
Immune Mechanisms Linking Obesity and Preeclampsia


Abstract
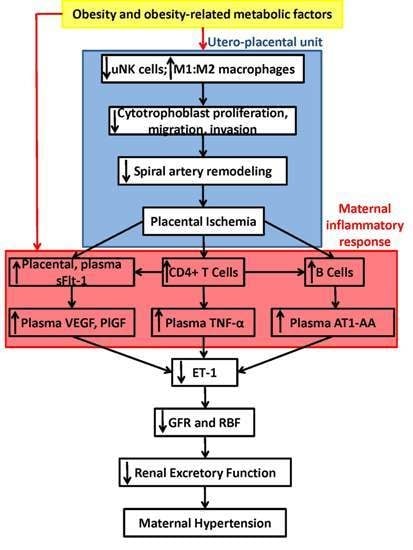
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
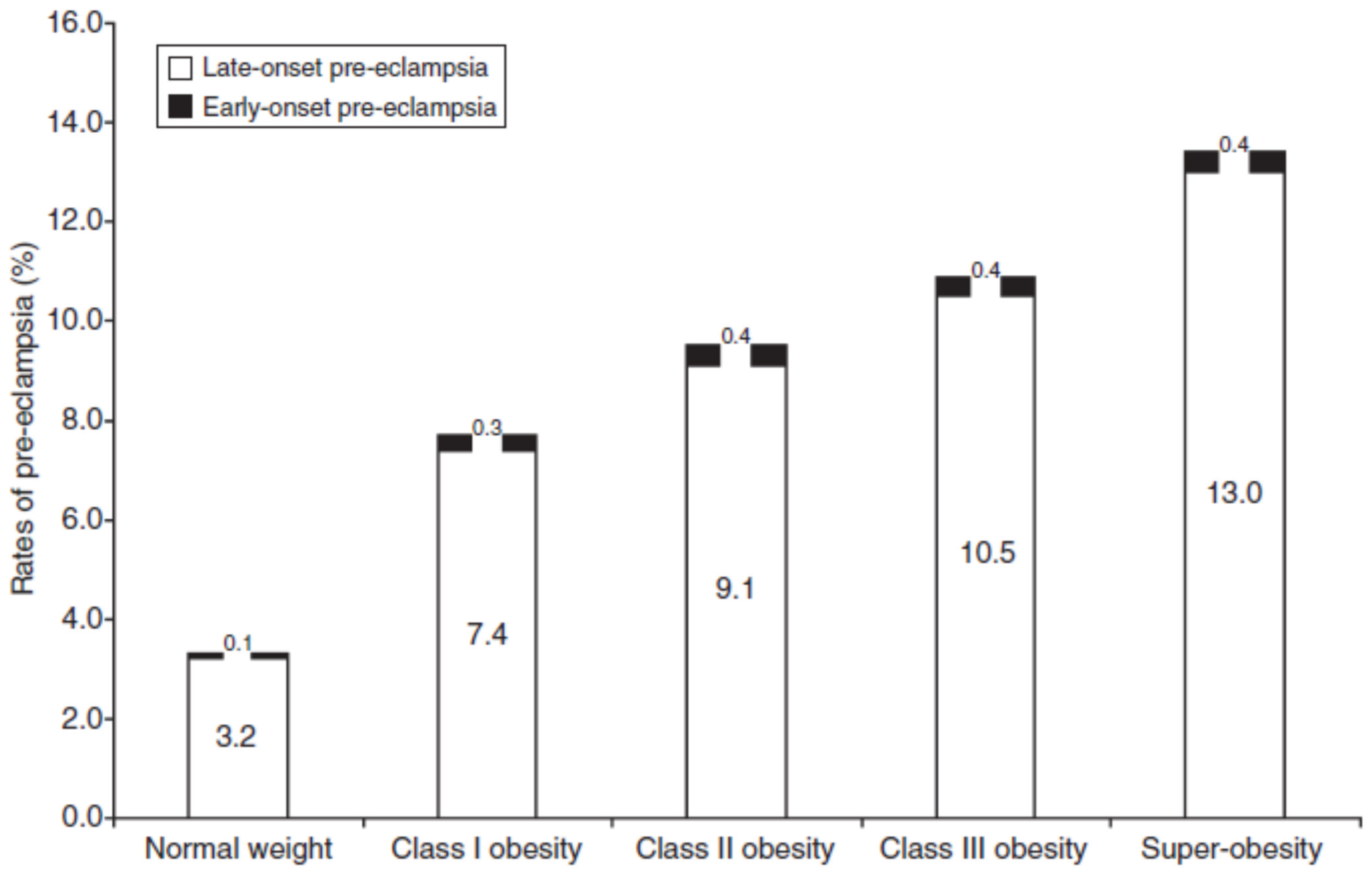
2. The Prevalence of PE and Obesity Are on the Rise
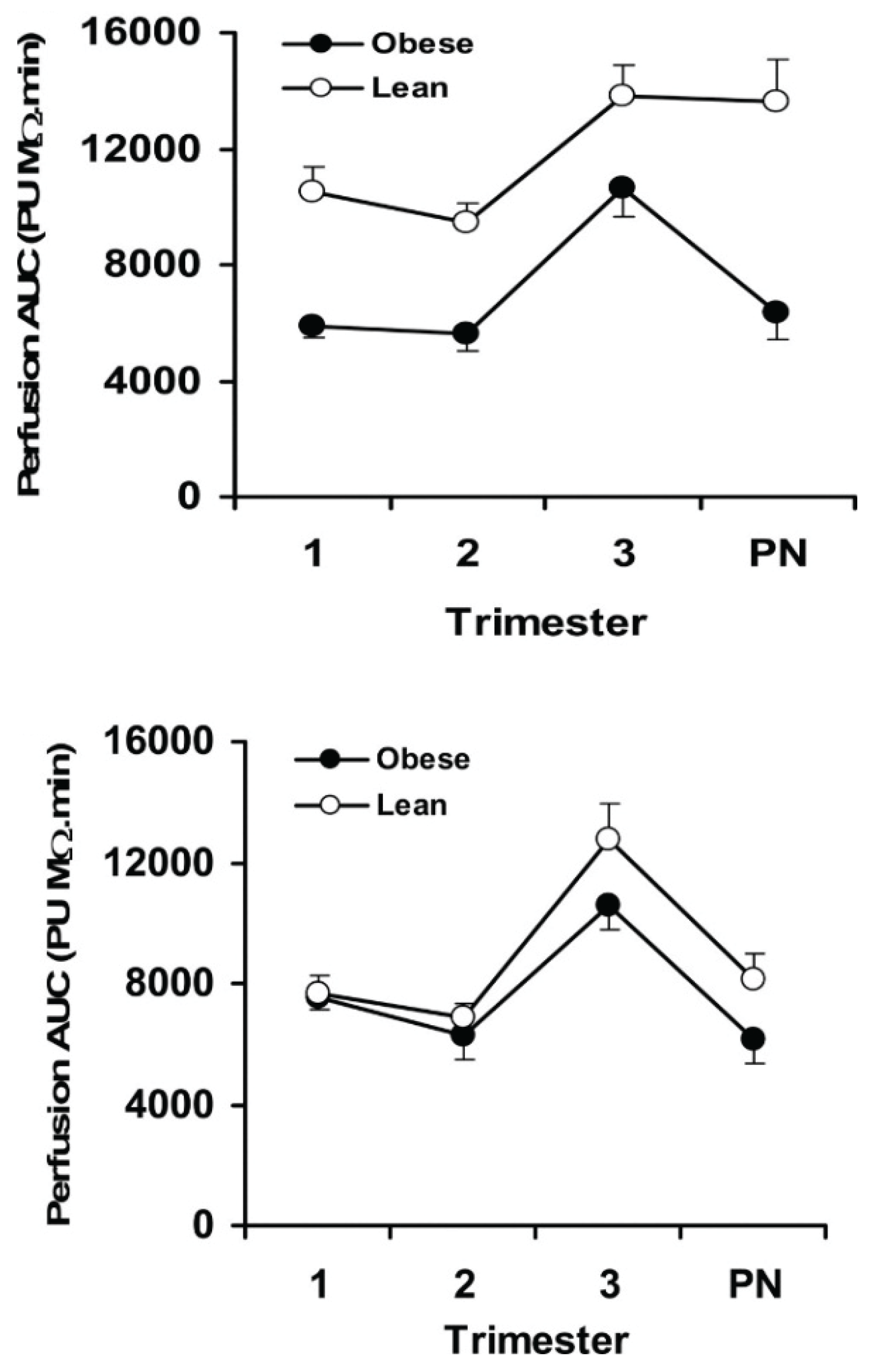
3. Obesity Increases Placental Inflammatory Cytokines, Immune Cells and Ischemia
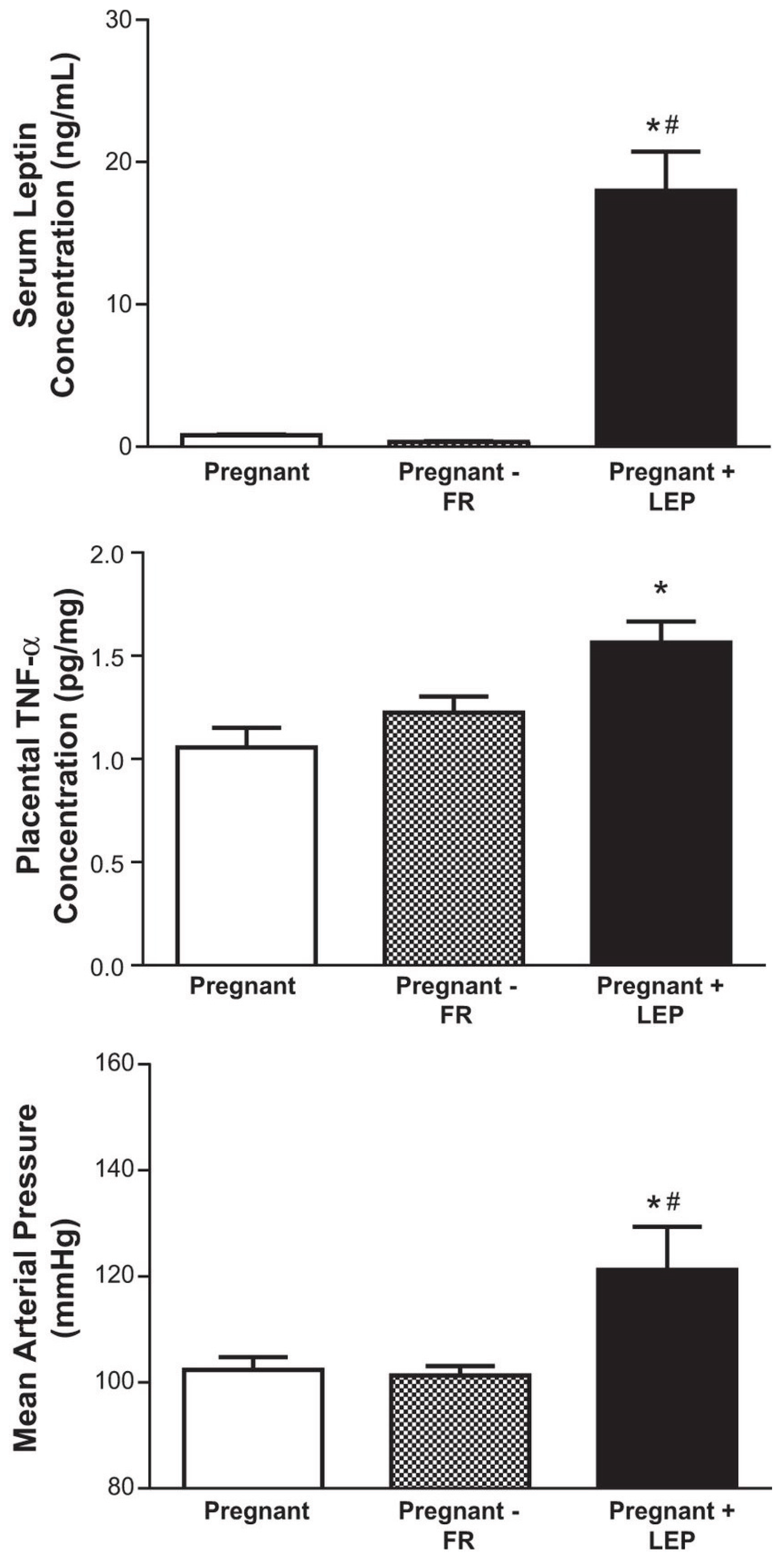
3.1. Inflammatory Cytokines
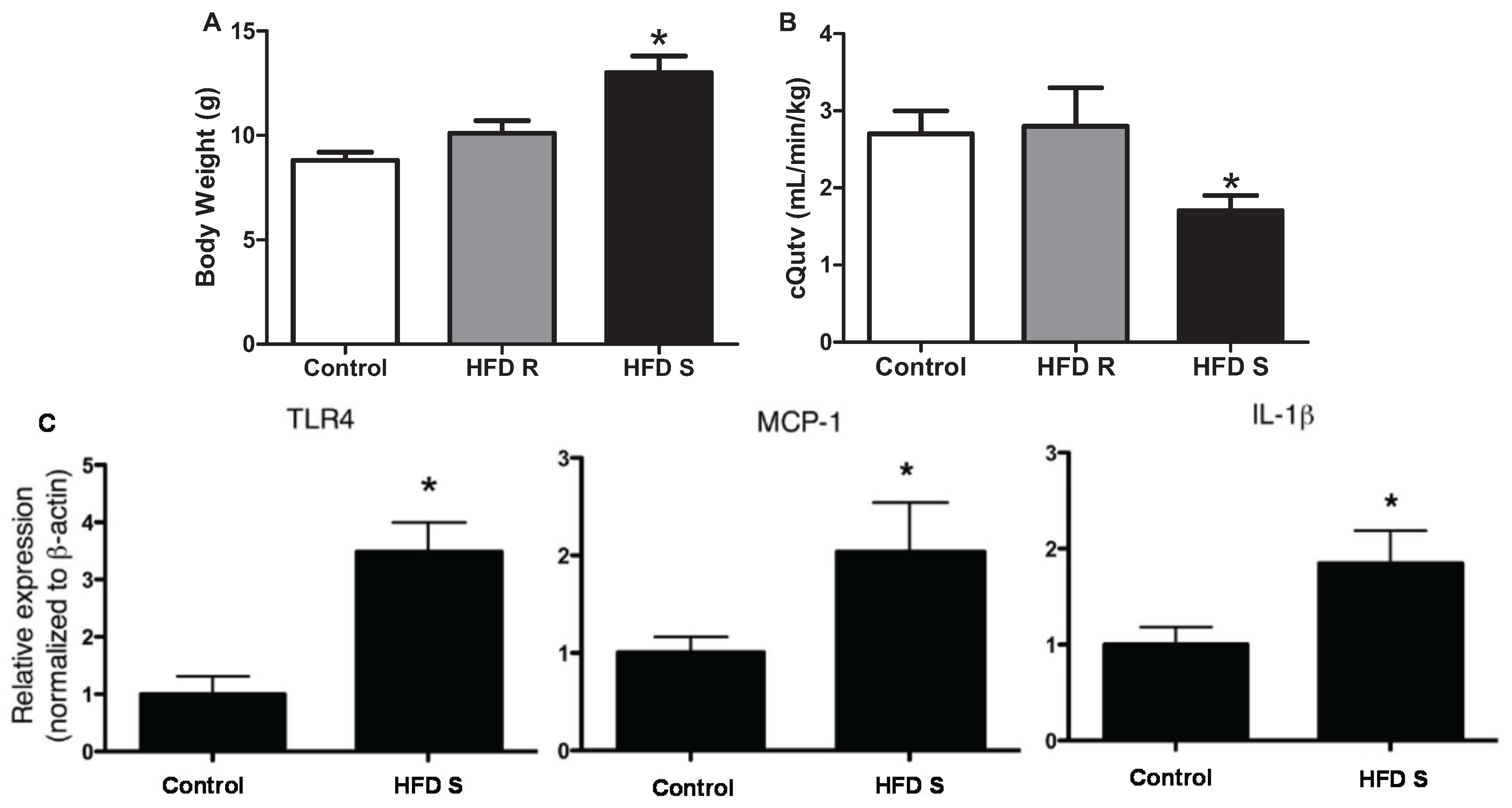
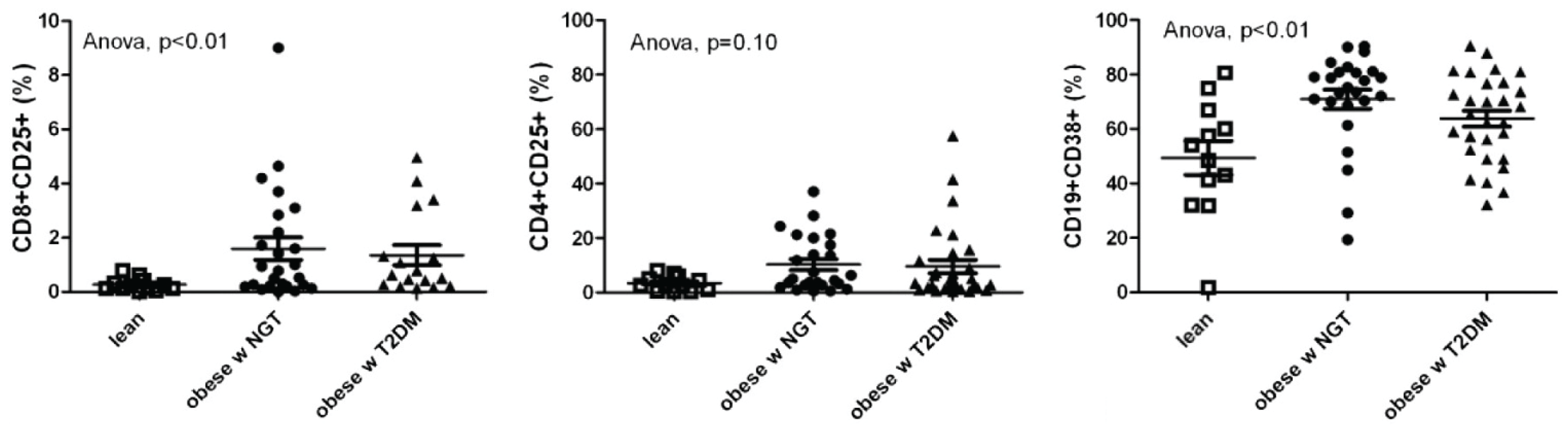
3.2. Immune Cells
Uterine Natural Killer Cells (uNKs)
3.3. Macrophages
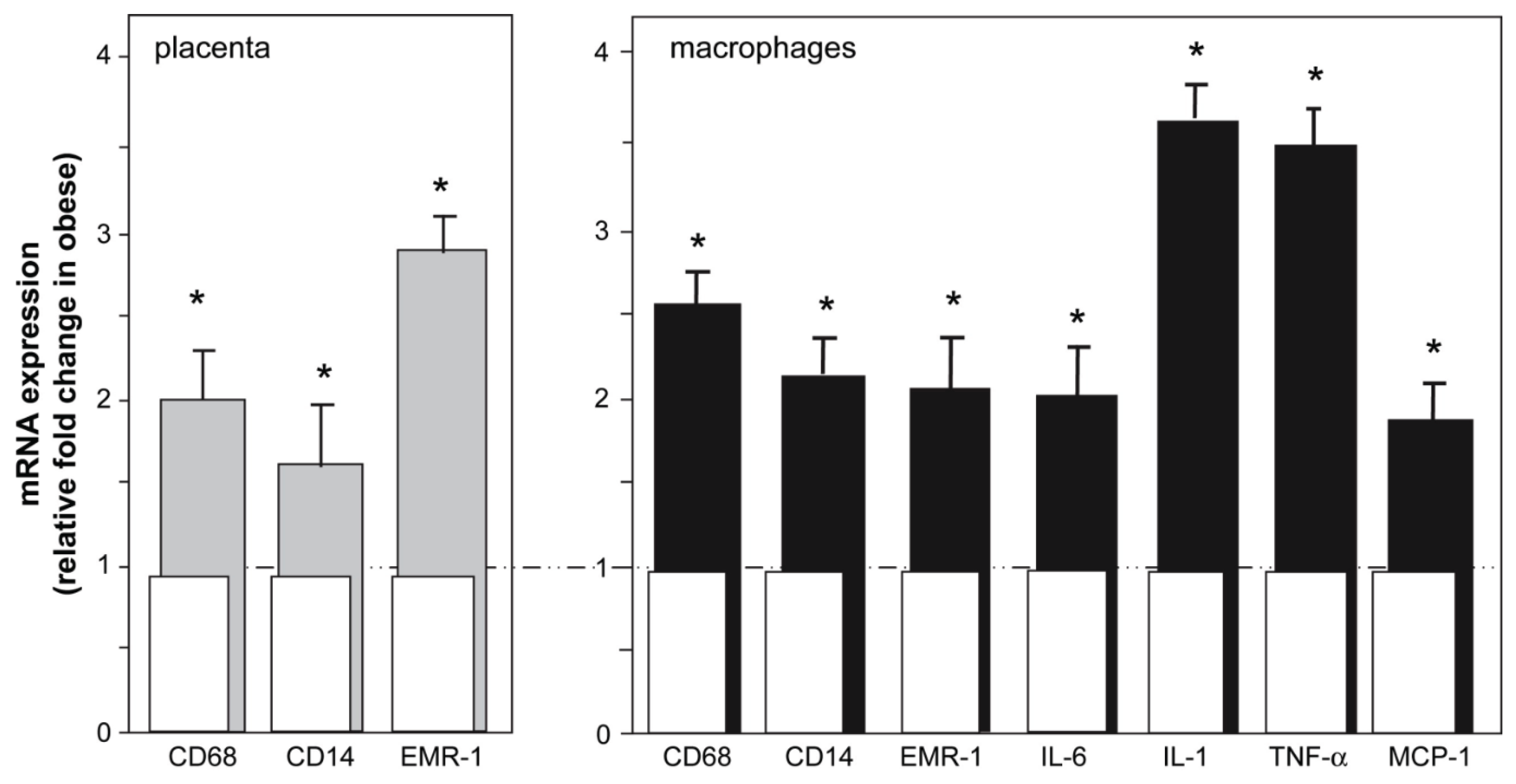
4. Effects of Obesity on Placental Ischemia-Induced Peripheral Inflammation, Vascular Dysfunction and Hypertension
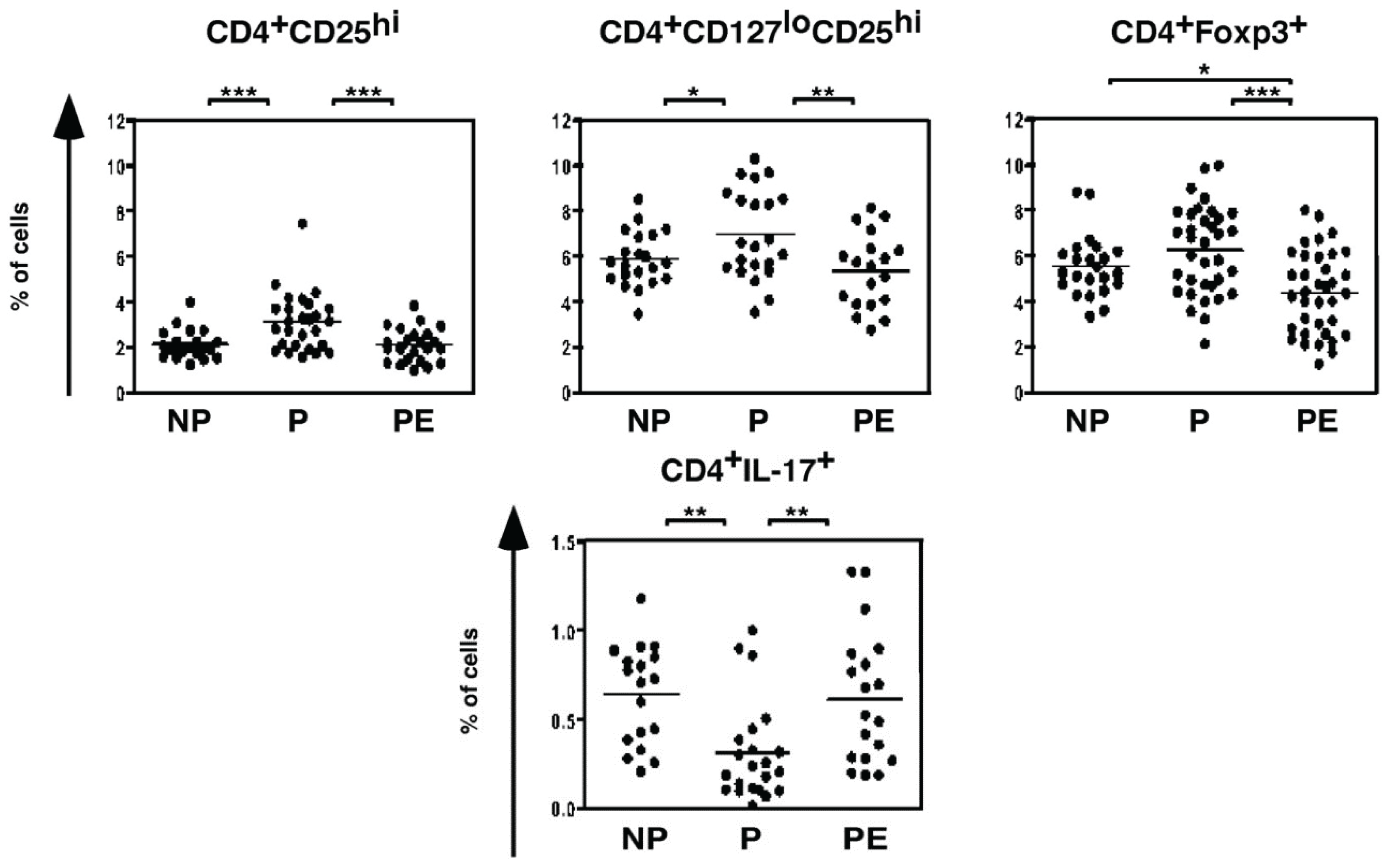
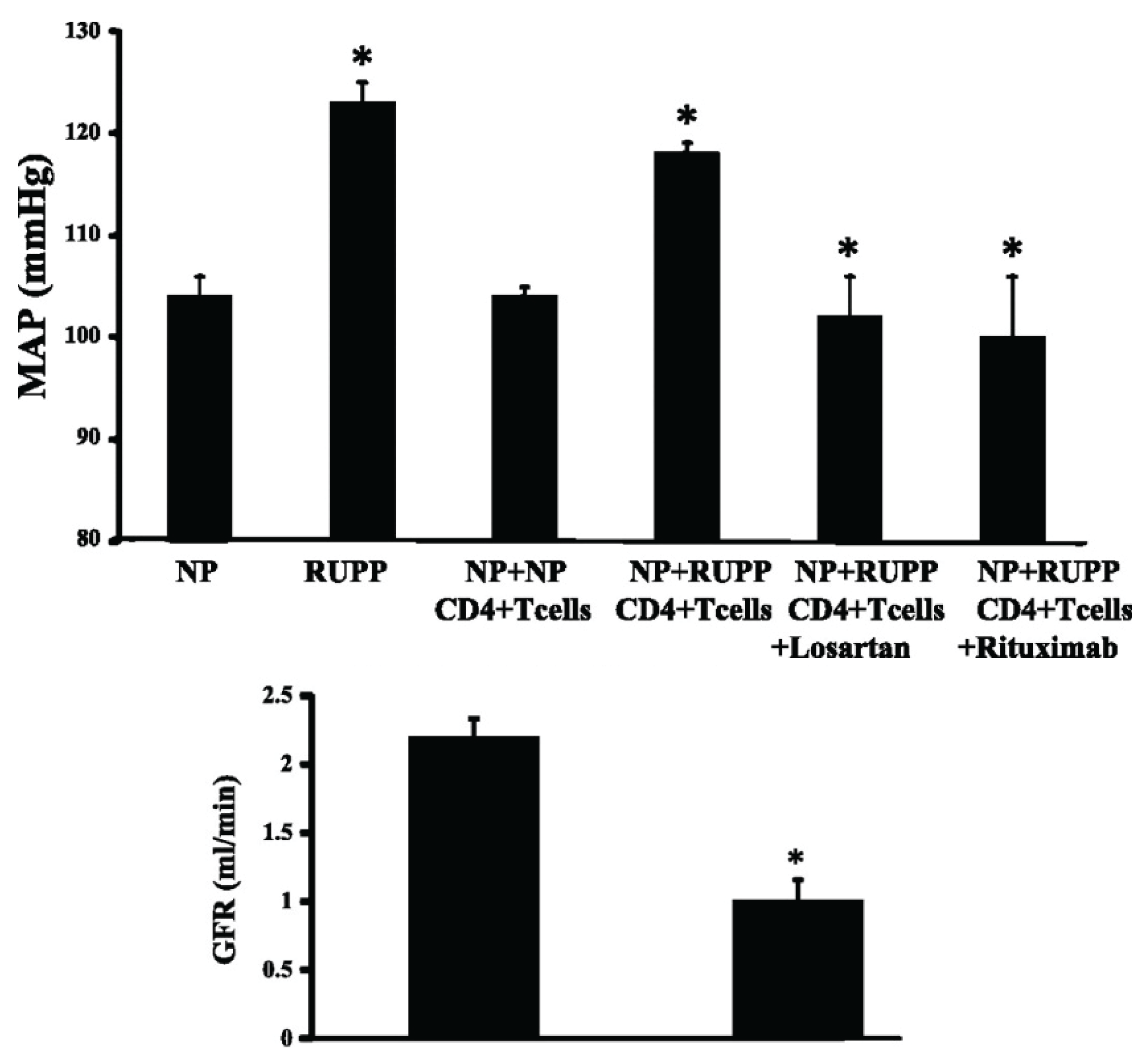
4.1. T lymphocytes
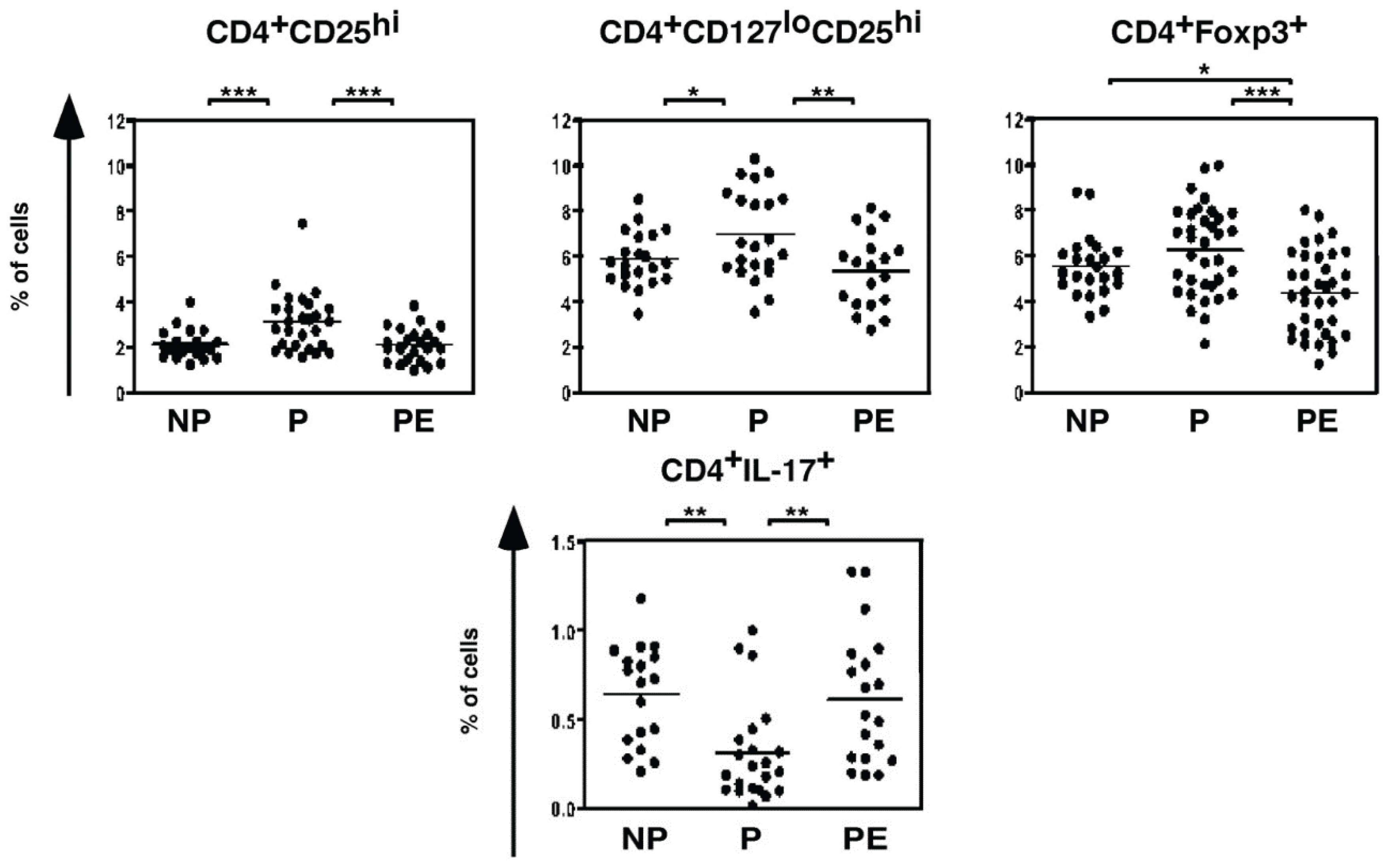
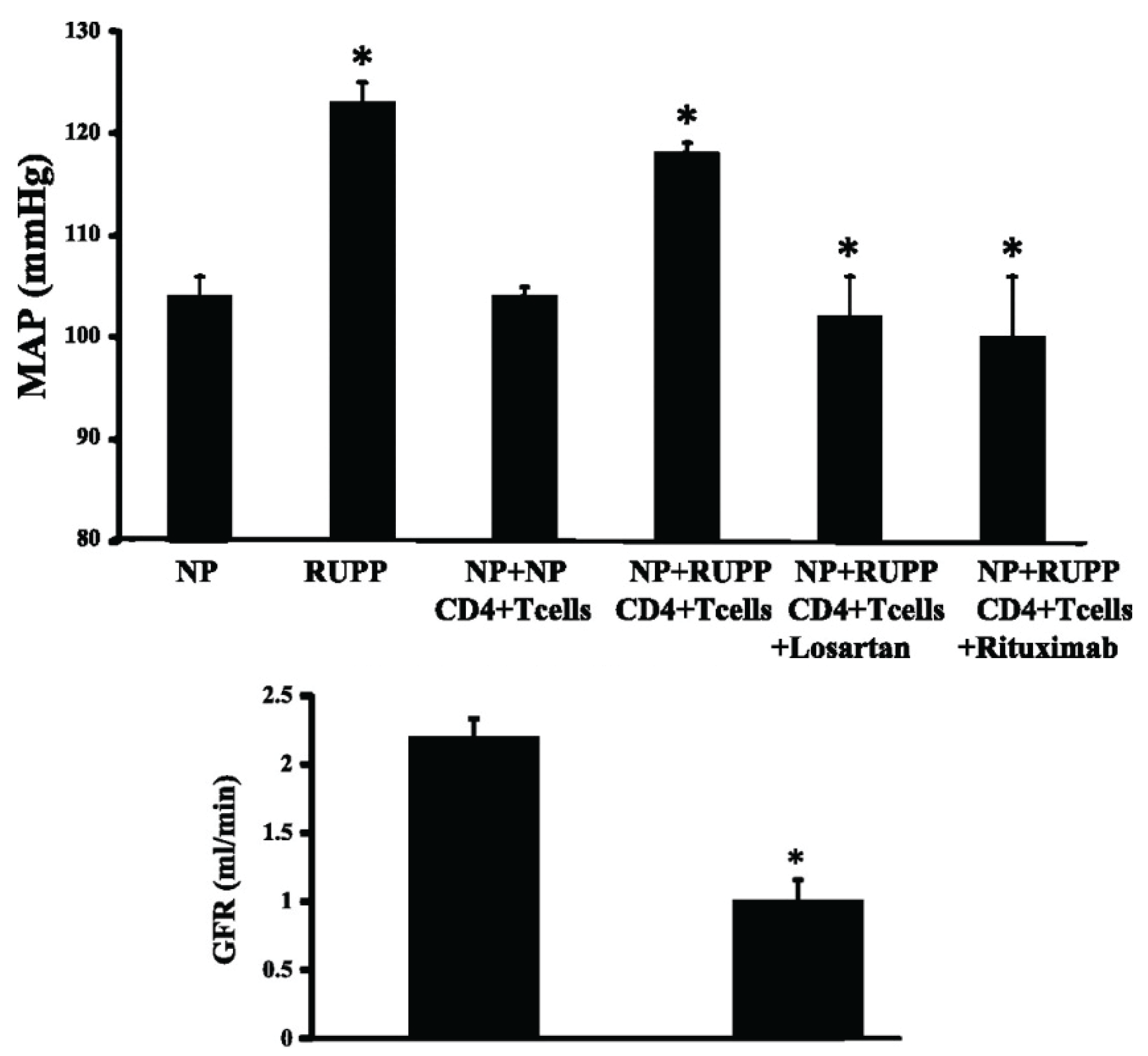
4.2. B lymphocytes
5. Impact of Obesity on Placental Ischemia-Induced Vascular Inflammation and Dysfunction
6. ET-1 as a Mechanism Linking Obesity, Inflammation and Hypertension in PE
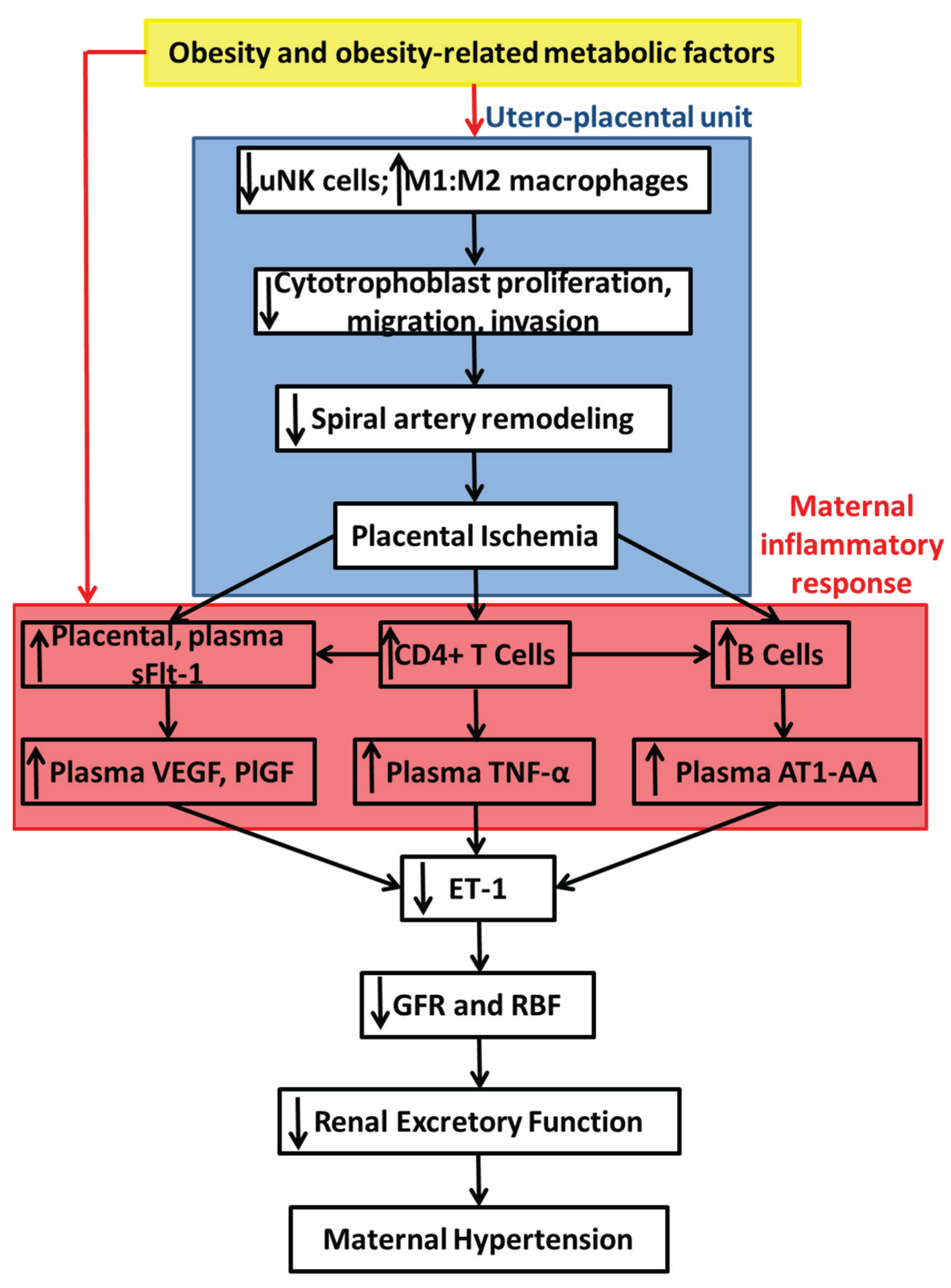
7. Summary of Proposed Mechanisms and Conclusions

Acknowledgments
Author Contributions
Conflicts of Interest
References
- American College of Obstetricians and Gynecologists; Task Force on Hypertension in Pregnancy. Hypertension in pregnancy. Report of the american college of obstetricians and gynecologists’ task force on hypertension in pregnancy. Obstet. Gynecol. 2013, 122, 1122–1131. [Google Scholar]
- Vogel, J.P.; Lee, A.C.; Souza, J.P. Maternal morbidity and preterm birth in 22 low- and middle-income countries: A secondary analysis of the who global survey dataset. BMC Pregnancy Childbirth 2014. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.S.; Wojdyla, D.; Say, L.; Gulmezoglu, A.M.; van Look, P.F. Who analysis of causes of maternal death: A systematic review. Lancet 2006, 367, 1066–1074. [Google Scholar] [CrossRef]
- Steegers, E.A.; von Dadelszen, P.; Duvekot, J.J.; Pijnenborg, R. Pre-eclampsia. Lancet 2010, 376, 631–644. [Google Scholar] [CrossRef]
- Hytten, F. Blood volume changes in normal pregnancy. Clin. Haematol. 1985, 14, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Osol, G.; Moore, L.G. Maternal uterine vascular remodeling during pregnancy. Microcirculation 2014, 21, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Whitley, G.S.; Cartwright, J.E. Cellular and molecular regulation of spiral artery remodelling: Lessons from the cardiovascular field. Placenta 2010, 31, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.K.; Vance, A.M.; Raj, R.S.; Mandala, M.; Linder, E.A.; Gokina, N.I. Impact of experimental diabetes on the maternal uterine vascular remodeling during rat pregnancy. Reprod. Sci. 2012, 19, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, S.J.; Khalil, R.A. Risk factors and mediators of the vascular dysfunction associated with hypertension in pregnancy. Cardiovasc. Hematol. Disord. Drug Targets 2010, 10, 33–52. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.M. Pathophysiology of ischemic placental disease. Semin. Perinatol. 2014, 38, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Ashton, S.V.; Whitley, G.S.; Dash, P.R.; Wareing, M.; Crocker, I.P.; Baker, P.N.; Cartwright, J.E. Uterine spiral artery remodeling involves endothelial apoptosis induced by extravillous trophoblasts through fas/fasl interactions. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Hunkapiller, N.M.; Gasperowicz, M.; Kapidzic, M.; Plaks, V.; Maltepe, E.; Kitajewski, J.; Cross, J.C.; Fisher, S.J. A role for notch signaling in trophoblast endovascular invasion and in the pathogenesis of pre-eclampsia. Development 2011, 138, 2987–2998. [Google Scholar] [CrossRef] [PubMed]
- Lyall, F.; Robson, S.C.; Bulmer, J.N. Spiral artery remodeling and trophoblast invasion in preeclampsia and fetal growth restriction: Relationship to clinical outcome. Hypertension 2013, 62, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.P.; Gerber, S.A.; Croy, B.A. Uterine natural killer cells pace early development of mouse decidua basalis. Mol. Hum. Reprod. 2014, 20, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Aplin, J.D.; Haigh, T.; Jones, C.J.; Church, H.J.; Vicovac, L. Development of cytotrophoblast columns from explanted first-trimester human placental villi: Role of fibronectin and integrin alpha5beta1. Biol. Reprod. 1999, 60, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Salomon, C.; Yee, S.W.; Mitchell, M.D.; Rice, G.E. The possible role of extravillous trophoblast-derived exosomes on the uterine spiral arterial remodeling under both normal and pathological conditions. Biomed. Res. Int. 2014. [Google Scholar] [CrossRef] [PubMed]
- Robson, A.; Harris, L.K.; Innes, B.A.; Lash, G.E.; Aljunaidy, M.M.; Aplin, J.D.; Baker, P.N.; Robson, S.C.; Bulmer, J.N. Uterine natural killer cells initiate spiral artery remodeling in human pregnancy. FASEB J. 2012, 26, 4876–4885. [Google Scholar] [CrossRef] [PubMed]
- Renaud, S.J.; Postovit, L.M.; Macdonald-Goodfellow, S.K.; McDonald, G.T.; Caldwell, J.D.; Graham, C.H. Activated macrophages inhibit human cytotrophoblast invasiveness in vitro. Biol. Reprod. 2005, 73, 237–243. [Google Scholar] [CrossRef] [PubMed]
- LaMarca, B.; Cornelius, D.; Wallace, K. Elucidating immune mechanisms causing hypertension during pregnancy. Physiology 2013, 28, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Tal, R. The role of hypoxia and hypoxia-inducible factor-1alpha in preeclampsia pathogenesis. Biol. Reprod. 2012. [Google Scholar] [CrossRef] [PubMed]
- Iriyama, T.; Wang, W.; Parchim, N.F.; Song, A.; Blackwell, S.C.; Sibai, B.M.; Kellems, R.E.; Xia, Y. Hypoxia-independent upregulation of placental hypoxia inducible factor-1alpha gene expression contributes to the pathogenesis of preeclampsia. Hypertension 2015, 65, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Losonczy, G.; Brown, G.; Venuto, R.C. Increased peripheral resistance during reduced uterine perfusion pressure hypertension in pregnant rabbits. Am. J. Med. Sci. 1992, 303, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.S.; Ryan, M.J.; LaMarca, B.B.; Sedeek, M.; Murphy, S.R.; Granger, J.P. Pathophysiology of hypertension during preeclampsia: Linking placental ischemia with endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H541–H550. [Google Scholar] [CrossRef] [PubMed]
- Hussein, W.; Lafayette, R.A. Renal function in normal and disordered pregnancy. Curr. Opin. Nephrol. Hypertens. 2014, 23, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Sholook, M.M.; Gilbert, J.S.; Sedeek, M.H.; Huang, M.; Hester, R.L.; Granger, J.P. Systemic hemodynamic and regional blood flow changes in response to chronic reductions in uterine perfusion pressure in pregnant rats. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2080–H2084. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ananth, C.V.; Keyes, K.M.; Wapner, R.J. Pre-eclampsia rates in the united states, 1980–2010: Age-period-cohort analysis. BMJ 2013. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.; Shaw, D. The worldwide epidemic of female obesity. Best Pract. Res. Clin. Obstet. Gynaecol. 2015, 29, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Alexandra, P.; Vassilios, B.; Alexandra, V.; George, K.; Vassiliki, L.; Chryssa, B. Population-based trends of pregnancy outcome in obese mothers: What has changed over 15 years. Obesity 2011, 19, 1861–1865. [Google Scholar] [CrossRef] [PubMed]
- Popkin, B.M.; Slining, M.M. New dynamics in global obesity facing low- and middle-income countries. Obes. Rev. 2013, 14, S11–S20. [Google Scholar] [CrossRef] [PubMed]
- Flegal, K.M.; Carroll, M.D.; Kit, B.K.; Ogden, C.L. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999–2010. JAMA 2012, 307, 491–497. [Google Scholar] [CrossRef] [PubMed]
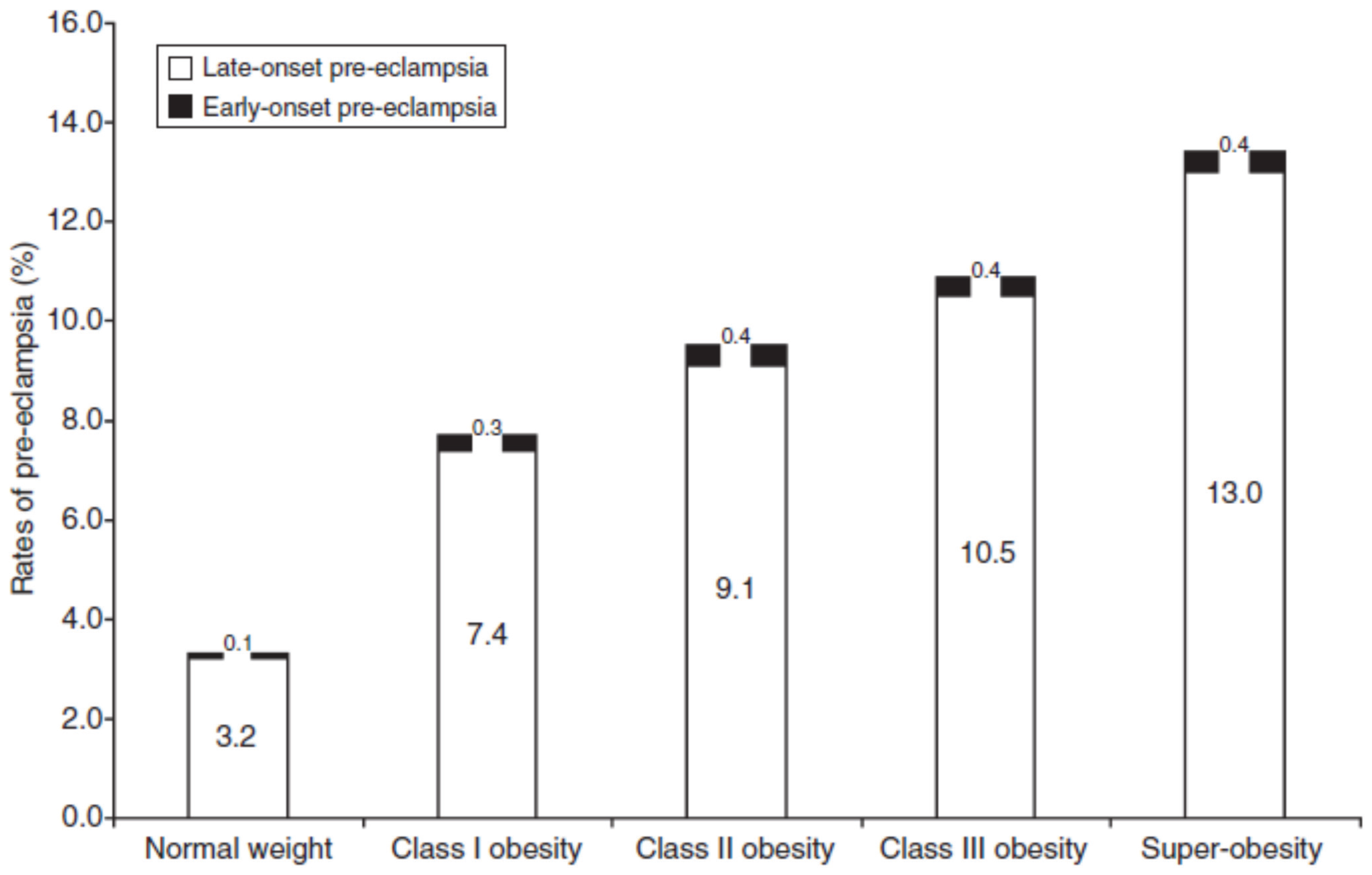
- Mbah, A.K.; Kornosky, J.L.; Kristensen, S.; August, E.M.; Alio, A.P.; Marty, P.J.; Belogolovkin, V.; Bruder, K.; Salihu, H.M. Super-obesity and risk for early and late pre-eclampsia. BJOG 2010, 117, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C.; Ahluwalia, N.; Brouns, F.; Buetler, T.; Clement, K.; Cunningham, K.; Esposito, K.; Jonsson, L.S.; Kolb, H.; Lansink, M.; et al. Dietary factors and low-grade inflammation in relation to overweight and obesity. Br. J. Nut. 2011, 106, S5–S78. [Google Scholar] [CrossRef] [PubMed]
- Romeo, G.R.; Lee, J.; Shoelson, S.E. Metabolic syndrome, insulin resistance, and roles of inflammation—Mechanisms and therapeutic targets. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1771–1776. [Google Scholar] [CrossRef] [PubMed]
- Womack, J.; Tien, P.C.; Feldman, J.; Shin, J.H.; Fennie, K.; Anastos, K.; Cohen, M.H.; Bacon, M.C.; Minkoff, H. Obesity and immune cell counts in women. Metabolism 2007, 56, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, L.; Lips, M.A.; Visser, A.; Pijl, H.; Ioan-Facsinay, A.; Toes, R.; Berends, F.J.; van Dijk, K.W.; Koning, F.; van Harmelen, V. Increased systemic and adipose tissue inflammation differentiates obese women with T2DM from obese women with normal glucose tolerance. Metabolism 2014, 63, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.; Rote, N.S.; Minium, J.; O’Leary V, B.; Kirwan, J.P. Obese reproductive-age women exhibit a proatherogenic inflammatory response during hyperglycemia. Obesity 2007, 15, 2436–2444. [Google Scholar] [CrossRef] [PubMed]
- Singer, K.; Maley, N.; Mergian, T.; DelProposto, J.; Cho, K.W.; Zamarron, B.F.; Martinez-Santibanez, G.; Geletka, L.; Muir, L.; Wachowiak, P.; et al. Differences in hematopoietic stem cells contribute to sexually dimorphic inflammatory responses to high fat diet induced obesity. J. Biol. Chem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.R.; Hui, H.; Kuvin, J.T.; Pandian, N.G.; Karas, R.H. Modestly overweight women have vascular endothelial dysfunction. Clin. Cardiol. 2009, 32, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Cola, M.S.; Gava, A.L.; Meyrelles, S.S.; Vasquez, E.C. Endothelial dysfunction of resistance vessels in female apolipoprotein e-deficient mice. Lipids Health Dis. 2010. [Google Scholar] [CrossRef] [PubMed]
- Meyrelles, S.S.; Peotta, V.A.; Pereira, T.M.; Vasquez, E.C. Endothelial dysfunction in the apolipoprotein e-deficient mouse: Insights into the influence of diet, gender and aging. Lipids Health Dis. 2011. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Wang, J.; Xue, D.; Zhu, Z.; Chen, Z.; Li, X.; Su, D.; Du, J. Why does a high-fat diet induce preeclampsia-like symptoms in pregnant rats. Neural Regen. Res. 2013, 8, 1872–1880. [Google Scholar] [PubMed]
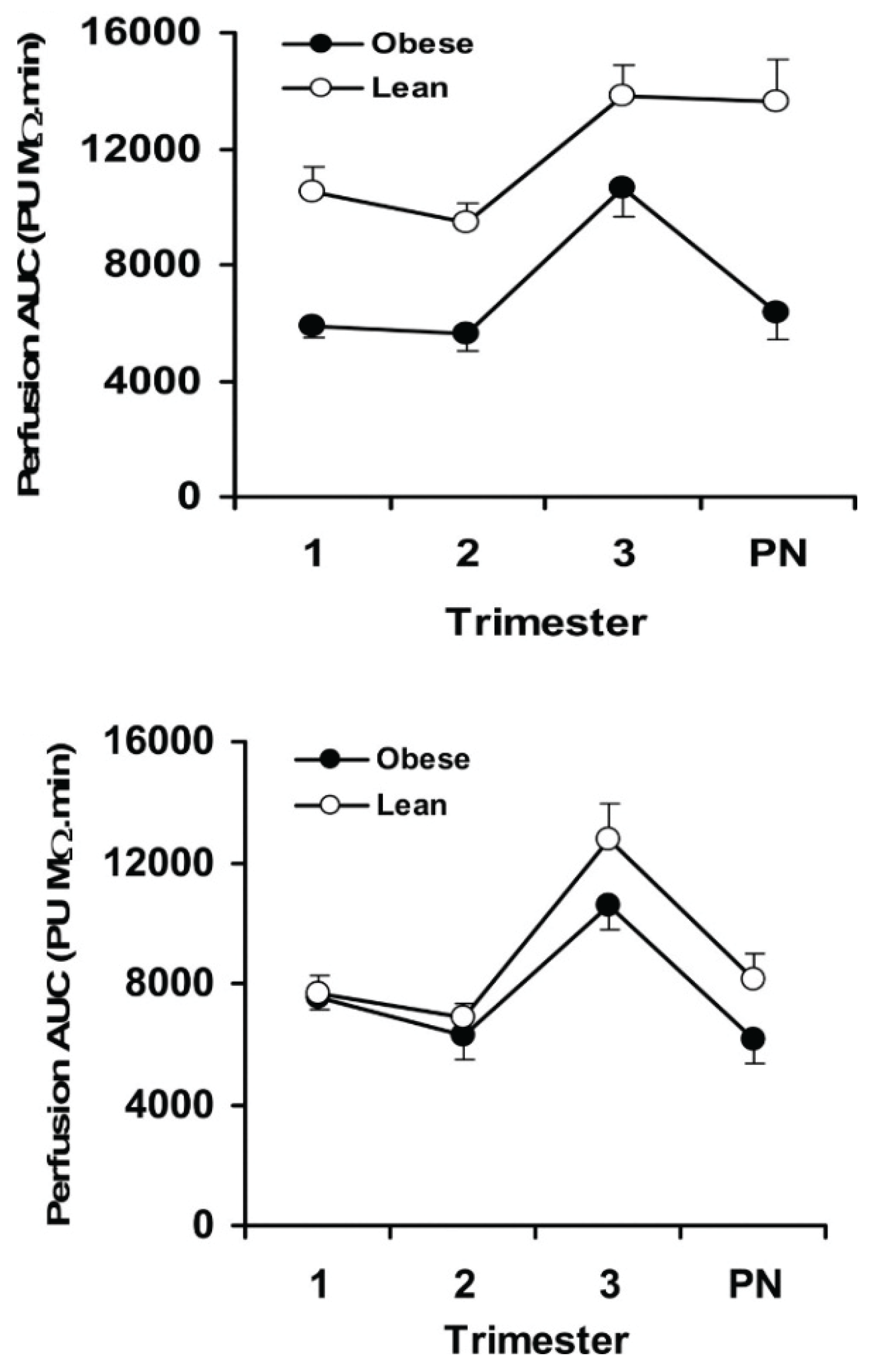
- Stewart, F.M.; Freeman, D.J.; Ramsay, J.E.; Greer, I.A.; Caslake, M.; Ferrell, W.R. Longitudinal assessment of maternal endothelial function and markers of inflammation and placental function throughout pregnancy in lean and obese mothers. J. Clin. Endocrinol. Metab. 2007, 92, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Hayward, C.E.; Cowley, E.J.; Mills, T.A.; Sibley, C.P.; Wareing, M. Maternal obesity impairs specific regulatory pathways in human myometrial arteries. Biol. Reprod. 2014. [Google Scholar] [CrossRef] [PubMed]
- Saftlas, A.F.; Levine, R.J.; Klebanoff, M.A.; Martz, K.L.; Ewell, M.G.; Morris, C.D.; Sibai, B.M. Abortion, changed paternity, and risk of preeclampsia in nulliparous women. Am. J. Epidemiol. 2003, 157, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Robillard, P.Y.; Hulsey, T.C.; Alexander, G.R.; Keenan, A.; de Caunes, F.; Papiernik, E. Paternity patterns and risk of preeclampsia in the last pregnancy in multiparae. J. Reprod. Immunol. 1993, 24, 1–12. [Google Scholar] [CrossRef]
- Robillard, P.Y.; Hulsey, T.C.; Perianin, J.; Janky, E.; Miri, E.H.; Papiernik, E. Association of pregnancy-induced hypertension with duration of sexual cohabitation before conception. Lancet 1994, 344, 973–975. [Google Scholar] [CrossRef]
- Robertson, S.A.; Ingman, W.V.; O’Leary, S.; Sharkey, D.J.; Tremellen, K.P. Transforming growth factor beta—A mediator of immune deviation in seminal plasma. J. Reprod. Immunol. 2002, 57, 109–128. [Google Scholar] [CrossRef]
- Aye, I.L.; Lager, S.; Ramirez, V.I.; Gaccioli, F.; Dudley, D.J.; Jansson, T.; Powell, T.L. Increasing maternal body mass index is associated with systemic inflammation in the mother and the activation of distinct placental inflammatory pathways. Biol. Reprod. 2014. [Google Scholar] [CrossRef] [PubMed]
- Saben, J.; Lindsey, F.; Zhong, Y.; Thakali, K.; Badger, T.M.; Andres, A.; Gomez-Acevedo, H.; Shankar, K. Maternal obesity is associated with a lipotoxic placental environment. Placenta 2014, 35, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.A.; Riley, S.C.; Reynolds, R.M.; Barr, S.; Evans, M.; Statham, A.; Hor, K.; Jabbour, H.N.; Norman, J.E.; Denison, F.C. Placental structure and inflammation in pregnancies associated with obesity. Placenta 2011, 32, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Hayes, E.K.; Tessier, D.R.; Percival, M.E.; Holloway, A.C.; Petrik, J.J.; Gruslin, A.; Raha, S. Trophoblast invasion and blood vessel remodeling are altered in a rat model of lifelong maternal obesity. Reprod Sci 2014, 21, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Hayes, E.K.; Lechowicz, A.; Petrik, J.J.; Storozhuk, Y.; Paez-Parent, S.; Dai, Q.; Samjoo, I.A.; Mansell, M.; Gruslin, A.; Holloway, A.C.; et al. Adverse fetal and neonatal outcomes associated with a life-long high fat diet: Role of altered development of the placental vasculature. PloS ONE 2012, 7, e33370. [Google Scholar] [CrossRef] [PubMed]
- Frias, A.E.; Morgan, T.K.; Evans, A.E.; Rasanen, J.; Oh, K.Y.; Thornburg, K.L.; Grove, K.L. Maternal high-fat diet disturbs uteroplacental hemodynamics and increases the frequency of stillbirth in a nonhuman primate model of excess nutrition. Endocrinology 2011, 152, 2456–2464. [Google Scholar] [CrossRef] [PubMed]
- Anim-Nyame, N.; Sooranna, S.R.; Steer, P.J.; Johnson, M.R. Longitudinal analysis of maternal plasma leptin concentrations during normal pregnancy and pre-eclampsia. Hum Reprod 2000, 15, 2033–2036. [Google Scholar] [CrossRef] [PubMed]
- Zeron, H.M.; Solorio, V.J.G.; Diaz, P.M.N.; Alanis, A.G.; Benitez, J.G.S.; Garcia, V.D.; Briones, C.E.; Gutierrez, E.D. Hyperleptinemia as a prognostic factor for preeclampsia: A cohort study. Acta Med. 2012, 55, 165–171. [Google Scholar]
- Ning, Y.; Williams, M.A.; Muy-Rivera, M.; Leisenring, W.M.; Luthy, D.A. Relationship of maternal plasma leptin and risk of pre-eclampsia: A prospective study. J. Matern. Fetal Neonatal. Med. 2004, 15, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Papastefanou, I.; Samolis, S.; Panagopoulos, P.; Tagia, M.; Bale, C.; Kouskoukis, A.; Galazios, G. Correlation between maternal first trimester plasma leptin levels and birth weight among normotensive and preeclamptic women. J. Matern. Fetal Neonatal Med. 2010, 23, 1435–1443. [Google Scholar] [CrossRef] [PubMed]
- Samolis, S.; Papastefanou, I.; Panagopoulos, P.; Galazios, G.; Kouskoukis, A.; Maroulis, G. Relation between first trimester maternal serum leptin levels and body mass index in normotensive and pre-eclamptic pregnancies—Role of leptin as a marker of pre-eclampsia: A prospective case-control study. Gynecol. Endocrinol. 2010, 26, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.D.; Ness, R.B.; Olsen, J.; Hougaard, D.M.; Skogstrand, K.; Roberts, J.M.; Haggerty, C.L. Serum leptin measured in early pregnancy is higher in women with preeclampsia compared with normotensive pregnant women. Hypertension 2015, 65, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Iwagaki, S.; Yokoyama, Y.; Tang, L.; Takahashi, Y.; Nakagawa, Y.; Tamaya, T. Augmentation of leptin and hypoxia-inducible factor 1alpha mRNAs in the pre-eclamptic placenta. Gynecol. Endocrinol. 2004, 18, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Lepercq, J.; Guerre-Millo, M.; Andre, J.; Cauzac, M.; Hauguel-de Mouzon, S. Leptin: A potential marker of placental insufficiency. Gynecol. Obstet Investig. 2003, 55, 151–155. [Google Scholar] [CrossRef]
- Mise, H.; Sagawa, N.; Matsumoto, T.; Yura, S.; Nanno, H.; Itoh, H.; Mori, T.; Masuzaki, H.; Hosoda, K.; Ogawa, Y.; et al. Augmented placental production of leptin in preeclampsia: Possible involvement of placental hypoxia. J. Clin. Endocrinol. Metab. 1998, 83, 3225–3229. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.J.; Davis, J.; Bryant-Greenwood, G. Systemic and placental leptin and its receptors in pregnancies associated with obesity. Reprod. Sci. 2015, 22, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Mazzucco, M.B.; Higa, R.; Capobianco, E.; Kurtz, M.; Jawerbaum, A.; White, V. Saturated fat-rich diet increases fetal lipids and modulates LPL and leptin receptor expression in rat placentas. J. Endocrinol. 2013, 217, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Bartha, J.L.; Romero-Carmona, R.; Escobar-Llompart, M.; Comino-Delgado, R. The relationships between leptin and inflammatory cytokines in women with pre-eclampsia. BJOG 2001, 108, 1272–1276. [Google Scholar] [PubMed]
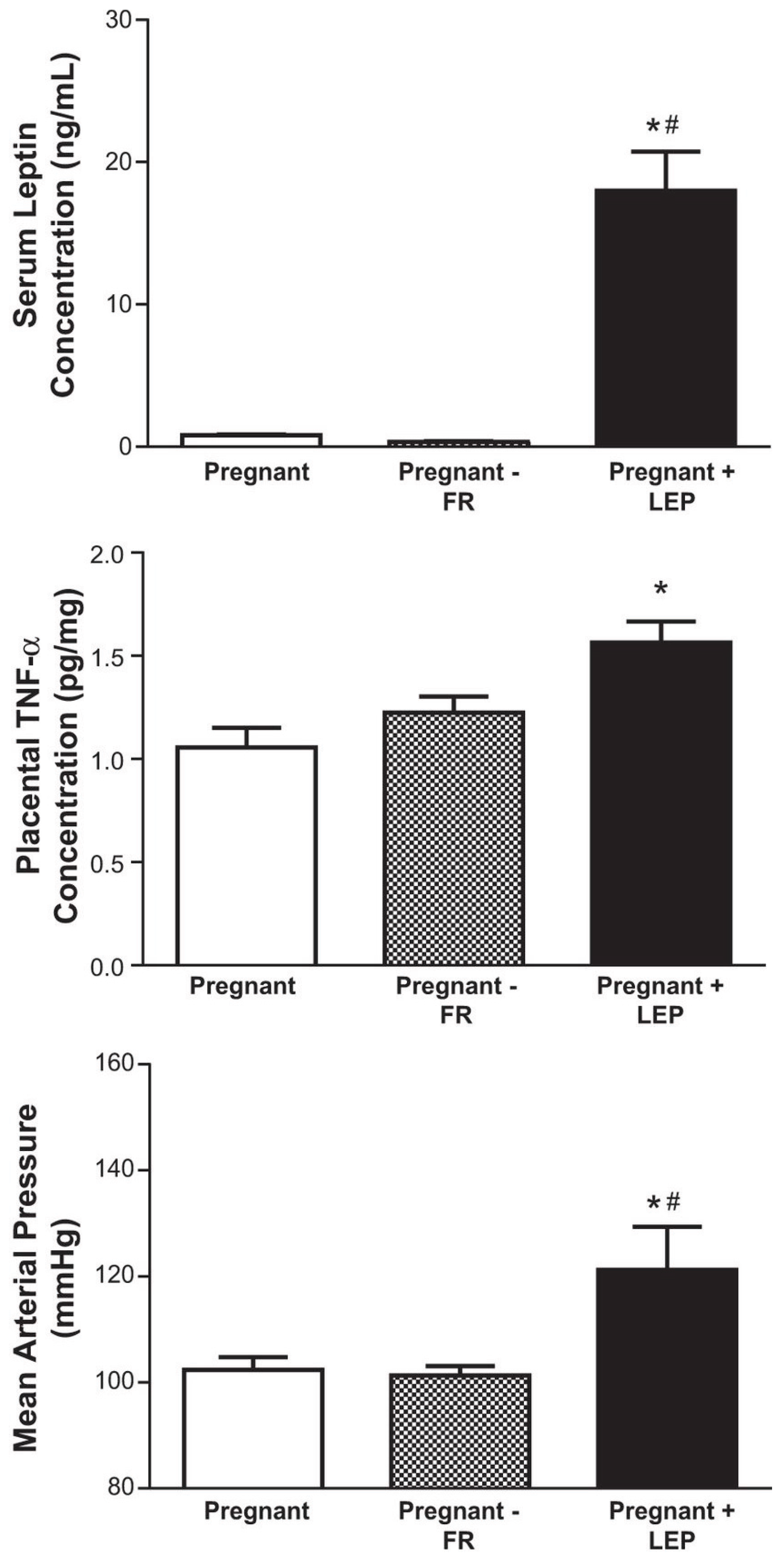
- Palei, A.C.; Spradley, F.T.; Granger, J.P. Chronic hyperleptinemia results in the development of hypertension in pregnant rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 38, R855–R861. [Google Scholar] [CrossRef] [PubMed]
- Lappas, M.; Permezel, M.; Rice, G.E. Leptin and adiponectin stimulate the release of proinflammatory cytokines and prostaglandins from human placenta and maternal adipose tissue via nuclear factor-kappaB, peroxisomal proliferator-activated receptor-gamma and extracellularly regulated kinase 1/2. Endocrinology 2005, 146, 3334–3342. [Google Scholar] [PubMed]
- Saben, J.; Zhong, Y.; Gomez-Acevedo, H.; Thakali, K.M.; Borengasser, S.J.; Andres, A.; Shankar, K. Early growth response protein-1 mediates lipotoxicity-associated placental inflammation: Role in maternal obesity. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E1–E14. [Google Scholar] [CrossRef] [PubMed]
- Faas, M.M.; Spaans, F.; de Vos, P. Monocytes and macrophages in pregnancy and pre-eclampsia. Front. Immunol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Wallace, A.E.; Fraser, R.; Cartwright, J.E. Extravillous trophoblast and decidual natural killer cells: A remodelling partnership. Hum. Reprod. Update 2012, 18, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.J.; Bulmer, J.N.; Searle, R.F.; Innes, B.A.; Robson, S.C. Altered decidual leucocyte populations in the placental bed in pre-eclampsia and foetal growth restriction: A comparison with late normal pregnancy. Reproduction 2009, 138, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Bulmer, J.N.; Williams, P.J.; Lash, G.E. Immune cells in the placental bed. Int. J. Dev. Biol. 2010, 54, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Le Bouteiller, P.; Siewiera, J.; Casart, Y.; Aguerre-Girr, M.; el Costa, H.; Berrebi, A.; Tabiasco, J.; Jabrane-Ferrat, N. The human decidual NK-cell response to virus infection: What can we learn from circulating NK lymphocytes? J. Reprod. Immunol. 2011, 88, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.; Goldman-Wohl, D.; Hamani, Y.; Avraham, I.; Greenfield, C.; Natanson-Yaron, S.; Prus, D.; Cohen-Daniel, L.; Arnon, T.I.; Manaster, I.; et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat. Med. 2006, 12, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- El Costa, H.; Tabiasco, J.; Berrebi, A.; Parant, O.; Aguerre-Girr, M.; Piccinni, M.P.; le Bouteiller, P. Effector functions of human decidual NK cells in healthy early pregnancy are dependent on the specific engagement of natural cytotoxicity receptors. J. Reprod. Immunol. 2009, 82, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Chazara, O.; Xiong, S.; Moffett, A. Maternal KIR and fetal HLA-C: A fine balance. J. Leukoc. Biol. 2011, 90, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.K. Review: Trophoblast-vascular cell interactions in early pregnancy: How to remodel a vessel. Placenta 2010, 31, S93–S98. [Google Scholar] [CrossRef] [PubMed]
- Guimond, M.J.; Luross, J.A.; Wang, B.; Terhorst, C.; Danial, S.; Croy, B.A. Absence of natural killer cells during murine pregnancy is associated with reproductive compromise in TgE26 mice. Biol. Reprod. 1997, 56, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Guimond, M.J.; Wang, B.; Croy, B.A. Engraftment of bone marrow from severe combined immunodeficient (SCID) mice reverses the reproductive deficits in natural killer cell-deficient tg epsilon 26 mice. J. Exp. Med. 1998, 187, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Ashkar, A.A.; di Santo, J.P.; Croy, B.A. Interferon gamma contributes to initiation of uterine vascular modification, decidual integrity, and uterine natural killer cell maturation during normal murine pregnancy. J. Exp. Med. 2000, 192, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Monk, J.M.; Leonard, S.; McBey, B.A.; Croy, B.A. Induction of murine spiral artery modification by recombinant human interferon-gamma. Placenta 2005, 26, 835–838. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.D.; Dunk, C.E.; Aplin, J.D.; Harris, L.K.; Jones, R.L. Evidence for immune cell involvement in decidual spiral arteriole remodeling in early human pregnancy. Am. J. Pathol. 2009, 174, 1959–1971. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Dutz, J.P.; MacCalman, C.D.; Yong, P.; Tan, R.; von Dadelszen, P. Decidual NK cells alter in vitro first trimester extravillous cytotrophoblast migration: A role for IFN-gamma. J. Immunol. 2006, 177, 8522–8530. [Google Scholar] [CrossRef] [PubMed]
- Kalkunte, S.S.; Mselle, T.F.; Norris, W.E.; Wira, C.R.; Sentman, C.L.; Sharma, S. Vascular endothelial growth factor C facilitates immune tolerance and endovascular activity of human uterine NK cells at the maternal-fetal interface. J. Immunol. 2009, 182, 4085–4092. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Nishikawa, K.; Morii, T.; Enomoto, M.; Narita, N.; Motoyoshi, K.; Ichijo, M. Cytokine production by CD16-CD56bright natural killer cells in the human early pregnancy decidua. Int. Immunol. 1993, 5, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Otun, H.A.; Lash, G.E.; Innes, B.A.; Bulmer, J.N.; Naruse, K.; Hannon, T.; Searle, R.F.; Robson, S.C. Effect of tumour necrosis factor-alpha in combination with interferon-gamma on first trimester extravillous trophoblast invasion. J. Reprod. Immunol. 2011, 88, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, M.; Stefanoska, I.; Radojcic, L.; Vicovac, L. Interleukin-8 (CXCL8) stimulates trophoblast cell migration and invasion by increasing levels of matrix metalloproteinase (MMP)2 and MMP9 and integrins alpha5 and beta1. Reproduction 2010, 139, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Parker, V.J.; Solano, M.E.; Arck, P.C.; Douglas, A.J. Diet-induced obesity may affect the uterine immune environment in early-mid pregnancy, reducing NK-cell activity and potentially compromising uterine vascularization. Int. J. Obes. 2014, 38, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Lynch, L.A.; O’Connell, J.M.; Kwasnik, A.K.; Cawood, T.J.; O’Farrelly, C.; O’Shea, D.B. Are natural killer cells protecting the metabolically healthy obese patient? Obesity 2009, 17, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Huebner, L.; Engeli, S.; Wrann, C.D.; Goudeva, L.; Laue, T.; Kielstein, H. Human NK cell subset functions are differentially affected by adipokines. PLoS ONE 2013, 8, e75703. [Google Scholar] [CrossRef] [PubMed]
- Orlova, E.G.; Shirshev, S.V. Leptin as an immunocorrecting agent during normal pregnancy. Bull. Exp. Biol. Med. 2009, 148, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Wrann, C.D.; Laue, T.; Hubner, L.; Kuhlmann, S.; Jacobs, R.; Goudeva, L.; Nave, H. Short-term and long-term leptin exposure differentially affect human natural killer cell immune functions. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E108–E116. [Google Scholar] [CrossRef] [PubMed]
- Haas, P.; Straub, R.H.; Bedoui, S.; Nave, H. Peripheral but not central leptin treatment increases numbers of circulating NK cells, granulocytes and specific monocyte subpopulations in non-endotoxaemic lean and obese LEW-rats. Regul. Pept. 2008, 151, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Lamas, B.; Goncalves-Mendes, N.; Nachat-Kappes, R.; Rossary, A.; Caldefie-Chezet, F.; Vasson, M.P.; Farges, M.C. Leptin modulates dose-dependently the metabolic and cytolytic activities of NK-92 cells. J. Cell Physiol. 2013, 228, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Nave, H.; Mueller, G.; Siegmund, B.; Jacobs, R.; Stroh, T.; Schueler, U.; Hopfe, M.; Behrendt, P.; Buchenauer, T.; Pabst, R.; et al. Resistance of janus kinase-2 dependent leptin signaling in natural killer (NK) cells: A novel mechanism of NK cell dysfunction in diet-induced obesity. Endocrinology 2008, 149, 3370–3378. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sun, R.; You, L.; Gao, C.; Tian, Z. Expression of leptin receptors and response to leptin stimulation of human natural killer cell lines. Biochem. Biophys. Res. Commun. 2003, 300, 247–252. [Google Scholar] [CrossRef]
- Gustafsson, C.; Mjosberg, J.; Matussek, A.; Geffers, R.; Matthiesen, L.; Berg, G.; Sharma, S.; Buer, J.; Ernerudh, J. Gene expression profiling of human decidual macrophages: Evidence for immunosuppressive phenotype. PLoS ONE 2008, 3, e2078. [Google Scholar] [CrossRef] [PubMed]
- Singh, U.; Nicholson, G.; Urban, B.C.; Sargent, I.L.; Kishore, U.; Bernal, A.L. Immunological properties of human decidual macrophages—A possible role in intrauterine immunity. Reproduction 2005, 129, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Nagamatsu, T.; Schust, D.J. The immunomodulatory roles of macrophages at the maternal-fetal interface. Reprod. Sci. 2010, 17, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, V.M.; Kim, Y.M.; Straszewski, S.L.; Romero, R.; Mor, G. Macrophages and apoptotic cell clearance during pregnancy. Am. J. Reprod. Immunol. 2004, 51, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Burk, M.R.; Troeger, C.; Brinkhaus, R.; Holzgreve, W.; Hahn, S. Severely reduced presence of tissue macrophages in the basal plate of pre-eclamptic placentae. Placenta 2001, 22, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Reister, F.; Frank, H.G.; Heyl, W.; Kosanke, G.; Huppertz, B.; Schroder, W.; Kaufmann, P.; Rath, W. The distribution of macrophages in spiral arteries of the placental bed in pre-eclampsia differs from that in healthy patients. Placenta 1999, 20, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Schonkeren, D.; van der Hoorn, M.L.; Khedoe, P.; Swings, G.; van Beelen, E.; Claas, F.; van Kooten, C.; de Heer, E.; Scherjon, S. Differential distribution and phenotype of decidual macrophages in preeclamptic versus control pregnancies. Am. J. Pathol. 2011, 178, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Hoshimoto, K.; Ohkura, T.; Inaba, N. Increased levels of macrophage colony-stimulating factor in the placenta and blood in preeclampsia. Am. J. Reprod. Immunol. 2002, 47, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Katabuchi, H.; Yih, S.; Ohba, T.; Matsui, K.; Takahashi, K.; Takeya, M.; Okamura, H. Characterization of macrophages in the decidual atherotic spiral artery with special reference to the cytology of foam cells. Med. Electron. Microsc. 2003, 36, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Keelan, J.A.; Mitchell, M.D. Placental cytokines and preeclampsia. Front. Biosci. 2007, 12, 2706–2727. [Google Scholar] [CrossRef] [PubMed]
- Reister, F.; Frank, H.G.; Kingdom, J.C.; Heyl, W.; Kaufmann, P.; Rath, W.; Huppertz, B. Macrophage-induced apoptosis limits endovascular trophoblast invasion in the uterine wall of preeclamptic women. Lab. Investig. 2001, 81, 1143–1152. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, O.V.; Sheveleva, T.S.; Selkov, S.A. In vitro expression of vascular endothelial growth factor and its receptors by placental macrophages. Bull. Exp. Biol. Med. 2012, 153, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Prins, J.R.; Faas, M.M.; Melgert, B.N.; Huitema, S.; Timmer, A.; Hylkema, M.N.; Erwich, J.J. Altered expression of immune-associated genes in first-trimester human decidua of pregnancies later complicated with hypertension or foetal growth restriction. Placenta 2012, 33, 453–455. [Google Scholar] [CrossRef] [PubMed]
- Melgert, B.N.; Spaans, F.; Borghuis, T.; Klok, P.A.; Groen, B.; Bolt, A.; de Vos, P.; van Pampus, M.G.; Wong, T.Y.; van Goor, H.; et al. Pregnancy and preeclampsia affect monocyte subsets in humans and rats. PLoS ONE 2012, 7, e45229. [Google Scholar] [CrossRef] [PubMed]
- Nagamatsu, T.; Schust, D.J. The contribution of macrophages to normal and pathological pregnancies. Am. J. Reprod. Immunol. 2010, 63, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Lockwood, C.J.; Basar, M.; Kayisli, U.A.; Guzeloglu-Kayisli, O.; Murk, W.; Wang, J.; de Paz, N.; Shapiro, J.P.; Masch, R.J.; Semerci, N.; et al. Interferon-gamma protects first-trimester decidual cells against aberrant matrix metalloproteinases 1, 3, and 9 expression in preeclampsia. Am. J. Pathol. 2014, 184, 2549–2559. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef] [PubMed]
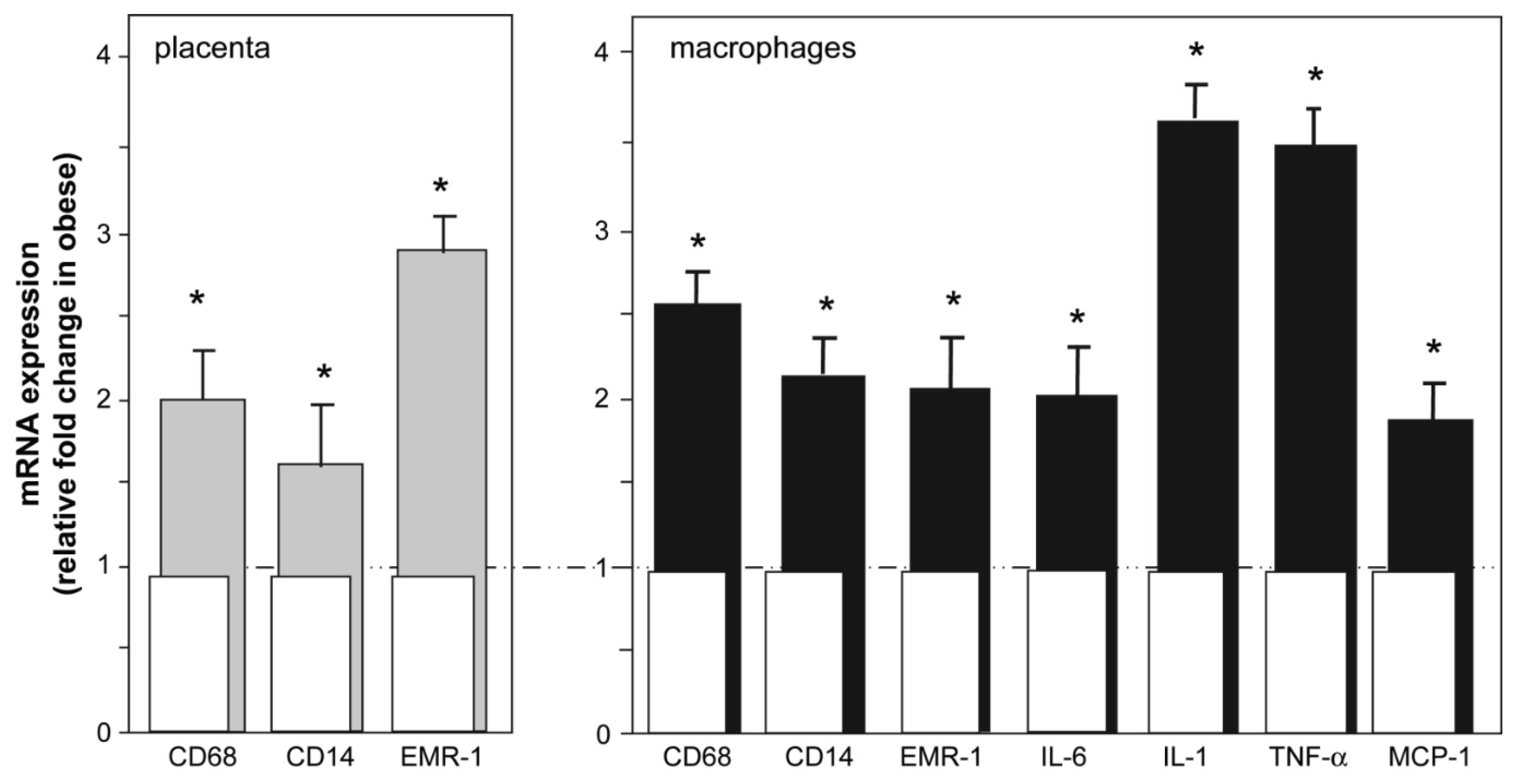
- Challier, J.C.; Basu, S.; Bintein, T.; Minium, J.; Hotmire, K.; Catalano, P.M.; Hauguel-de Mouzon, S. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta 2008, 29, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.J.; Du, M.; Nathanielsz, P.W.; Ford, S.P. Maternal obesity up-regulates inflammatory signaling pathways and enhances cytokine expression in the mid-gestation sheep placenta. Placenta 2010, 31, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Farley, D.; Tejero, M.E.; Comuzzie, A.G.; Higgins, P.B.; Cox, L.; Werner, S.L.; Jenkins, S.L.; Li, C.; Choi, J.; Dick, E.J., Jr.; et al. Feto-placental adaptations to maternal obesity in the baboon. Placenta 2009, 30, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Benyo, D.F.; Smarason, A.; Redman, C.W.; Sims, C.; Conrad, K.P. Expression of inflammatory cytokines in placentas from women with preeclampsia. J. Clin. Endocrinol. Metab. 2001, 86, 2505–2512. [Google Scholar] [CrossRef] [PubMed]
- Peracoli, M.T.; Bannwart, C.F.; Cristofalo, R.; Borges, V.T.; Costa, R.A.; Witkin, S.S.; Peracoli, J.C. Increased reactive oxygen species and tumor necrosis factor-alpha production by monocytes are associated with elevated levels of uric acid in pre-eclamptic women. Am. J. Reprod. Immunol. 2011, 66, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Leahy, P.; Challier, J.C.; Minium, J.; Catalano, P.; Hauguel-de Mouzon, S. Molecular phenotype of monocytes at the maternal-fetal interface. Am. J. Obstet. Gynecol. 2011, 205, e261–e268. [Google Scholar] [CrossRef] [PubMed]
- Staff, A.C.; Johnsen, G.M.; Dechend, R.; Redman, C.W. Preeclampsia and uteroplacental acute atherosis: Immune and inflammatory factors. J. Reprod. Immunol. 2014, 101–102, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Harsem, N.K.; Roald, B.; Braekke, K.; Staff, A.C. Acute atherosis in decidual tissue: Not associated with systemic oxidative stress in preeclampsia. Placenta 2007, 28, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Preston-Hurlburt, P.; Janeway, C.A., Jr. A human homologue of the drosophila toll protein signals activation of adaptive immunity. Nature 1997, 388, 394–397. [Google Scholar] [PubMed]
- Han, G.M.; O’Neil-Andersen, N.J.; Zurier, R.B.; Lawrence, D.A. CD4+CD25HIGH T cell numbers are enriched in the peripheral blood of patients with rheumatoid arthritis. Cell. Immunol. 2008, 253, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Longhi, M.S.; Wang, P.; Vergani, D.; Ma, Y. Human CD4+CD25(high)CD127 (low/neg) regulatory T cells. Methods Mol. Biol. 2012, 806, 287–299. [Google Scholar] [PubMed]
- Santner-Nanan, B.; Peek, M.J.; Khanam, R.; Richarts, L.; Zhu, E.; de St Groth, B.F.; Nanan, R. Systemic increase in the ratio between Foxp3+ and IL-17-producing CD4+ T cells in healthy pregnancy but not in preeclampsia. J. Immunol. 2009, 183, 7023–7030. [Google Scholar] [CrossRef] [PubMed]
- Granger, J.P.; LaMarca, B.B.; Cockrell, K.; Sedeek, M.; Balzi, C.; Chandler, D.; Bennett, W. Reduced uterine perfusion pressure (RUPP) model for studying cardiovascular-renal dysfunction in response to placental ischemia. Methods Mol. Med. 2006, 122, 383–392. [Google Scholar] [PubMed]
- Tam Tam, K.B.; George, E.; Cockrell, K.; Arany, M.; Speed, J.; Martin, J.N., Jr.; Lamarca, B.; Granger, J.P. Endothelin type a receptor antagonist attenuates placental ischemia-induced hypertension and uterine vascular resistance. Am. J. Obstet. Gynecol. 2011, 204, e331–e334. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.; Richards, S.; Dhillon, P.; Weimer, A.; Edholm, E.S.; Bengten, E.; Wilson, M.; Martin, J.N., Jr.; LaMarca, B. CD4+ T-helper cells stimulated in response to placental ischemia mediate hypertension during pregnancy. Hypertension 2011, 57, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.; Novotny, S.; Heath, J.; Moseley, J.; Martin, J.N., Jr.; Owens, M.Y.; LaMarca, B. Hypertension in response to CD4(+) T cells from reduced uterine perfusion pregnant rats is associated with activation of the endothelin-1 system. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R144–R149. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.; Cornelius, D.C.; Scott, J.; Heath, J.; Moseley, J.; Chatman, K.; LaMarca, B. CD4+ T cells are important mediators of oxidative stress that cause hypertension in response to placental ischemia. Hypertension 2014, 64, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- Luppi, P.; Haluszczak, C.; Betters, D.; Richard, C.A.; Trucco, M.; DeLoia, J.A. Monocytes are progressively activated in the circulation of pregnant women. J. Leukoc. Biol. 2002, 72, 874–884. [Google Scholar] [PubMed]
- Wallace, F.A.; Miles, E.A.; Evans, C.; Stock, T.E.; Yaqoob, P.; Calder, P.C. Dietary fatty acids influence the production of th1- but not th2-type cytokines. J. Leukoc. Biol. 2001, 69, 449–457. [Google Scholar] [PubMed]
- Tao, S.Y.; Gallaher, M.; Roberts, J.M. [9-or]: Obese women who develop preeclampsia exhibit a different metabolic profile early in pregnancy compared to obese women with uncomplicated pregnancies. Pregnancy Hypertens. 2015. [Google Scholar] [CrossRef]
- Novotny, S.R.; Wallace, K.; Heath, J.; Moseley, J.; Dhillon, P.; Weimer, A.; Wallukat, G.; Herse, F.; Wenzel, K.; Martin, J.N., Jr.; et al. Activating autoantibodies to the angiotensin II type 1 receptor play an important role in mediating hypertension in response to adoptive transfer of CD4+ T lymphocytes from placental ischemic rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R1197–R1201. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Hoch, N.E.; Brown, K.A.; McCann, L.A.; Rahman, A.; Dikalov, S.; Goronzy, J.; Weyand, C.; Harrison, D.G. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 2007, 204, 2449–2460. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, C.; Das, S.; Lund, H.; Mattson, D.L. T lymphocytes mediate hypertension and kidney damage in Dahl salt-sensitive rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1136–R1142. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, C.; Lund, H.; Mattson, D.L. High dietary protein exacerbates hypertension and renal damage in Dahl ss rats by increasing infiltrating immune cells in the kidney. Hypertension 2011, 57, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Vaziri, N.D. Hypertension promotes integrin expression and reactive oxygen species generation by circulating leukocytes. Kidney Int. 2005, 67, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Gurantz, D.; Cowling, R.T.; Villarreal, F.J.; Greenberg, B.H. Tumor necrosis factor-alpha upregulates angiotensin II type 1 receptors on cardiac fibroblasts. Circ. Res. 1999, 85, 272–279. [Google Scholar] [CrossRef] [PubMed]
- LaMarca, B.; Wallukat, G.; Llinas, M.; Herse, F.; Dechend, R.; Granger, J.P. Autoantibodies to the angiotensin type I receptor in response to placental ischemia and tumor necrosis factor alpha in pregnant rats. Hypertension 2008, 52, 1168–1172. [Google Scholar] [CrossRef] [PubMed]
- LaMarca, B.D.; Gilbert, J.; Granger, J.P. Recent progress toward the understanding of the pathophysiology of hypertension during preeclampsia. Hypertension 2008, 51, 982–988. [Google Scholar] [CrossRef] [PubMed]
- LaMarca, B.B.; Cockrell, K.; Sullivan, E.; Bennett, W.; Granger, J.P. Role of endothelin in mediating tumor necrosis factor-induced hypertension in pregnant rats. Hypertension 2005, 46, 82–86. [Google Scholar] [CrossRef] [PubMed]
- LaMarca, B.; Parrish, M.; Ray, L.F.; Murphy, S.R.; Roberts, L.; Glover, P.; Wallukat, G.; Wenzel, K.; Cockrell, K.; Martin, J.N., Jr.; et al. Hypertension in response to autoantibodies to the angiotensin II type 1 receptor (AT1-AA) in pregnant rats: Role of endothelin-1. Hypertension 2009, 54, 905–909. [Google Scholar] [CrossRef] [PubMed]
- LaMarca, B.D.; Alexander, B.T.; Gilbert, J.S.; Ryan, M.J.; Sedeek, M.; Murphy, S.R.; Granger, J.P. Pathophysiology of hypertension in response to placental ischemia during pregnancy: A central role for endothelin? Gender Med. 2008, 5, S133–S138. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, H.P.; Chen, X.; Li, M.Q. Gestational diabetes induces chronic hypoxia stress and excessive inflammatory response in murine placenta. Int. J. Clin. Exp. Pathol. 2013, 6, 650–659. [Google Scholar] [PubMed]
- Moreno, R.; Sobotzik, J.M.; Schultz, C.; Schmitz, M.L. Specification of the NF-kappab transcriptional response by p65 phosphorylation and TNF-induced nuclear translocation of IKK epsilon. Nucl. Acids Res. 2010, 38, 6029–6044. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E. Nuclear factor-kappaB and its role in sepsis-associated organ failure. J. Infect. Dis. 2003, 187, S364–S369. [Google Scholar] [CrossRef] [PubMed]
- Schaefer-Graf, U.M.; Meitzner, K.; Ortega-Senovilla, H.; Graf, K.; Vetter, K.; Abou-Dakn, M.; Herrera, E. Differences in the implications of maternal lipids on fetal metabolism and growth between gestational diabetes mellitus and control pregnancies. Diabet. Med. 2011, 28, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Bomba-Opon, D.; Wielgos, M.; Szymanska, M.; Bablok, L. Effects of free fatty acids on the course of gestational diabetes mellitus. Neuro Endocrinol. Lett. 2006, 27, 277–280. [Google Scholar] [PubMed]
- Weiss, J.L.; Malone, F.D.; Emig, D.; Ball, R.H.; Nyberg, D.A.; Comstock, C.H.; Saade, G.; Eddleman, K.; Carter, S.M.; Craigo, S.D.; et al. Obesity, obstetric complications and cesarean delivery rate—A population-based screening study. Am. J. Obstet. Gynecol. 2004, 190, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Bryson, C.L.; Ioannou, G.N.; Rulyak, S.J.; Critchlow, C. Association between gestational diabetes and pregnancy-induced hypertension. Am. J. Epidemiol. 2003, 158, 1148–1153. [Google Scholar] [CrossRef] [PubMed]
- Desoye, G.; Hauguel-de Mouzon, S. The human placenta in gestational diabetes mellitus. The insulin and cytokine network. Diabetes Care 2007, 30, S120–S126. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhao, Y.H.; Chen, Y.P.; Yuan, X.L.; Wang, J.; Zhu, H.; Lu, C.M. Maternal circulating concentrations of tumor necrosis factor-alpha, leptin, and adiponectin in gestational diabetes mellitus: A systematic review and meta-analysis. Sci. World J. 2014, 2014. Article ID 926932. [Google Scholar] [CrossRef] [PubMed]
- Guermouche, B.; Yessoufou, A.; Soulimane, N.; Merzouk, H.; Moutairou, K.; Hichami, A.; Khan, N.A. N-3 fatty acids modulate t-cell calcium signaling in obese macrosomic rats. Obes. Res. 2004, 12, 1744–1753. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Wu, Q.Y.; Du, J.J.; Zeng, J.Y.; Li, T.T.; Xu, C.Q.; Sun, Y.H. Calcium-sensing receptor in the T lymphocyte enhanced the apoptosis and cytokine secretion in sepsis. Mol. Immunol. 2015, 63, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.; Mehta, S.; Bharrhan, S.; Chen, Y.; Achkar, J.M.; Casadevall, A.; Flynn, J. The role of B cells and humoral immunity in mycobacterium tuberculosis infection. Semin. Immunol. 2014, 26, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Fettke, F.; Schumacher, A.; Costa, S.D.; Zenclussen, A.C. B cells: The old new players in reproductive immunology. Front. Immunol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.H. Historical overview of immunological tolerance. Cold Spring Harb. Perspect. Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- LaMarca, B.; Wallace, K.; Herse, F.; Wallukat, G.; Martin, J.N., Jr.; Weimer, A.; Dechend, R. Hypertension in response to placental ischemia during pregnancy: Role of B lymphocytes. Hypertension 2011, 57, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Brewer, J.; Liu, R.; Lu, Y.; Scott, J.; Wallace, K.; Wallukat, G.; Moseley, J.; Herse, F.; Dechend, R.; Martin, J.N., Jr.; et al. Endothelin-1, oxidative stress, and endogenous angiotensin II: Mechanisms of angiotensin II type I receptor autoantibody-enhanced renal and blood pressure response during pregnancy. Hypertension 2013, 62, 886–892. [Google Scholar] [CrossRef] [PubMed]
- LaMarca, B.; Wallace, K.; Granger, J. Role of angiotensin II type I receptor agonistic autoantibodies (AT1-AA) in preeclampsia. Curr. Opin. Pharmacol. 2011, 11, 175–179. [Google Scholar] [CrossRef] [PubMed]
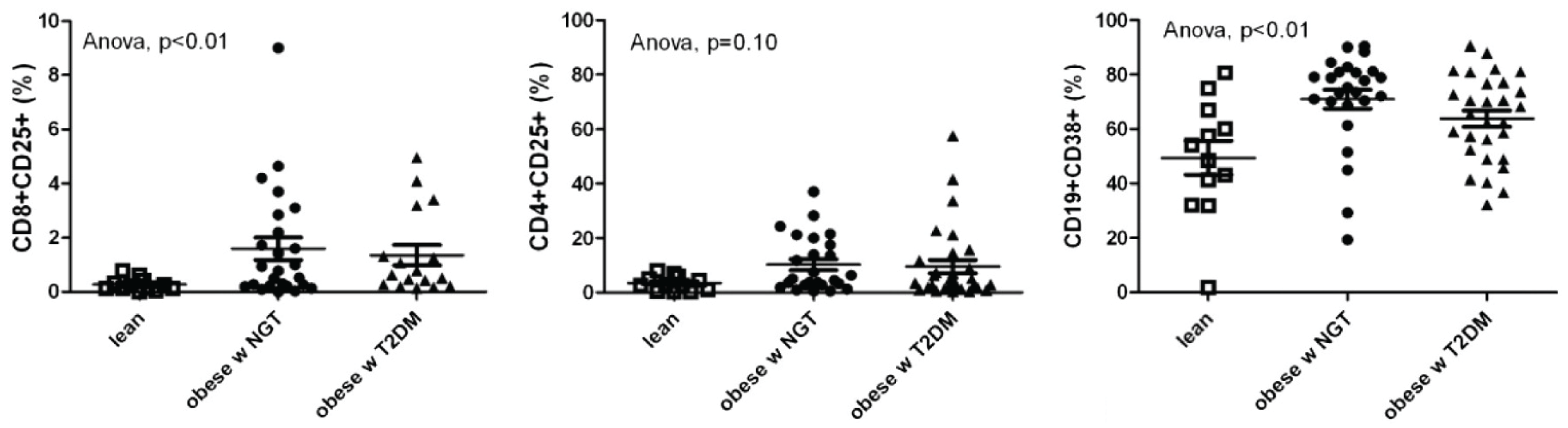
- Sen, S.; Iyer, C.; Klebenov, D.; Histed, A.; Aviles, J.A.; Meydani, S.N. Obesity impairs cell-mediated immunity during the second trimester of pregnancy. Am. J. Obstet. Gynecol. 2013, 208, e131–e138. [Google Scholar] [CrossRef] [PubMed]
- Arai, S.; Maehara, N.; Iwamura, Y.; Honda, S.; Nakashima, K.; Kai, T.; Ogishi, M.; Morita, K.; Kurokawa, J.; Mori, M.; et al. Obesity-associated autoantibody production requires aim to retain the immunoglobulin m immune complex on follicular dendritic cells. Cell Rep. 2013, 3, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Muller-Fielitz, H.; Lau, M.; Johren, O.; Stellmacher, F.; Schwaninger, M.; Raasch, W. Blood pressure response to angiotensin II is enhanced in obese zucker rats and is attributed to an aldosterone-dependent mechanism. Br. J. Pharmacol. 2012, 166, 2417–2429. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.; Halliwill, J.R.; Joyner, M.J.; Jensen, M.D. Vascular response to angiotensin II in upper body obesity. Hypertension 2004, 44, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.D.; Khan, I.Y.; Lakasing, L.; Dekou, V.; O’Brien-Coker, I.; Mallet, A.I.; Hanson, M.A.; Poston, L. Uterine artery function in pregnant rats fed a diet supplemented with animal lard. Exp. Physiol. 2003, 88, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, M.; Micheletti, A.; Cassatella, M.A. Modulation of human neutrophil survival and antigen expression by activated CD4+ and CD8+ T cells. J. Leukoc. Biol. 2010, 88, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
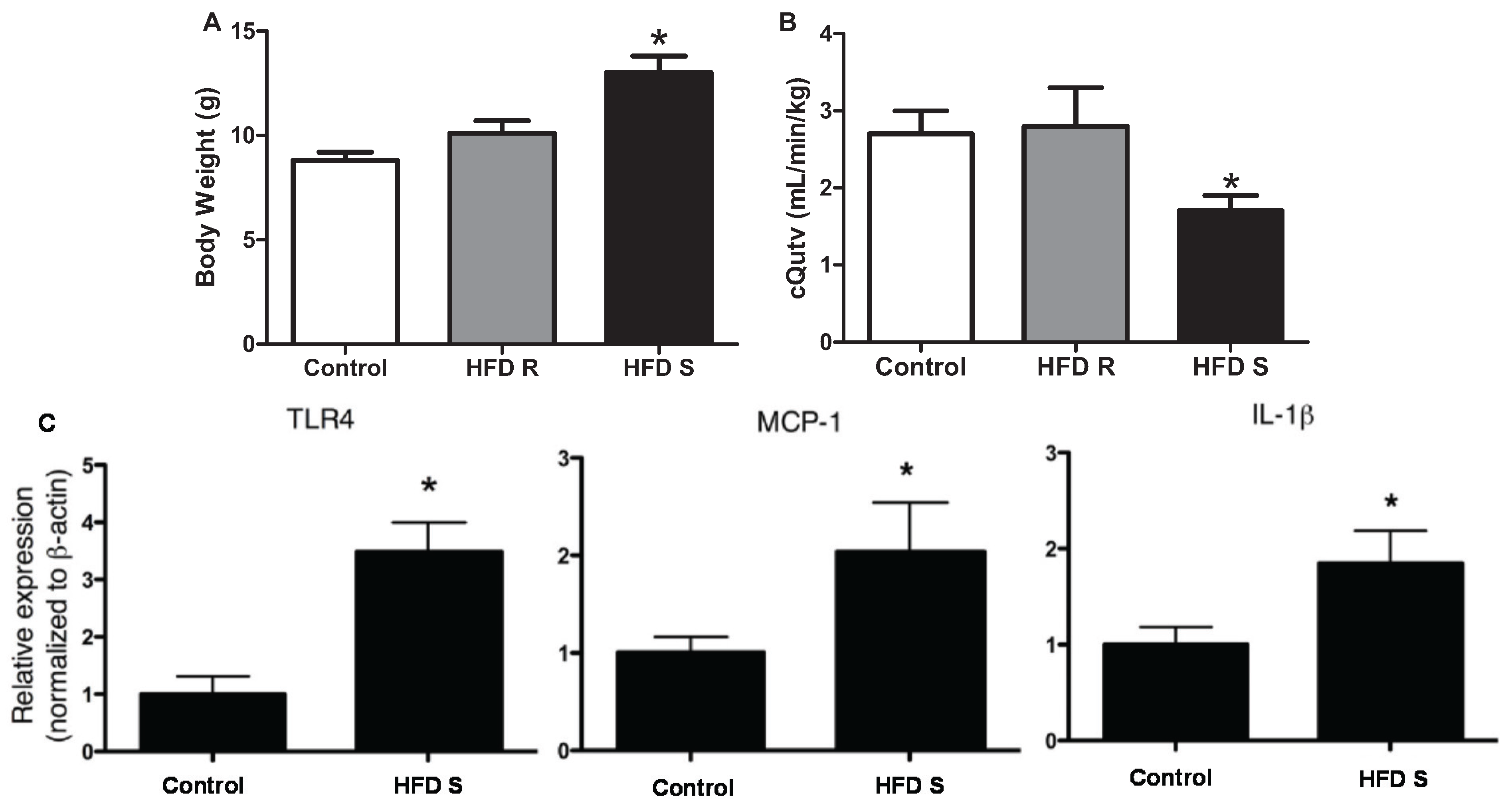
- Shah, T.J.; Leik, C.E.; Walsh, S.W. Neutrophil infiltration and systemic vascular inflammation in obese women. Reprod. Sci. 2010, 17, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Shukla, J.; Walsh, S.W. Neutrophil release of myeloperoxidase in systemic vasculature of obese women may put them at risk for preeclampsia. Reprod. Sci. 2015, 22, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J.; Nilsson, L.M.; Chen, Y.W.; Molkentin, J.D.; Erlinge, D.; Gomez, M.F. High glucose activates nuclear factor of activated T cells in native vascular smooth muscle. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Barquiel, B.; Herranz, L.; Grande, C.; Castro-Dufourny, I.; Llaro, M.; Parra, P.; Burgos, M.A.; Pallardo, L.F. Body weight, weight gain and hyperglycaemia are associated with hypertensive disorders of pregnancy in women with gestational diabetes. Diabetes Metab. 2014, 40, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Gallos, I.D.; Sivakumar, K.; Kilby, M.D.; Coomarasamy, A.; Thangaratinam, S.; Vatish, M. Pre-eclampsia is associated with, and preceded by, hypertriglyceridaemia: A meta-analysis. BJOG 2013, 120, 1321–1332. [Google Scholar] [CrossRef] [PubMed]
- Rajasingam, D.; Seed, P.T.; Briley, A.L.; Shennan, A.H.; Poston, L. A prospective study of pregnancy outcome and biomarkers of oxidative stress in nulliparous obese women. Am. J. Obstet. Gynecol. 2009, 200, e391–e399. [Google Scholar] [CrossRef] [PubMed]
- Leiva, A.; de Medina, C.D.; Salsoso, R.; Saez, T.; san Martin, S.; Abarzua, F.; Farias, M.; Guzman-Gutierrez, E.; Pardo, F.; Sobrevia, L. Maternal hypercholesterolemia in pregnancy associates with umbilical vein endothelial dysfunction: Role of endothelial nitric oxide synthase and arginase II. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2444–2453. [Google Scholar] [CrossRef] [PubMed]
- Chyu, K.Y.; Lio, W.M.; Dimayuga, P.C.; Zhou, J.; Zhao, X.; Yano, J.; Trinidad, P.; Honjo, T.; Cercek, B.; Shah, P.K. Cholesterol lowering modulates t cell function in vivo and in vitro. PLoS ONE 2014, 9, e92095. [Google Scholar] [CrossRef] [PubMed]
- Denison, F.C.; Roberts, K.A.; Barr, S.M.; Norman, J.E. Obesity, pregnancy, inflammation, and vascular function. Reproduction 2010, 140, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Pausova, Z.; Deslauriers, B.; Gaudet, D.; Tremblay, J.; Kotchen, T.A.; Larochelle, P.; Cowley, A.W.; Hamet, P. Role of tumor necrosis factor-alpha gene locus in obesity and obesity-associated hypertension in French canadians. Hypertension 2000, 36, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, G.S.; Reuter, S.; Hillebrand, U.; Amler, S.; Konig, M.; Larger, E.; Oberleithner, H.; Brand, E.; Pavenstadt, H.; Brand, M. The soluble VEGF receptor sFlt1 contributes to endothelial dysfunction in CKD. J. Am. Soc. Nephrol. 2009, 20, 2235–2245. [Google Scholar] [CrossRef] [PubMed]
- Veron, D.; Villegas, G.; Aggarwal, P.K.; Bertuccio, C.; Jimenez, J.; Velazquez, H.; Reidy, K.; Abrahamson, D.R.; Moeckel, G.; Kashgarian, M.; et al. Acute podocyte vascular endothelial growth factor (VEGF-A) knockdown disrupts alphaVbeta3 integrin signaling in the glomerulus. PLoS ONE 2012, 7, e40589. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.E.; Min, J.Y.; Merchan, J.; Lim, K.H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Invest. 2003, 111, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.J.; Maynard, S.E.; Qian, C.; Lim, K.H.; England, L.J.; Yu, K.F.; Schisterman, E.F.; Thadhani, R.; Sachs, B.P.; Epstein, F.H.; et al. Circulating angiogenic factors and the risk of preeclampsia. N. Engl. J. Med. 2004, 350, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Huckle, W.R.; Roche, R.I. Post-transcriptional control of expression of sFlt-1, an endogenous inhibitor of vascular endothelial growth factor. J. Cell. Biochem. 2004, 93, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Rana, S.; Karumanchi, S.A. Preeclampsia: The role of angiogenic factors in its pathogenesis. Physiology 2009, 24, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Spradley, F.T.; Palei, A.C.; Granger, J.P. Obese melanocortin-4 receptor-deficient rats exhibit augmented angiogenic balance and vasorelaxation during pregnancy. Physiol. Rep. 2013, 1, e00081. [Google Scholar] [CrossRef] [PubMed]
- Warrington, J.P.; George, E.M.; Palei, A.C.; Spradley, F.T.; Granger, J.P. Recent advances in the understanding of the pathophysiology of preeclampsia. Hypertension 2013, 62, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.S.; Babcock, S.A.; Granger, J.P. Hypertension produced by reduced uterine perfusion in pregnant rats is associated with increased soluble FMS-like tyrosine kinase-1 expression. Hypertension 2007, 50, 1142–1147. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.S.; Verzwyvelt, J.; Colson, D.; Arany, M.; Karumanchi, S.A.; Granger, J.P. Recombinant vascular endothelial growth factor 121 infusion lowers blood pressure and improves renal function in rats with placentalischemia-induced hypertension. Hypertension 2010, 55, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.R.; LaMarca, B.B.; Parrish, M.; Cockrell, K.; Granger, J.P. Control of soluble fms-like tyrosine-1 (sFlt-1) production response to placental ischemia/hypoxia: Role of tumor necrosis factor-alpha. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R130–R135. [Google Scholar] [CrossRef] [PubMed]
- Parrish, M.R.; Murphy, S.R.; Rutland, S.; Wallace, K.; Wenzel, K.; Wallukat, G.; Keiser, S.; Ray, L.F.; Dechend, R.; Martin, J.N.; et al. The effect of immune factors, tumor necrosis factor-alpha, and agonistic autoantibodies to the angiotensin ii type i receptor on soluble fms-like tyrosine-1 and soluble endoglin production in response to hypertension during pregnancy. Am. J. Hypertens. 2010, 23, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.M.; Bodnar, L.M.; Patrick, T.E.; Powers, R.W. The role of obesity in preeclampsia. Pregnancy Hypertens. 2011, 1, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, L.M.; Catov, J.M.; Klebanoff, M.A.; Ness, R.B.; Roberts, J.M. Prepregnancy body mass index and the occurrence of severe hypertensive disorders of pregnancy. Epidemiology 2007, 18, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, L.M.; Ness, R.B.; Markovic, N.; Roberts, J.M. The risk of preeclampsia rises with increasing prepregnancy body mass index. Ann. Epidemiol. 2005, 15, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.; Kettyle, E.; Sandler, L.; Ecker, J.L.; Roberts, J.; Thadhani, R. Obesity and preeclampsia: The potential role of inflammation. Obstet. Gynecol. 2001, 98, 757–762. [Google Scholar] [CrossRef]
- Zera, C.A.; Seely, E.W.; Wilkins-Haug, L.E.; Lim, K.H.; Parry, S.I.; McElrath, T.F. The association of body mass index with serum angiogenic markers in normal and abnormal pregnancies. Am. J. Obstet. Gynecol. 2014, 211, e241–e247. [Google Scholar] [CrossRef] [PubMed]
- Suwaki, N.; Masuyama, H.; Nakatsukasa, H.; Masumoto, A.; Sumida, Y.; Takamoto, N.; Hiramatrsu, Y. Hypoadiponectinemia and circulating angiogenic factors in overweight patients complicated with pre-eclampsia. Am. J. Obstet. Gynecol. 2006, 195, 1687–1692. [Google Scholar] [CrossRef] [PubMed]
- Kohan, D.E.; Rossi, N.F.; Inscho, E.W.; Pollock, D.M. Regulation of blood pressure and salt homeostasis by endothelin. Physiol. Rev. 2011, 91, 1–77. [Google Scholar] [CrossRef] [PubMed]
- Nova, A.; Sibai, B.M.; Barton, J.R.; Mercer, B.M.; Mitchell, M.D. Maternal plasma level of endothelin is increased in preeclampsia. Am. J. Obstet. Gynecol 1991, 165, 724–727. [Google Scholar] [CrossRef]
- Pollock, D.M. Dissecting the complex physiology of endothelin: New lessons from genetic models. Hypertension 2010, 56, 31–33. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.T.; Rinewalt, A.N.; Cockrell, K.L.; Massey, M.B.; Bennett, W.A.; Granger, J.P. Endothelin type a receptor blockade attenuates the hypertension in response to chronic reductions in uterine perfusion pressure. Hypertension 2001, 37, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Bridges, J.P.; Gilbert, J.S.; Colson, D.; Gilbert, S.A.; Dukes, M.P.; Ryan, M.J.; Granger, J.P. Oxidative stress contributes to soluble fms-like tyrosine kinase-1 induced vascular dysfunction in pregnant rats. Am. J. Hypertens. 2009, 22, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.R.; LaMarca, B.B.; Cockrell, K.; Granger, J.P. Role of endothelin in mediating soluble fms-like tyrosine kinase 1-induced hypertension in pregnant rats. Hypertension 2010, 55, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Lankhorst, S.; Kappers, M.H.; van Esch, J.H.; Danser, A.H.; van den Meiracker, A.H. Hypertension during vascular endothelial growth factor inhibition: Focus on nitric oxide, endothelin-1, and oxidative stress. Antioxid. Redox Signal. 2014, 20, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Star, G.P.; Giovinazzo, M.; Lamoureux, E.; Langleben, D. Effects of vascular endothelial growth factor on endothelin-1 production by human lung microvascular endothelial cells in vitro. Life Sci. 2014, 118, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.R.; LaMarca, B.; Cockrell, K.; Arany, M.; Granger, J.P. L-arginine supplementation abolishes the blood pressure and endothelin response to chronic increases in plasma sFlt-1 in pregnant rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R259–R263. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.J.; Jones, O.B.; Hall, J.E. Inhibition of no synthesis enhances chronic cardiovascular and renal actions of leptin. Hypertension 2001, 37, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Hurst, J.W.; Fuster, V.; Walsh, R.A.; Harrington, R.A. Hurst’s the heart., 13th ed.; McGraw-Hill: New York, NY, USA, 2011. [Google Scholar]
- Hall, J.E.; Granger, J.P.; do Carmo, J.M.; da Silva, A.A.; Dubinion, J.; George, E.; Hamza, S.; Speed, J.; Hall, M.E. Hypertension: Physiology and pathophysiology. Compr. Physiol. 2012, 2, 2393–2442. [Google Scholar] [PubMed]
- Trovati, M.; Doronzo, G.; Barale, C.; Vaccheris, C.; Russo, I.; Cavalot, F. Leptin and vascular smooth muscle cells. Curr. Pharm. Des. 2013, 20, 625–634. [Google Scholar] [CrossRef]
- Juan, C.C.; Chuang, T.Y.; Lien, C.C.; Lin, Y.J.; Huang, S.W.; Kwok, C.F.; Ho, L.T. Leptin increases endothelin type a receptor levels in vascular smooth muscle cells. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E481–E487. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Adiarto, S.; Emoto, N.; Iwasa, N.; Yokoyama, M. Obesity-induced upregulation of myocardial endothelin-1 expression is mediated by leptin. Biochem. Biophys. Res. Commun. 2007, 353, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Quehenberger, P.; Exner, M.; Sunder-Plassmann, R.; Ruzicka, K.; Bieglmayer, C.; Endler, G.; Muellner, C.; Speiser, W.; Wagner, O. Leptin induces endothelin-1 in endothelial cells in vitro. Circul. Res. 2002, 90, 711–718. [Google Scholar] [CrossRef]
- Da Silva, A.A.; Kuo, J.J.; Tallam, L.S.; Hall, J.E. Role of endothelin-1 in blood pressure regulation in a rat model of visceral obesity and hypertension. Hypertension 2004, 43, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Molvarec, A.; Szarka, A.; Walentin, S.; Beko, G.; Karadi, I.; Prohaszka, Z.; Rigo, J., Jr. Serum leptin levels in relation to circulating cytokines, chemokines, adhesion molecules and angiogenic factors in normal pregnancy and preeclampsia. Reprod. Biol. Endocrinol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Lisonkova, S.; Sabr, Y.; Mayer, C.; Young, C.; Skoll, A.; Joseph, K.S. Maternal morbidity associated with early-onset and late-onset preeclampsia. Obstet. Gynecol. 2014, 124, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Raymond, D.; Peterson, E. A critical review of early-onset and late-onset preeclampsia. Obstet. Gynecol. Surv. 2011, 66, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Verlohren, S.; Melchiorre, K.; Khalil, A.; Thilaganathan, B. Uterine artery doppler, birth weight and timing of onset of pre-eclampsia: Providing insights into the dual etiology of late-onset pre-eclampsia. Ultrasound Obstet. Gynecol. 2014, 44, 293–298. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spradley, F.T.; Palei, A.C.; Granger, J.P. Immune Mechanisms Linking Obesity and Preeclampsia. Biomolecules 2015, 5, 3142-3176. https://doi.org/10.3390/biom5043142
Spradley FT, Palei AC, Granger JP. Immune Mechanisms Linking Obesity and Preeclampsia. Biomolecules. 2015; 5(4):3142-3176. https://doi.org/10.3390/biom5043142
Chicago/Turabian StyleSpradley, Frank T., Ana C. Palei, and Joey P. Granger. 2015. "Immune Mechanisms Linking Obesity and Preeclampsia" Biomolecules 5, no. 4: 3142-3176. https://doi.org/10.3390/biom5043142
APA StyleSpradley, F. T., Palei, A. C., & Granger, J. P. (2015). Immune Mechanisms Linking Obesity and Preeclampsia. Biomolecules, 5(4), 3142-3176. https://doi.org/10.3390/biom5043142