
Roles of Chemokines and Chemokine Receptors in Obesity-Associated Insulin Resistance and Nonalcoholic Fatty Liver Disease


Abstract
:1. Introduction
2. Immune Cell Activity during Obesity-Induced Inflammation and Insulin Resistance
2.1. Macrophages as a Major Player in Obesity-Induced Insulin Resistance
2.2. Macrophage Plasticity and Polarization
2.3. The Role of Other Immune Cells during Insulin Resistance
3. Regulation of Insulin Resistance and Inflammation in Adipose Tissue by the Chemokine System
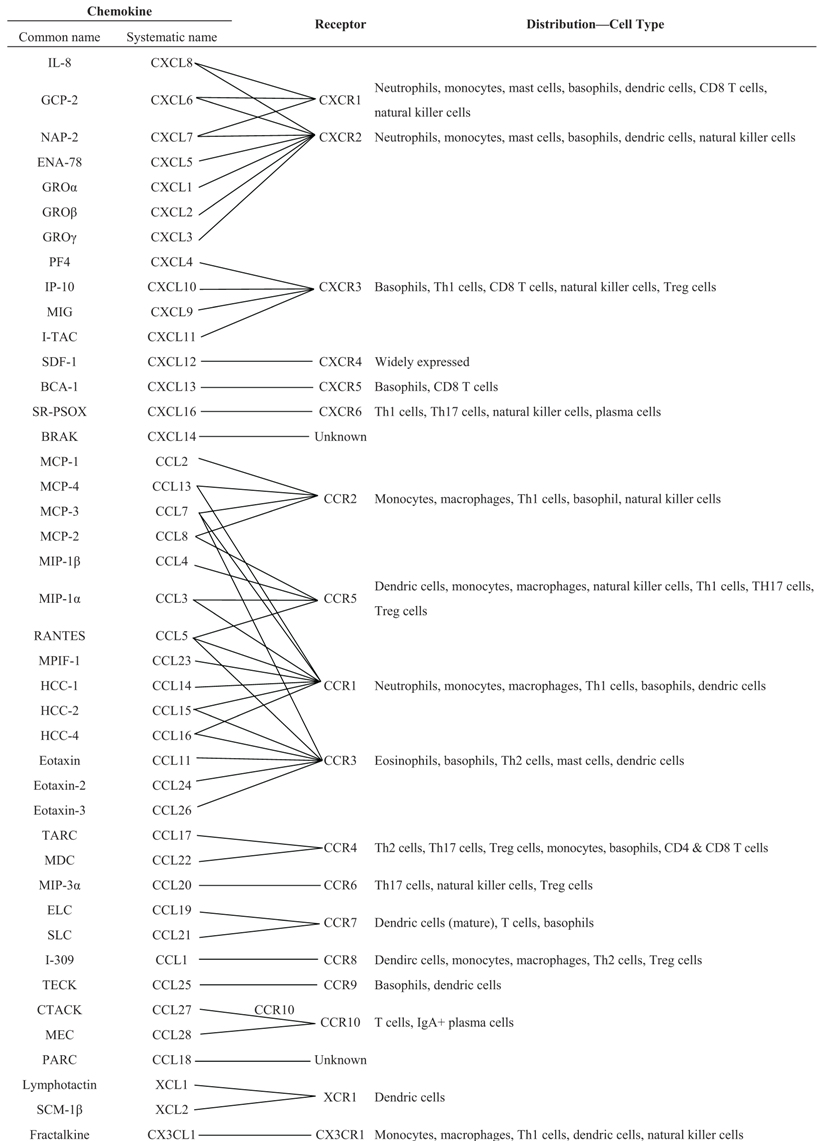
3.1. Classification of Chemokines and Chemokine Receptors

{kind=link}
{kind=link}
{kind=link}
 |
3.2. Role of the MCP-1-CCR2 System in ATM Recruitment
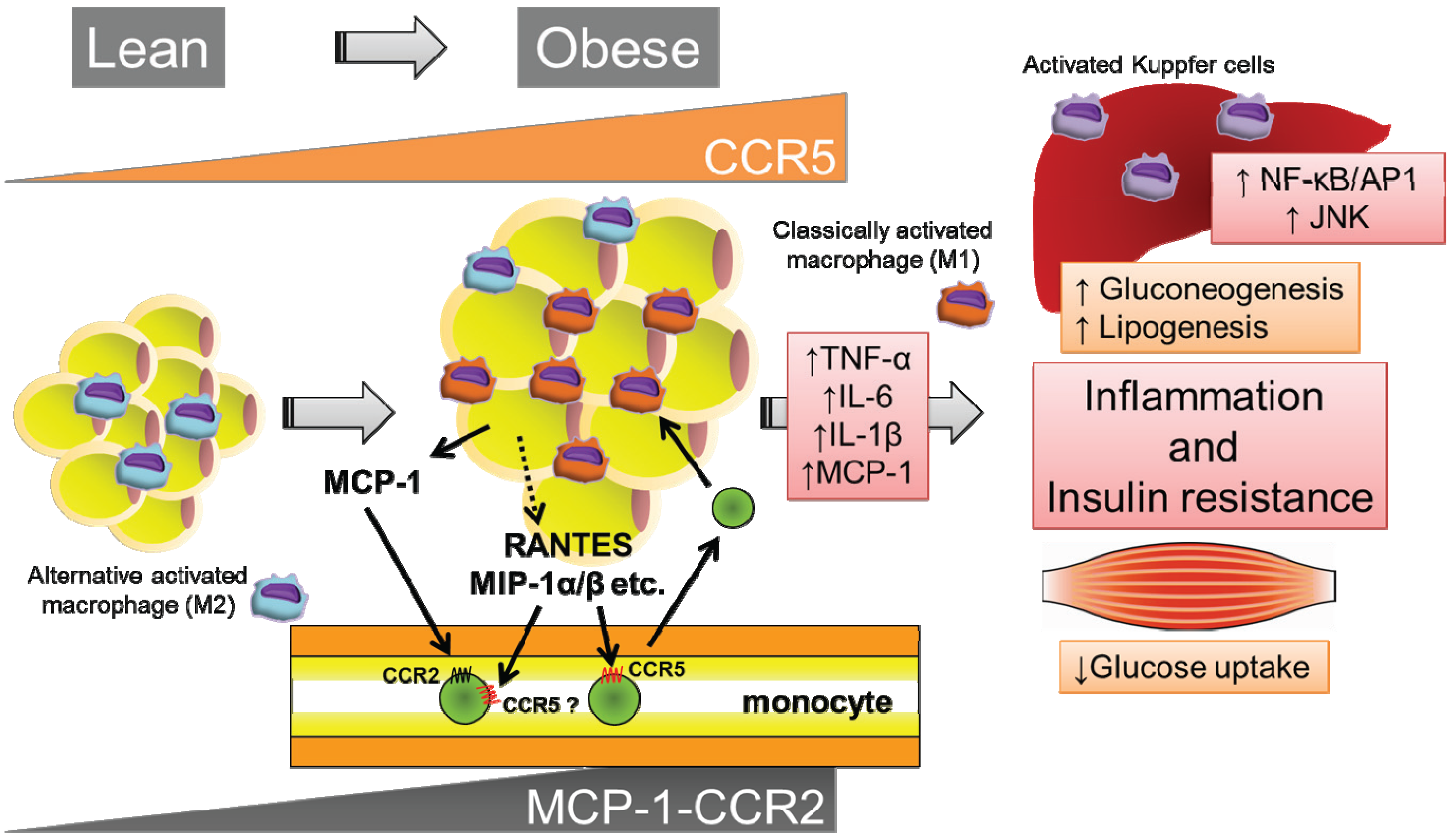
3.3. CCR5 Regulates Insulin Resistance and the Recruitment and Polarization of ATMs
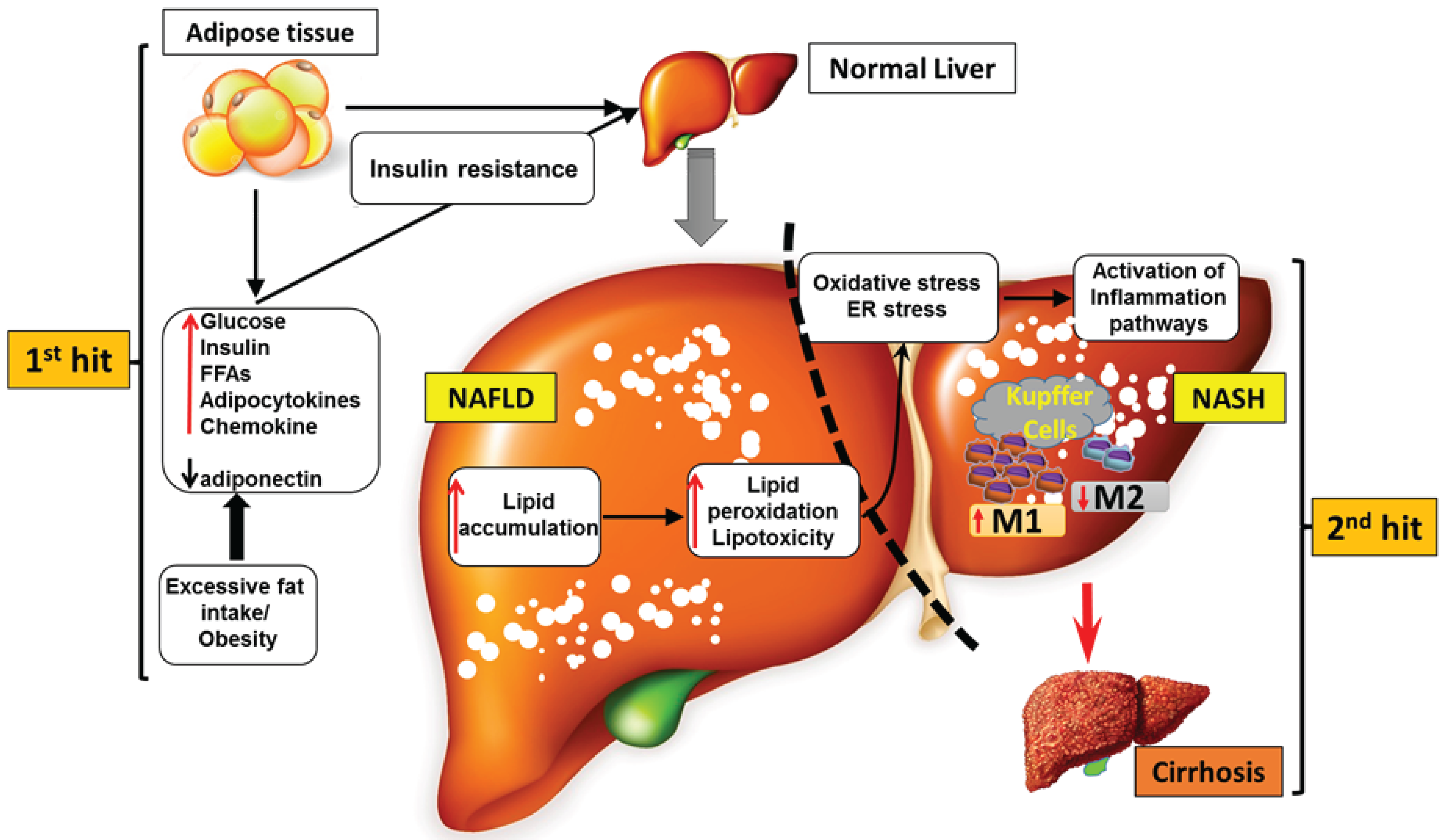
4. Role of the Chemokine System during the Progression from NAFLD to NASH
4.1. Progression of NAFLD to NASH
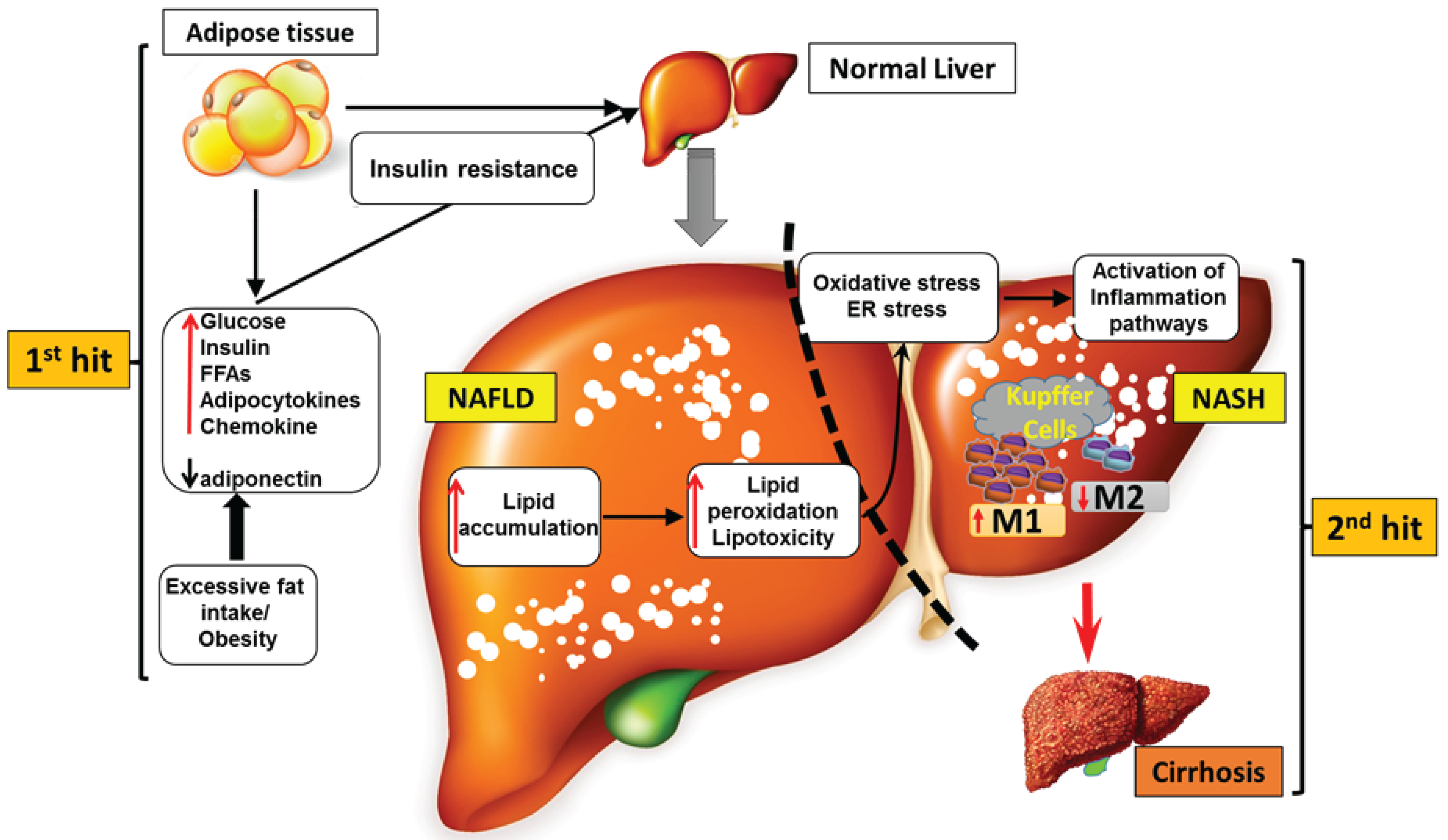
4.2. CCRs in NAFLD and NASH
4.3. The Polarization of Liver M1/M2 Macrophage Regulates Insulin Resistance and NASH
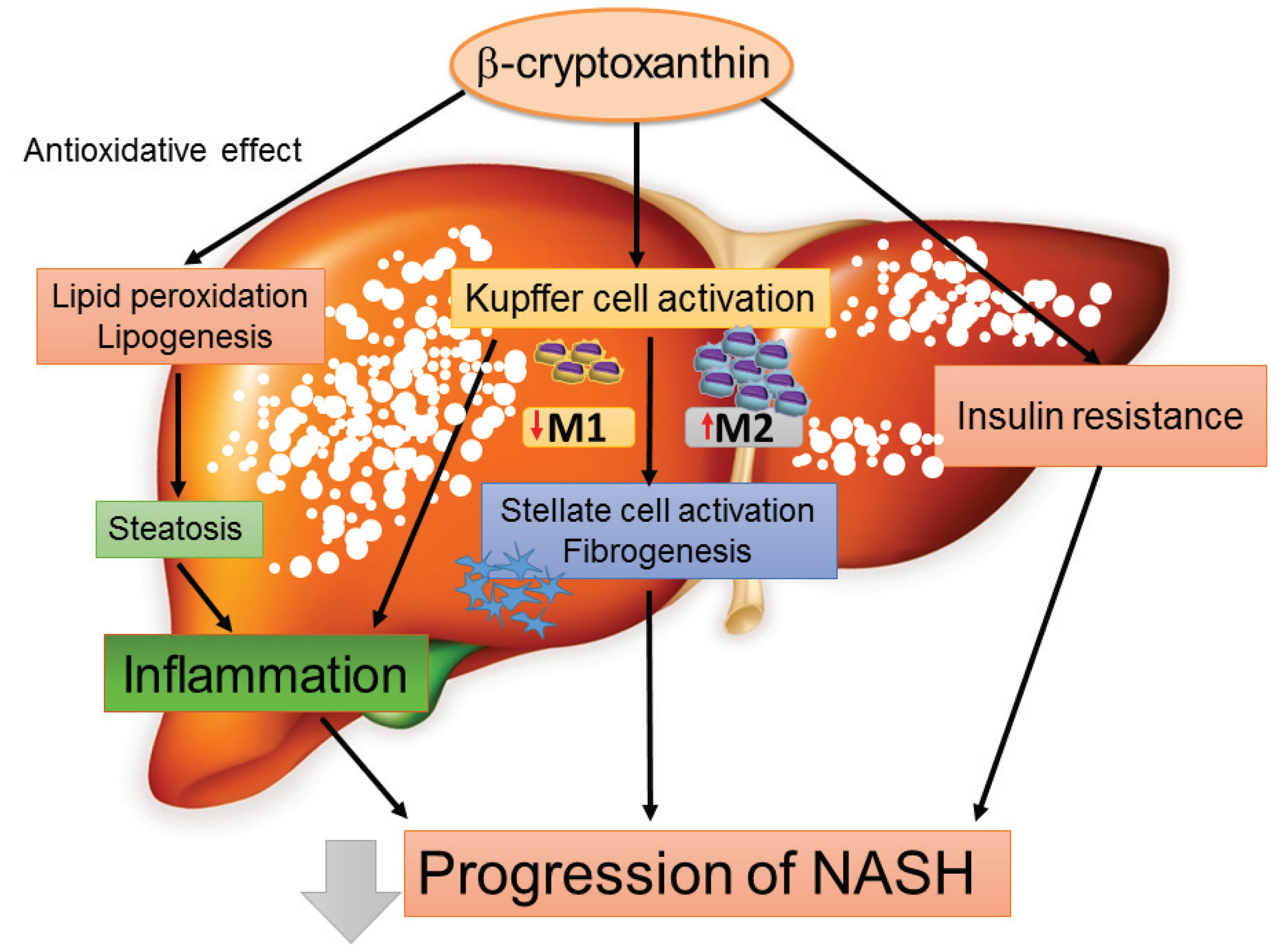
4.4. Chemokines as a Therapeutic Target for the Treatment of NASH: The Emerging Role of β-Cryptoxanthin
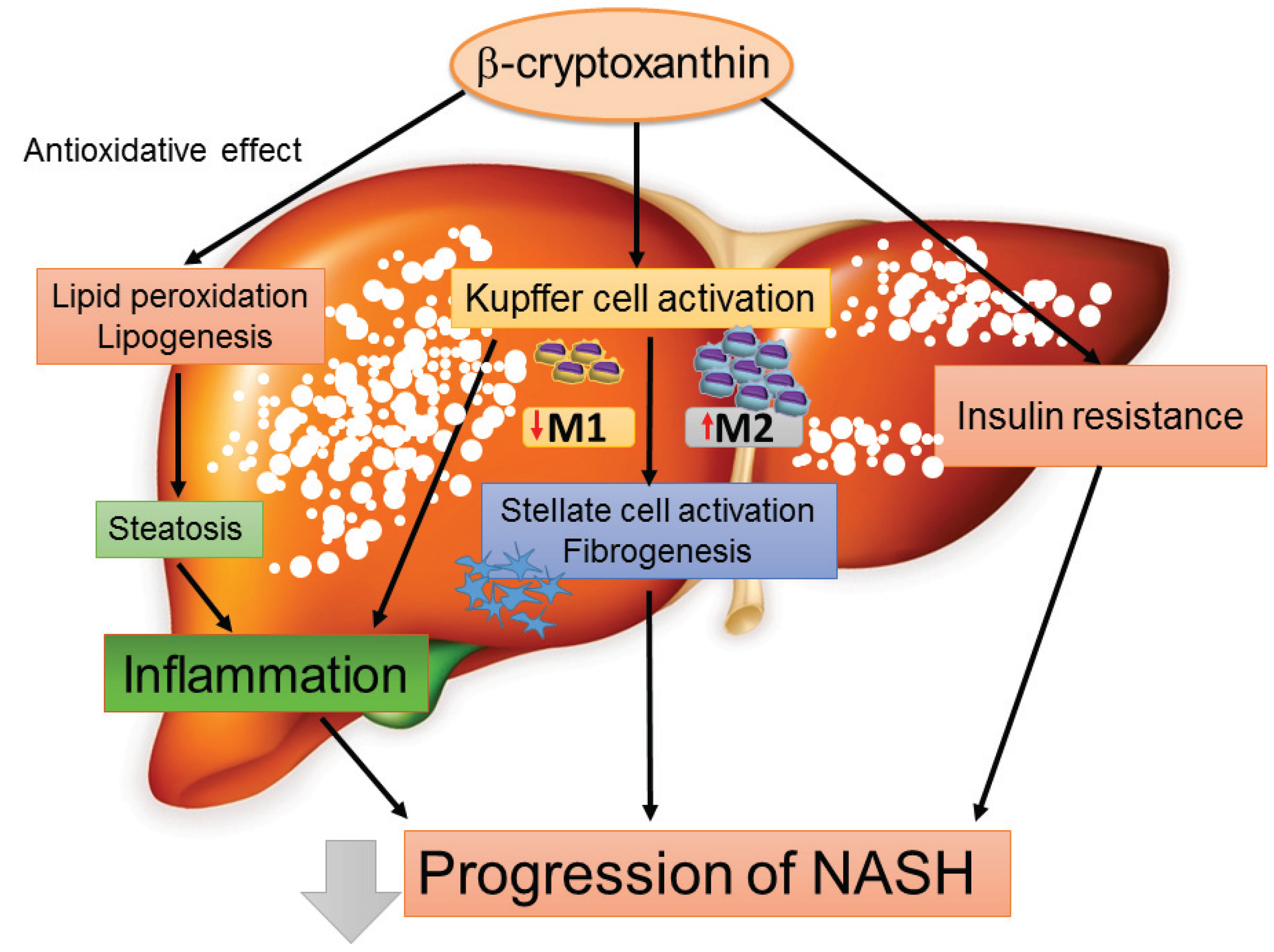
5. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hotamisligil, G.S.; Erbay, E. Nutrient sensing and inflammation in metabolic diseases. Nat. Rev. Immunol. 2008, 8, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Wajchenberg, B.L. Subcutaneous and visceral adipose tissue: Their relation to the metabolic syndrome. Endocr. Rev. 2000, 21, 697–738. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Invest. 2006, 116, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Siiteri, P.K. Adipose tissue as a source of hormones. Am. J. Clin. Nutr. 1987, 45, 277–282. [Google Scholar] [PubMed]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Neels, J.G.; Olefsky, J.M. Inflamed fat: What starts the fire? J. Clin. Invest. 2006, 116, 33–35. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Wang, Y.; Keshaw, H.; Xu, L.Y.; Lam, K.S.; Cooper, G.J. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J. Clin. Invest. 2003, 112, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Obstfeld, A.E.; Sugaru, E.; Thearle, M.; Francisco, A.M.; Gayet, C.; Ginsberg, H.N.; Ables, E.V.; Ferrante, A.W., Jr. CC chemokine receptor 2 (CCR2) regulates the hepatic recruitment of myeloid cells that promote obesity-induced hepatic steatosis. Diabetes 2010, 59, 916–925. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest. 2007, 117, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E. Chemokine receptors: Multifaceted therapeutic targets. Nat. Rev. Immunol. 2002, 2, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Bonecchi, R.; Locati, M. Tuning inflammation and immunity by chemokine sequestration: Decoys and more. Nat. Rev. Immunol. 2006, 6, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Bachelerie, F.; Ben-Baruch, A.; Burkhardt, A.M.; Combadiere, C.; Farber, J.M.; Graham, G.J.; Horuk, R.; Sparre-Ulrich, A.H.; Locati, M.; Luster, A.D.; et al. International Union of Basic and Clinical Pharmacology. [corrected]. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol. Rev. 2014, 66, 1–79. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; Hunter, D.; Huber, R.; Lemieux, J.; Slaymaker, S.; Vaddi, K.; Charo, I.; Leibel, R.L.; Ferrante, A.W., Jr. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J. Clin. Invest. 2006, 116, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. Mcp-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Invest. 2006, 116, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Metlakunta, A.; Dedousis, N.; Zhang, P.; Sipula, I.; Dube, J.J.; Scott, D.K.; O’Doherty, R.M. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes 2010, 59, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.G.; Ko, H.J.; Cho, Y.R.; Kim, H.J.; Ma, Z.; Yu, T.Y.; Friedline, R.H.; Kurt-Jones, E.; Finberg, R.; Fischer, M.A.; et al. Interleukin-10 prevents diet-induced insulin resistance by attenuating macrophage and cytokine response in skeletal muscle. Diabetes 2009, 58, 2525–2535. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Eagle, A.R.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; DelProposto, J.B.; Westcott, D.J.; Saltiel, A.R. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 2008, 57, 3239–3246. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.; Favelyukis, S.; Nguyen, A.K.; Reichart, D.; Scott, P.A.; Jenn, A.; Liu-Bryan, R.; Glass, C.K.; Neels, J.G.; Olefsky, J.M. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J. Biol. Chem. 2007, 282, 35279–35292. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Sun, S.; Xu, A.; Bhargava, P.; Yang, L.; Lam, K.S.; et al. Activation of natural killer T cells promotes M2 Macrophage polarization in adipose tissue and improves systemic glucose tolerance via interleukin-4(IL-4)/STAT6 protein signaling axis in obesity. J Biol Chem 2012, 287, 13561–13571. [Google Scholar] [CrossRef] [PubMed]
- Cintra, D.E.; Pauli, J.R.; Araujo, E.P.; Moraes, J.C.; de Souza, C.T.; Milanski, M.; Morari, J.; Gambero, A.; Saad, M.J.; Velloso, L.A. Interleukin-10 is a protective factor against diet-induced insulin resistance in liver. J. Hepatol. 2008, 48, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Eto, K.; Yamashita, H.; Ohsugi, M.; Otsu, M.; Hara, K.; Ueki, K.; Sugiura, S.; et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 2009, 15, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Winer, S.; Chan, Y.; Paltser, G.; Truong, D.; Tsui, H.; Bahrami, J.; Dorfman, R.; Wang, Y.; Zielenski, J.; Mastronardi, F.; et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat. Med. 2009, 15, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Feuerer, M.; Herrero, L.; Cipolletta, D.; Naaz, A.; Wong, J.; Nayer, A.; Lee, J.; Goldfine, A.B.; Benoist, C.; Shoelson, S.; et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009, 15, 930–939. [Google Scholar] [CrossRef] [PubMed]
- DeFuria, J.; Belkina, A.C.; Jagannathan-Bogdan, M.; Snyder-Cappione, J.; Carr, J.D.; Nersesova, Y.R.; Markham, D.; Strissel, K.J.; Watkins, A.A.; Zhu, M.; et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc. Natl. Acad. Sci. USA 2013, 110, 5133–5138. [Google Scholar] [CrossRef] [PubMed]
- Winer, D.A.; Winer, S.; Shen, L.; Wadia, P.P.; Yantha, J.; Paltser, G.; Tsui, H.; Wu, P.; Davidson, M.G.; Alonso, M.N.; et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat. Med. 2011, 17, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Molofsky, A.B.; Liang, H.E.; Ricardo-Gonzalez, R.R.; Jouihan, H.A.; Bando, J.K.; Chawla, A.; Locksley, R.M. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 2011, 332, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, S.; Oh, D.Y.; Bandyopadhyay, G.; Li, D.; Xu, J.; McNelis, J.; Lu, M.; Li, P.; Yan, Q.; Zhu, Y.; et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 2012, 18, 1407–1412. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Divoux, A.; Sun, J.; Zhang, J.; Clement, K.; Glickman, J.N.; Sukhova, G.K.; Wolters, P.J.; Du, J.; Gorgun, C.Z.; et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat. Med. 2009, 15, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Nomura, Y.; Matsuda, A.; Ohmori, K.; Matsuda, H. Mast cells function as an alternative modulator of adipogenesis through 15-deoxy-delta-12, 14-prostaglandin J2. Am. J. Physiol. Cell Physiol. 2011, 301, C1360–C1367. [Google Scholar] [CrossRef] [PubMed]
- Baggiolini, M. Chemokines and leukocyte traffic. Nature 1998, 392, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Gerard, C.; Rollins, B.J. Chemokines and disease. Nat. Immunol. 2001, 2, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Kamei, N.; Tobe, K.; Suzuki, R.; Ohsugi, M.; Watanabe, T.; Kubota, N.; Ohtsuka-Kowatari, N.; Kumagai, K.; Sakamoto, K.; Kobayashi, M.; et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J. Biol. Chem. 2006, 281, 26602–26614. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Sugimoto, M.; Murayama, T.; Ueda, Y.; Kanamori, H.; Ono, K.; Ariyasu, H.; Akamizu, T.; Kita, T.; Yokode, M.; et al. Inhibition of CCR2 ameliorates insulin resistance and hepatic steatosis in db/db mice. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2195–2201. [Google Scholar] [CrossRef] [PubMed]
- Inouye, K.E.; Shi, H.; Howard, J.K.; Daly, C.H.; Lord, G.M.; Rollins, B.J.; Flier, J.S. Absence of CC chemokine ligand 2 does not limit obesity-associated infiltration of macrophages into adipose tissue. Diabetes 2007, 56, 2242–2250. [Google Scholar] [CrossRef] [PubMed]
- Kirk, E.A.; Sagawa, Z.K.; McDonald, T.O.; O’Brien, K.D.; Heinecke, J.W. Monocyte chemoattractant protein deficiency fails to restrain macrophage infiltration into adipose tissue. Diabetes 2008, 57, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Chavey, C.; Lazennec, G.; Lagarrigue, S.; Clape, C.; Iankova, I.; Teyssier, J.; Annicotte, J.S.; Schmidt, J.; Mataki, C.; Yamamoto, H.; et al. CXC ligand 5 is an adipose-tissue derived factor that links obesity to insulin resistance. Cell. Metab. 2009, 9, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, J.; Yoon, J.H.; Ghim, J.; Yea, K.; Song, P.; Park, S.; Lee, A.; Hong, C.P.; Jang, M.S.; et al. CXCL12 secreted from adipose tissue recruits macrophages and induces insulin resistance in mice. Diabetologia 2014, 57, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Kitade, H.; Sawamoto, K.; Nagashimada, M.; Inoue, H.; Yamamoto, Y.; Sai, Y.; Takamura, T.; Yamamoto, H.; Miyamoto, K.; Ginsberg, H.N.; et al. CCR5 plays a critical role in obesity-induced adipose tissue inflammation and insulin resistance by regulating both macrophage recruitment and M1/M2 status. Diabetes 2012, 61, 1680–1690. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Yang, L.; van Rooijen, N.; Ohnishi, H.; Seki, E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1310–G1321. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; de Minicis, S.; Inokuchi, S.; Taura, K.; Miyai, K.; van Rooijen, N.; Schwabe, R.F.; Brenner, D.A. CCR2 promotes hepatic fibrosis in mice. Hepatology 2009, 50, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; de Minicis, S.; Gwak, G.Y.; Kluwe, J.; Inokuchi, S.; Bursill, C.A.; Llovet, J.M.; Brenner, D.A.; Schwabe, R.F. CCR1 and CCR5 promote hepatic fibrosis in mice. J. Clin. Invest. 2009, 119, 1858–1870. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ghosh, S.; Perrard, X.D.; Feng, L.; Garcia, G.E.; Perrard, J.L.; Sweeney, J.F.; Peterson, L.E.; Chan, L.; Smith, C.W.; et al. T-cell accumulation and regulated on activation, normal T cell expressed and secreted upregulation in adipose tissue in obesity. Circulation 2007, 115, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Braunersreuther, V.; Viviani, G.L.; Mach, F.; Montecucco, F. Role of cytokines and chemokines in non-alcoholic fatty liver disease. World J. Gastroenterol. 2012, 18, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Bahcecioglu, I.H.; Yalniz, M.; Ataseven, H.; Ilhan, N.; Ozercan, I.H.; Seckin, D.; Sahin, K. Levels of serum hyaluronic acid, TNF-alpha and IL-8 in patients with nonalcoholic steatohepatitis. Hepatogastroenterology 2005, 52, 1549–1553. [Google Scholar] [PubMed]
- Wasmuth, H.E.; Lammert, F.; Zaldivar, M.M.; Weiskirchen, R.; Hellerbrand, C.; Scholten, D.; Berres, M.L.; Zimmermann, H.; Streetz, K.L.; Tacke, F.; et al. Antifibrotic effects of CXCL9 and its receptor CXCR3 in livers of mice and humans. Gastroenterology 2009, 137, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Invernizzi, P.; Mantovani, A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology 2014, 59, 2034–2042. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: A protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Louvet, A.; Teixeira-Clerc, F.; Chobert, M.N.; Deveaux, V.; Pavoine, C.; Zimmer, A.; Pecker, F.; Mallat, A.; Lotersztajn, S. Cannabinoid CB2 receptors protect against alcoholic liver disease by regulating Kupffer cell polarization in mice. Hepatology 2011, 54, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, M.; Nakamura, M.; Ikoma, Y.; Yano, M.; Ogawa, K.; Matsumoto, H.; Kato, M.; Ohshima, M.; Nagao, A. High serum carotenoids are inversely associated with serum gamma-glutamyltransferase in alcohol drinkers within normal liver function. J. Epidemiol. 2005, 15, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, M.; Nakamura, M.; Ikoma, Y.; Yano, M.; Ogawa, K.; Matsumoto, H.; Kato, M.; Ohshima, M.; Nagao, A. The homeostasis model assessment-insulin resistance index is inversely associated with serum carotenoids in non-diabetic subjects. J. Epidemiol. 2006, 16, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Katsuura, S.; Imamura, T.; Bando, N.; Yamanishi, R. Beta-carotene and beta-cryptoxanthin but not lutein evoke redox and immune changes in RAW264 murine macrophages. Mol. Nutr. Food Res. 2009, 53, 1396–1405. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, K.; Morimoto, S.; Shirakura, Y.; Mukai, K.; Sugiyama, T.; Tokuji, Y.; Ohnishi, M. Mechanism of visceral fat reduction in Tsumura Suzuki obese, diabetes (TSOD) mice orally administered beta-cryptoxanthin from Satsuma mandarin oranges (Citrus unshiu Marc). J. Agric. Food Chem. 2011, 59, 12342–12351. [Google Scholar] [CrossRef] [PubMed]
- Kobori, M.; Ni, Y.; Takahashi, Y.; Watanabe, N.; Sugiura, M.; Ogawa, K.; Nagashimada, M.; Kaneko, S.; Naito, S.; Ota, T. Beta-cryptoxanthin alleviates diet-induced nonalcoholic steatohepatitis by suppressing inflammatory gene expression in mice. PLoS ONE 2014, 9, e98294. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Nagashimada, M.; Zhan, L.; Nagata, N.; Kobori, M.; Sugiura, M.; Ogawa, K.; Kaneko, S.; Ota, T. Prevention and reversal of lipotoxicity-induced hepatic insulin resistance and steatohepatitis in mice by an antioxidant carotenoid, beta-cryptoxanthin. Endocrinology 2015, 156, 987–999. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, L.; Kitade, H.; Ni, Y.; Ota, T. Roles of Chemokines and Chemokine Receptors in Obesity-Associated Insulin Resistance and Nonalcoholic Fatty Liver Disease. Biomolecules 2015, 5, 1563-1579. https://doi.org/10.3390/biom5031563
Xu L, Kitade H, Ni Y, Ota T. Roles of Chemokines and Chemokine Receptors in Obesity-Associated Insulin Resistance and Nonalcoholic Fatty Liver Disease. Biomolecules. 2015; 5(3):1563-1579. https://doi.org/10.3390/biom5031563
Chicago/Turabian StyleXu, Liang, Hironori Kitade, Yinhua Ni, and Tsuguhito Ota. 2015. "Roles of Chemokines and Chemokine Receptors in Obesity-Associated Insulin Resistance and Nonalcoholic Fatty Liver Disease" Biomolecules 5, no. 3: 1563-1579. https://doi.org/10.3390/biom5031563
APA StyleXu, L., Kitade, H., Ni, Y., & Ota, T. (2015). Roles of Chemokines and Chemokine Receptors in Obesity-Associated Insulin Resistance and Nonalcoholic Fatty Liver Disease. Biomolecules, 5(3), 1563-1579. https://doi.org/10.3390/biom5031563