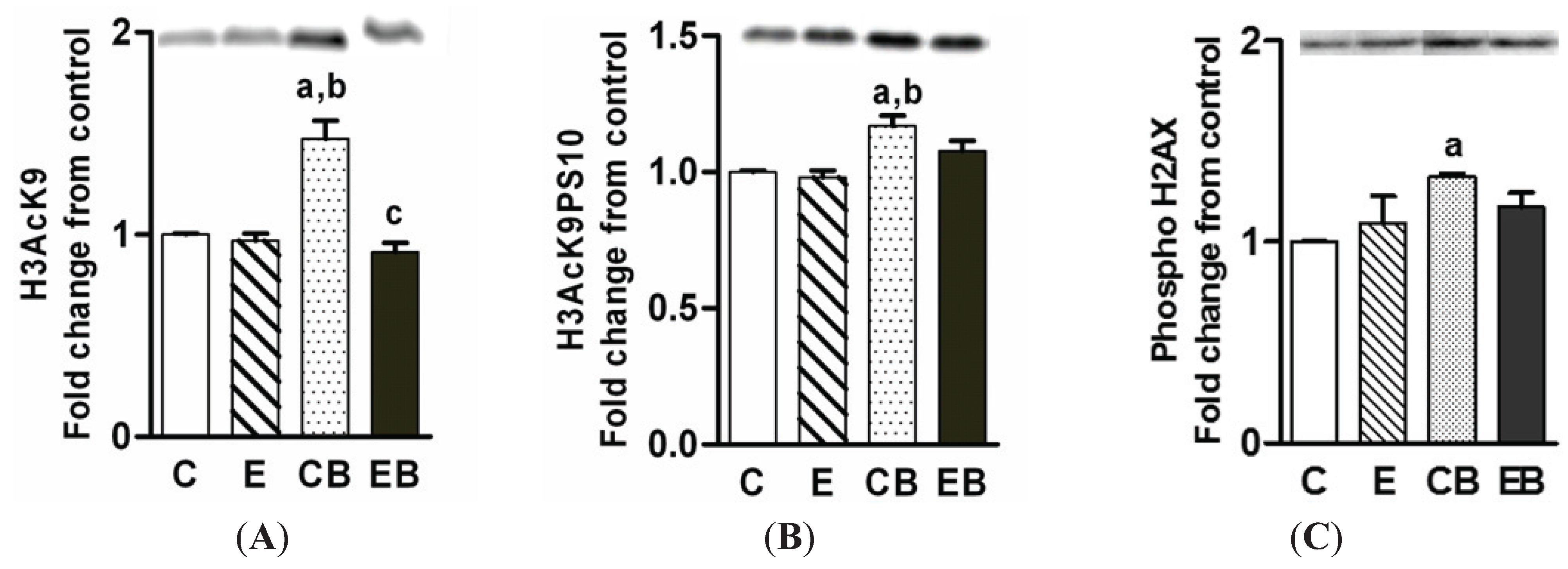
2.4. Increased H3K9 Acetylation, H3K9 Acetylation/S10 Phosphorylation and H2AX Phosphorylation
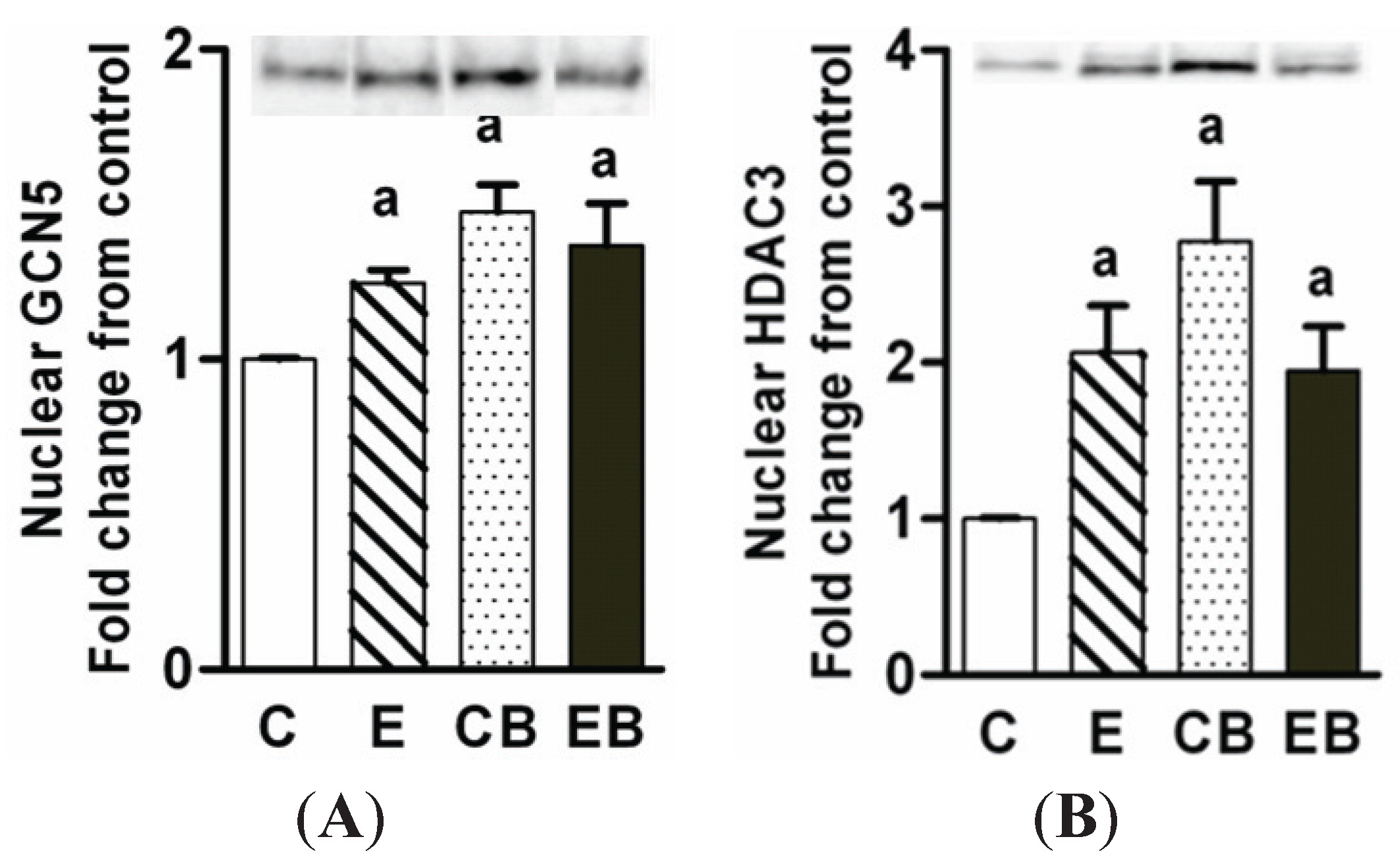
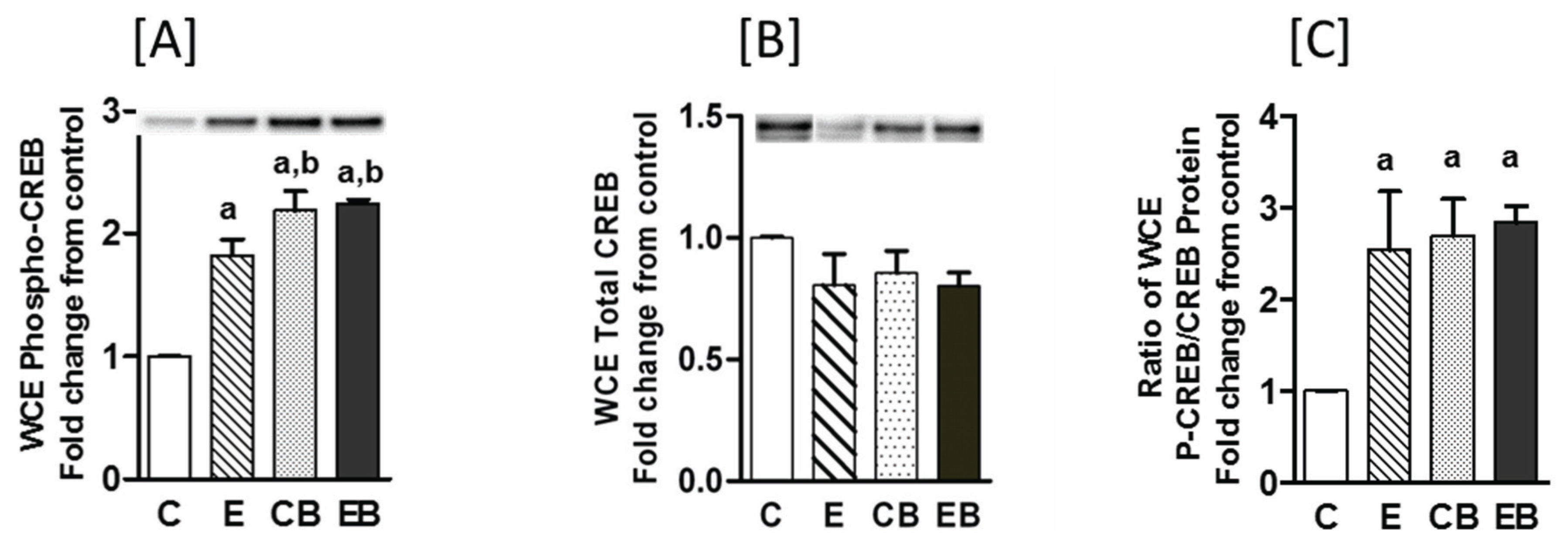
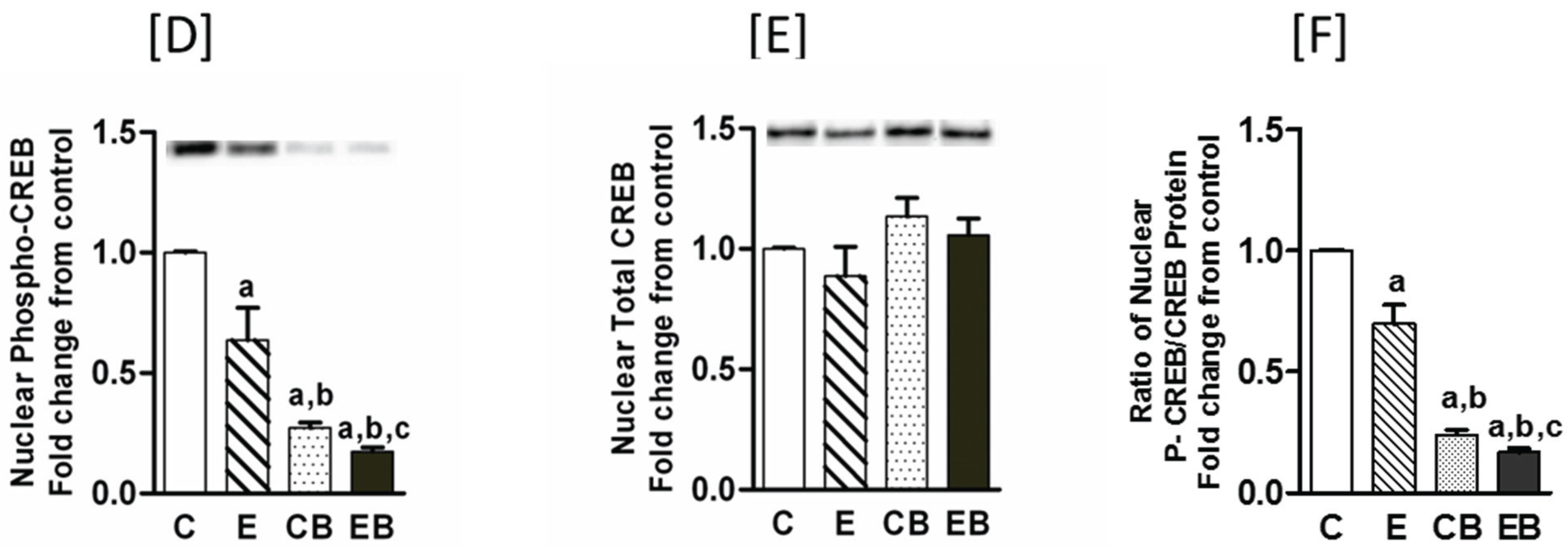
2.5. Increased Nuclear Levels of GCN5, HDAC3 and Decreased Levels of Phospho-CREB in Chronic Ethanol, Binge and Chronic Ethanol-Binge Groups
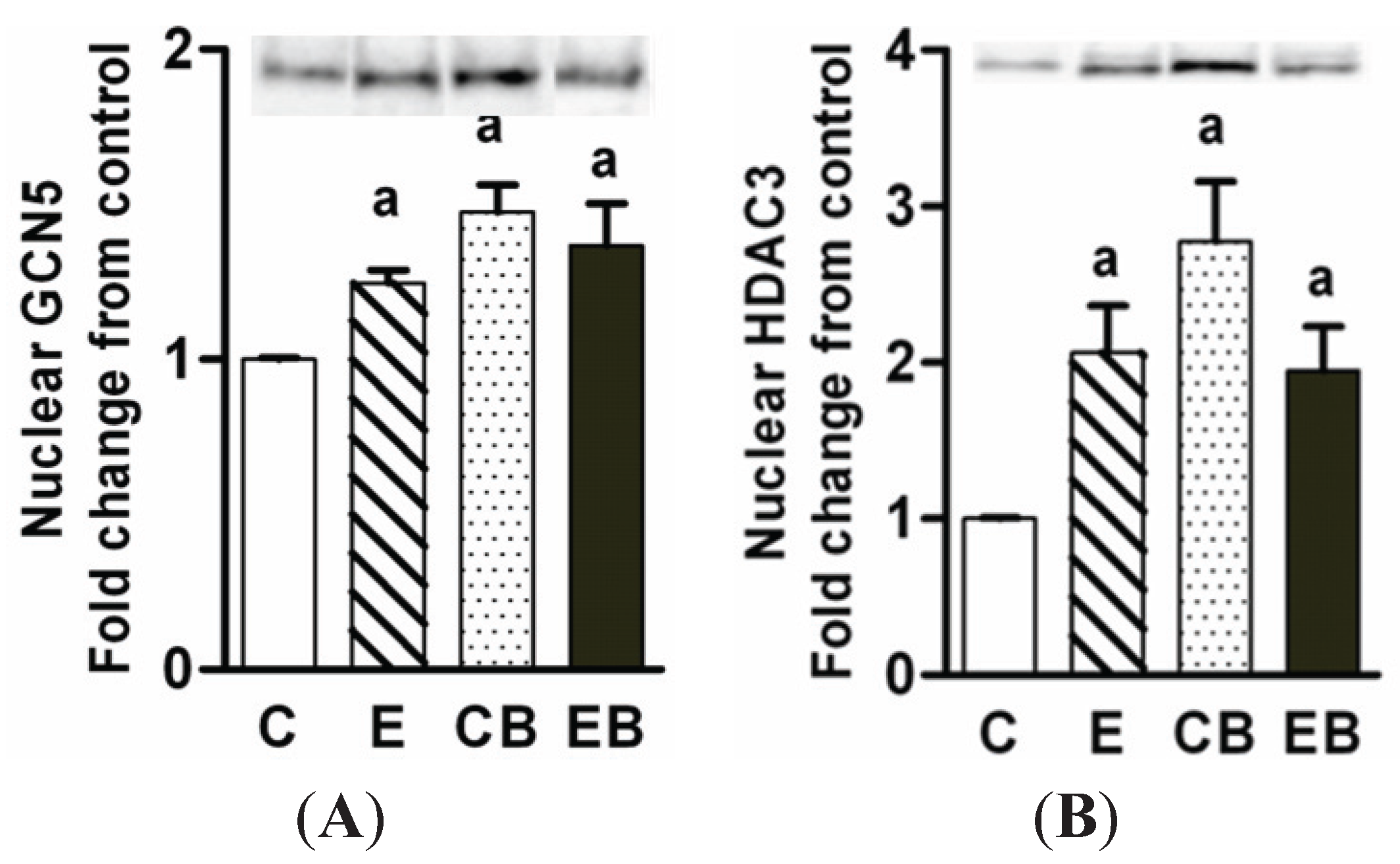
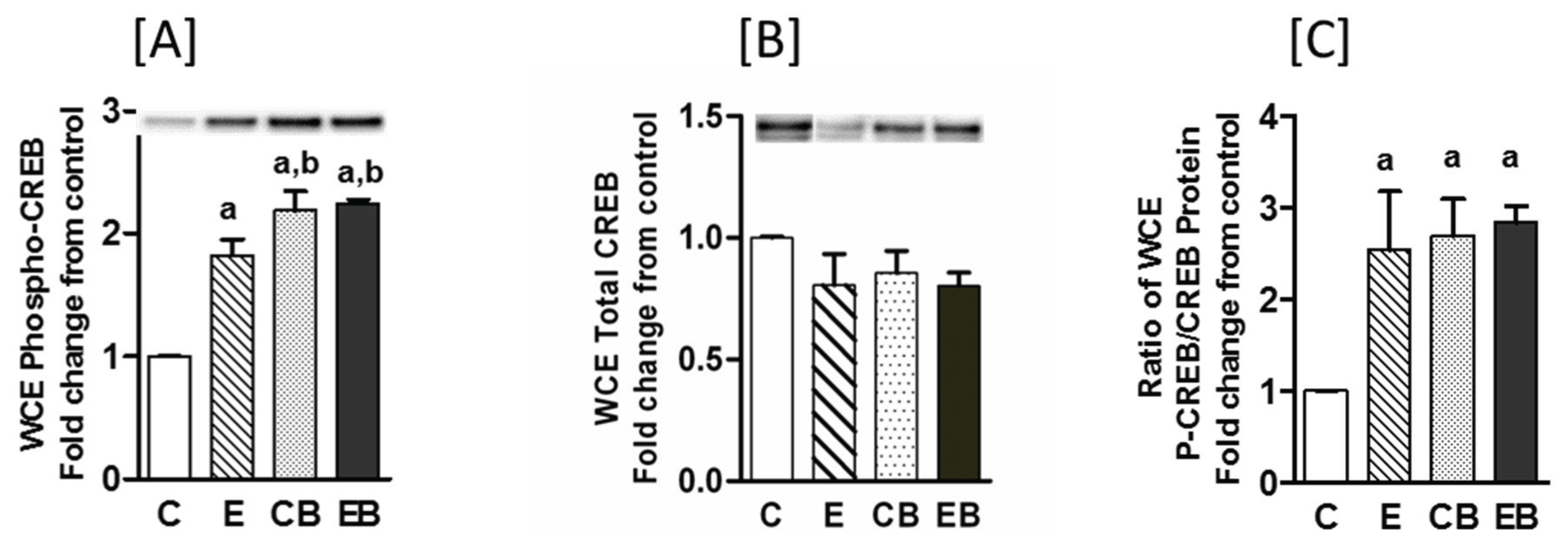
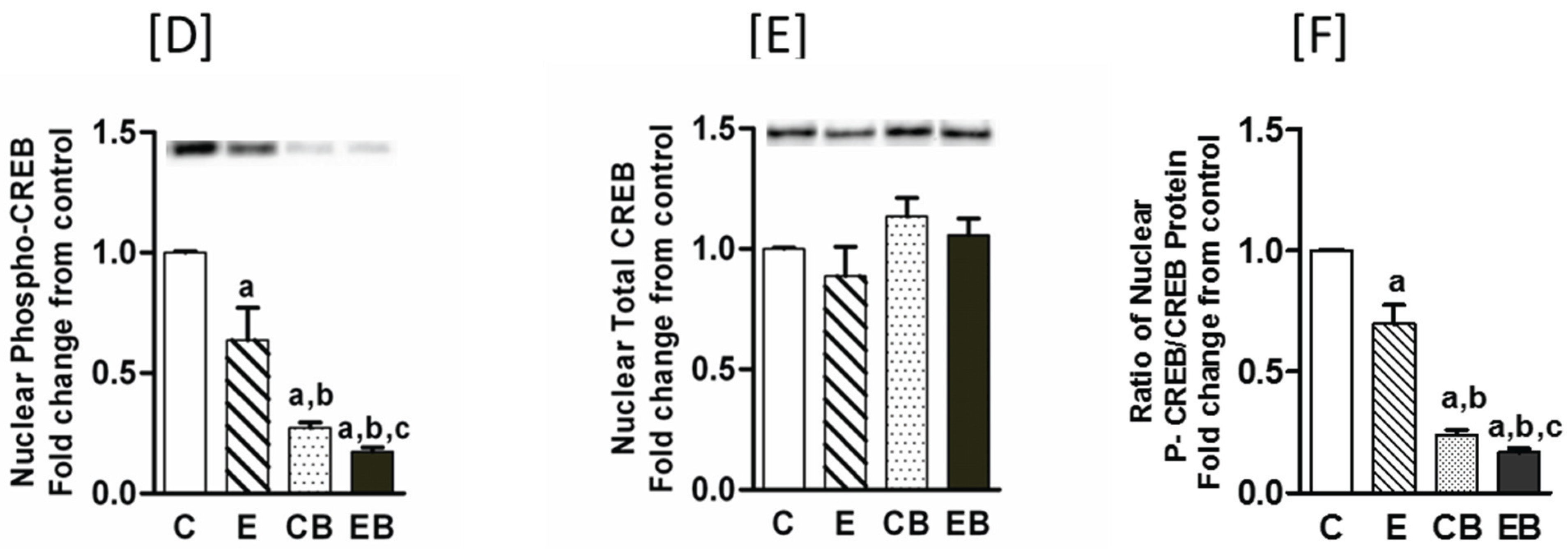
3. Discussion
4. Experimental Section
4.1. Reagents
4.2. Experimental Design for Chronic Ethanol Feeding and Binge Administration
4.3. Biochemical and Histological Studies
4.4. Nuclear Isolation and Immunoblot Analysis
4.5. Data Analysis
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Shukla, S.D.; Pruett, S.B.; Szabo, G.; Arteel, G.E. Binge ethanol and liver: New molecular developments. Alcohol Clin. Exp. Res. 2013, 37, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Zakhari, S.; Li, T.K. Determinants of alcohol use and abuse: Impact of quantity and frequency patterns on liver disease. Hepatology 2007, 46, 2032–2039. [Google Scholar] [CrossRef] [PubMed]
- Rehm, J.; Mathers, C.; Popova, S.; Thavorncharoensap, M.; Teerawattananon, Y.; Patra, J. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet 2009, 373, 2223–2233. [Google Scholar] [CrossRef]
- Mathurin, P.; Deltenre, P. Effect of binge drinking on the liver: An alarming public health issue? Gut 2009, 58, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Grucza, R.A.; Norberg, K.E.; Bierut, L.J. Binge drinking among youths and young adults in the United States: 1979–2006. J. Am. Acad. Child Adolesc. Psychiatry 2009, 48, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Courtney, K.E.; Polich, J. Binge drinking in young adults: Data, definitions, and determinants. Psychol. Bull. 2009, 135, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Ceccanti, M.; Attili, A.; Balducci, G.; Attilia, F.; Giacomelli, S.; Rotondo, C.; Sasso, G.F.; Xirouchakis, E.; Attilia, M.L. Acute alcoholic hepatitis. J. Clin. Gastroenterol. 2006, 40, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Mathurin, P.; Beuzin, F.; Louvet, A.; Carrié-Ganne, N.; Balian, A.; Trinchet, J.C.; Dalsoglio, D.; Prevot, S.; Naveau, S. Fibrosis progression occurs in a subgroup of heavy drinkers with typical histological features. Aliment. Pharmacol. Ther. 2007, 25, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Crosse, K.I.; Anania, F.K. Alcoholic hepatitis. Curr. Treat Options Gastroenterol. 2002, 5, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Jia, Z.; Misra, H.; Li, Y.R. Oxidative stress and redox signaling mechanisms of alcoholic liver disease: Updated experimental and clinical evidence. J. Dig. Dis. 2012, 13, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.K. Methionine metabolic pathway in alcoholic liver injury. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Borea, P.A.; Varani, K.; Wilder, T.; Yee, H.; Chiriboga, L.; Blackburn, M.R.; Azzena, G.; Resta, G.; Cronstein, B.N. Adenosine signaling contributes to ethanol-induced fatty liver in mice. J. Clin. Invest. 2009, 119, 582–594. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Fernandez, P.; Wilder, T.; Yee, H.; Chiriboga, L.; Chan, E.S.; Cronstein, B.N. Ecto-5'-nucleotidase (CD73)-mediated extracellular adenosine production plays a critical role in hepatic fibrosis. FASEB J. 2008, 22, 2263–2272. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.D.; Aroor, A.R. Epigenetic effects of ethanol on liver and gastrointestinal injury. World J. Gastroenterol. 2006, 12, 5265–5271. [Google Scholar] [PubMed]
- Shukla, S.D.; Lim, R.W. Epigenetic effects of ethanol on the liver and gastrointestinal system. Alcohol Res. Curr. Rev. 2013, 35, 47–55. [Google Scholar]
- Patel, D.J.; Wang, Z. Readout of epigenetic modifications. Annu. Rev. Biochem. 2013, 82, 81–118. [Google Scholar] [CrossRef] [PubMed]
- Hübner, M.R.; Eckersley-Maslin, M.A.; Spector, D.L. Chromatin organization and transcriptional regulation. Curr. Opin. Genet Dev. 2013, 23, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Pal-Bhadra, M.; Bhadra, U.; Jackson, D.E.; Mamatha, L.; Park, P.H.; Shukla, S.D. Distinct methylation patterns in histone H3 at Lys-4 and Lys-9 correlate with up- and down-regulation of genes by ethanol in hepatocytes. Life Sci. 2007, 81, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.D.; Restrepo, R.; Fish, P.; Lim, R.W.; Ibdah, J.A. Different mechanisms for histone acetylation by ethanol and its metabolite acetate in rat primary hepatocytes. J. Pharmacol. Exp. Ther. 2015, 354, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Shukla, S.D. Acute in vivo effect of ethanol (binge drinking) on histone H3 modifications in rat tissues. Alcohol Alcohol. 2006, 41, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Kirpich, I.; Zhang, J.; Gobejishvili, L.; Kharebava, G.; Barker, D.; Ghare, S.; Joshi-Barve, S.; McClain, C.J.; Barve, S. Binge ethanol-induced HDAC3 down-regulates Cpt1α expression leading to hepatic steatosis and injury. Alcohol Clin. Exp. Res. 2013, 37, 1920–1929. [Google Scholar] [CrossRef] [PubMed]
- James, T.T.; Aroor, A.R.; Lim, R.W.; Shukla, S.D. Histone H3 phosphorylation (Ser10, Ser28) and phosphoacetylation (K9S10) are differentially associated with gene expression in liver of rats treated in vivo with acute ethanol. J. Pharmacol. Exp. Ther. 2012, 340, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Aroor, A.R.; James, T.T.; Jackson, D.E.; Shukla, S.D. Differential changes in MAP kinases, histone modifications, and liver injury in rats acutely treated with ethanol. Alcohol Clin. Exp. Res. 2010, 34, 1543–1551. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Bardag-Gorce, F.; Oliva, J.; Dedes, J.; French, B.A.; French, S.W. Gene expression modifications in the liver caused by binge drinking and S-adenosylmethionine feeding. The role of epigenetic changes. Genes Nutr. 2010, 5, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Esfandiari, F.; Medici, V.; Wong, D.H.; Jose, S.; Dolatshahi, M.; Quinlivan, E.; Dayal, S.; Lentz, S.R.; Tsukamoto, H.; Zhang, Y.H.; et al. Epigenetic regulation of hepatic endoplasmic reticulum stress pathways in the ethanol-fed cystathionine beta synthase-deficient mouse. Hepatology 2010, 51, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Park, P.H.; Lim, R.W.; Shukla, S.D. Gene-selective histone H3 acetylation in the absence of increase in global histone acetylation in liver of rats chronically fed alcohol. Alcohol Alcohol. 2012, 47, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.D.; Velazquez, J.; French, S.W.; Lu, S.C.; Ticku, M.K.; Zakhari, S. Emerging role of epigenetics in the actions of alcohol. Alcohol Clin. Exp. Res. 2008, 32, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Cao, Q.; Liang, X.; Ajmo, J.M.; Ness, G.C. Mammalian sirtuin 1 is involved in the protective action of dietary saturated fat against alcoholic fatty liver in mice. J. Nutr. 2008, 138, 497–501. [Google Scholar] [PubMed]
- Aroor, A.R.; Jackson, D.E.; Shukla, S.D. Elevated activation of ERK1 and ERK2 accompany enhanced liver injury following alcohol binge in chronically ethanol-fed rats. Alcohol Clin. Exp. Res. 2011, 35, 2128–2138. [Google Scholar] [CrossRef] [PubMed]
- Aroor, A.R.; Restrepo, R.J.; Kharbanda, K.K.; Shukla, S.D. Epigenetic histone modifications in a clinically relevant rat model of chronic ethanol-binge-mediated liver injury. Hepatol. Int. 2014, 8, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Ji, C.; Kaplowitz, N. Differences in betaine-homocysteine methyl transferase expression, endoplasmic reticulum stress response, and liver injury between alcohol-fed mice and rats. Hepatology 2010, 51, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Arteel, G.E. Animal models of alcoholic liver disease. Dig. Dis. 2010, 28, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Aroor, A.R.; Roy, L.J.; Restrepo, R.J.; Mooney, B.P.; Shukla, S.D. A proteomic analysis of liver after ethanol binge in chronically ethanol treated rats. Proteome Sci. 2012. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Park, J.H.; Kim, S.J.; Kwon, S.J.; Kwon, J. A cooperative activation loop among SWI/SNF, gamma-H2AX and H3 acetylation for DNA double-strand break repair. EMBO J. 2010, 29, 1434–1445. [Google Scholar] [CrossRef] [PubMed]
- Szerlong, H.J.; Prenni, J.E.; Nyborg, J.K.; Hansen, J.C. Activator-dependent p300 acetylation of chromatin in vitro: Enhancement of transcription by disruption of repressive nucleosome-nucleosome interactions. J. Biol. Chem. 2010, 285, 31954–31964. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, M.; Pandey, R.S.; Clemens, D.L.; Davis, J.W.; Lim, R.W.; Shukla, S.D. Knock down of GCN5 histone acetyltransferase by siRNA decreases ethanol-induced histone acetylation and affects differential expression of genes in human hepatoma cells. Alcohol 2011, 45, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Schiltz, R.L.; Mizzen, C.A.; Vassilev, A.; Cook, R.G.; Allis, C.D.; Nakatani, Y. Overlapping but distinct patterns of histone acetylation by the human coactivators p300 and PCAF within nucleosomal substrates. J. Biol. Chem. 1999, 274, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Aroor, A.R.; Jackson, D.E.; Shukla, S.D. Dysregulated phosphorylation and nuclear translocation of cyclic AMP response element binding protein (CREB) in rat liver after chronic ethanol binge. Eur. J. Pharmacol. 2012, 679, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Iimuro, Y.; Bradford, B.U.; Yamashina, S.; Rusyn, I.; Nakagami, M.; Enomoto, N.; Kono, H.; Frey, W.; Forman, D.; Brenner, D.; et al. The glutathione precursor l-2-oxothiazolidine-4-carboxylic acid protects against liver injury due to chronic enteral ethanol exposure in the rat. Hepatology 2000, 31, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.K.; Todero, S.L.; King, A.L.; Osna, N.A.; McVicker, B.L.; Tuma, D.J.; Wisecarver, J.L.; Bailey, S.M. Betaine treatment attenuates chronic ethanol-induced hepatic steatosis and alterations to the mitochondrial respiratory chain proteome. Int. J. Hepatol. 2012. [Google Scholar] [CrossRef] [PubMed]
- El Gazzar, M.; Yoza, B.K.; Chen, X.; Hu, J.; Hawkins, G.A.; McCall, C.E. G9a and HP1 couple histone and DNA methylation to TNFα transcription silencing during endotoxin tolerance. J. Biol. Chem. 2008, 283, 32198–32208. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Chen, J.; Santiago, F.S.; Janes, M.; Kavurma, M.M.; Chong, B.H.; Pimanda, J.E.; Khachigian, L.M. Phosphorylation and acetylation of histone H3 and autoregulation by early growth response 1 mediate interleukin 1β induction of early growth response 1 transcription. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Brandon-Warner, E.; Schrum, L.W.; Schmidt, C.M.; McKillop, I.H. Rodent models of alcoholic liver disease: Of mice and men. Alcohol 2012, 46, 715–725. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shukla, S.D.; Aroor, A.R.; Restrepo, R.; Kharbanda, K.K.; Ibdah, J.A. In Vivo Acute on Chronic Ethanol Effects in Liver: A Mouse Model Exhibiting Exacerbated Injury, Altered Metabolic and Epigenetic Responses. Biomolecules 2015, 5, 3280-3294. https://doi.org/10.3390/biom5043280
Shukla SD, Aroor AR, Restrepo R, Kharbanda KK, Ibdah JA. In Vivo Acute on Chronic Ethanol Effects in Liver: A Mouse Model Exhibiting Exacerbated Injury, Altered Metabolic and Epigenetic Responses. Biomolecules. 2015; 5(4):3280-3294. https://doi.org/10.3390/biom5043280
Chicago/Turabian StyleShukla, Shivendra D., Annayya R. Aroor, Ricardo Restrepo, Kusum K. Kharbanda, and Jamal A. Ibdah. 2015. "In Vivo Acute on Chronic Ethanol Effects in Liver: A Mouse Model Exhibiting Exacerbated Injury, Altered Metabolic and Epigenetic Responses" Biomolecules 5, no. 4: 3280-3294. https://doi.org/10.3390/biom5043280
APA StyleShukla, S. D., Aroor, A. R., Restrepo, R., Kharbanda, K. K., & Ibdah, J. A. (2015). In Vivo Acute on Chronic Ethanol Effects in Liver: A Mouse Model Exhibiting Exacerbated Injury, Altered Metabolic and Epigenetic Responses. Biomolecules, 5(4), 3280-3294. https://doi.org/10.3390/biom5043280