
Mammalian Cell Surface Display as a Novel Method for Developing Engineered Lectins with Novel Characteristics

Abstract
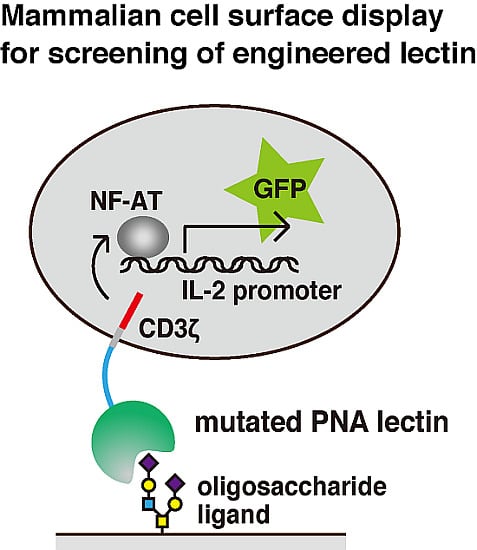
:
1. Introduction
2. Results
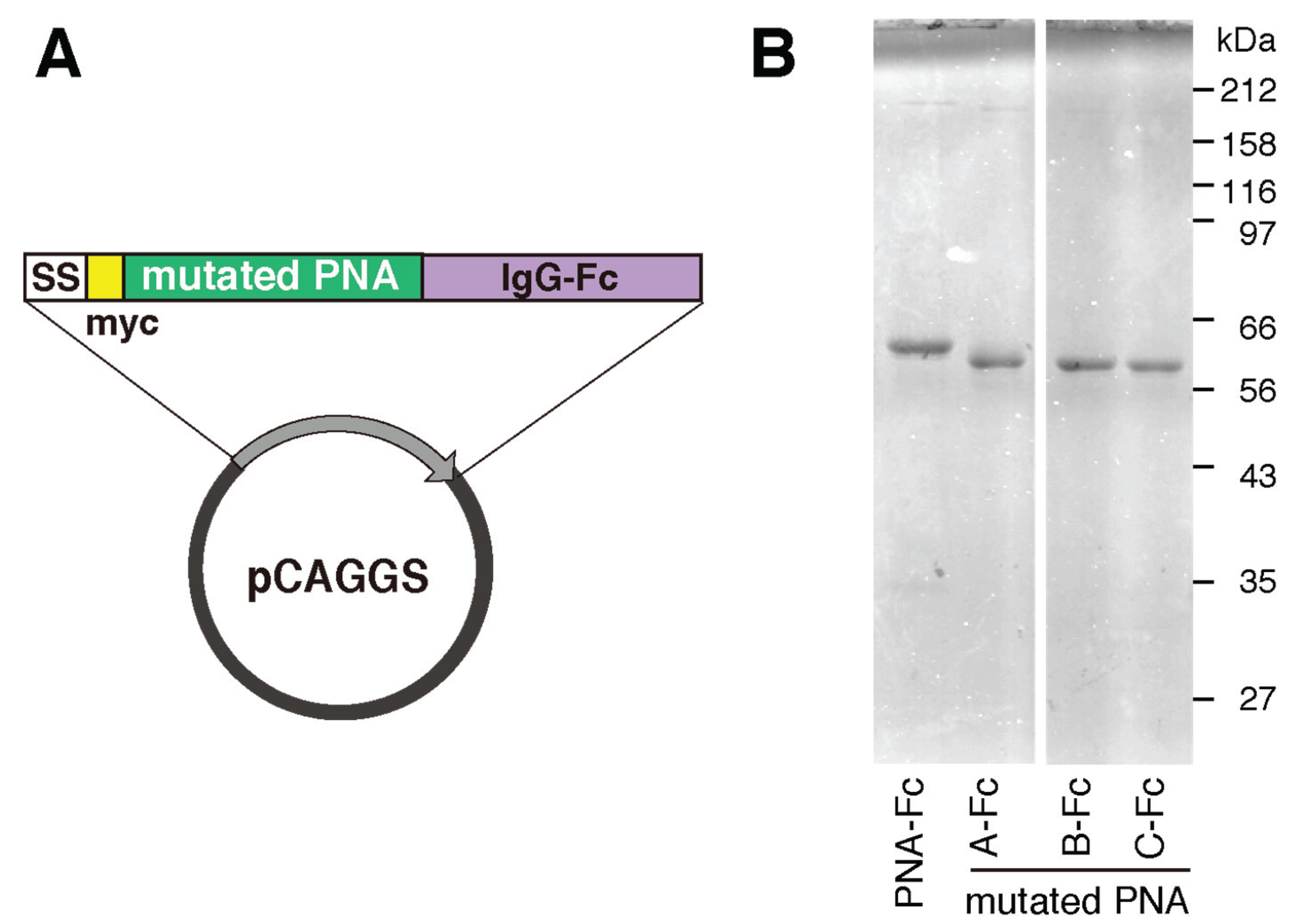
2.1. Construction of Mutated PNA Library Plasmids

{kind=link}
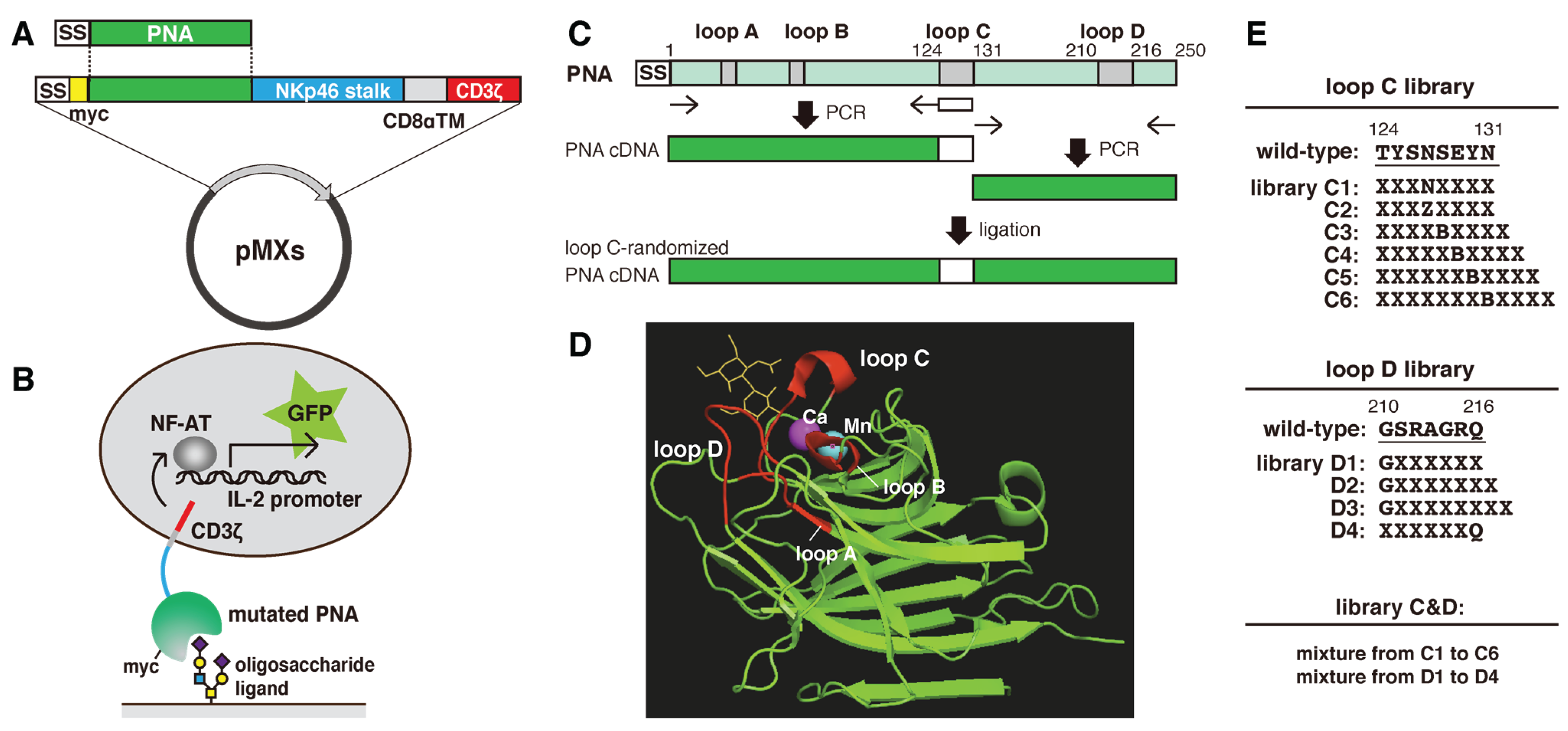{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Library Name | Colony-Forming Unit |
|---|---|
| C1 | 4.0 × 106 |
| C2 | 3.0 × 106 |
| C3 | 1.8 × 106 |
| C4 | 1.6 × 106 |
| C5 | 1.5 × 106 |
| C6 | 2.0 × 106 |
| D1 | 1.4 × 105 |
| D2 | 1.1 × 105 |
| D3 | 1.0 × 105 |
| D4 | 3.3 × 105 |
| C&D | 5.0 × 106 |
| Total | 1.96 × 107 |

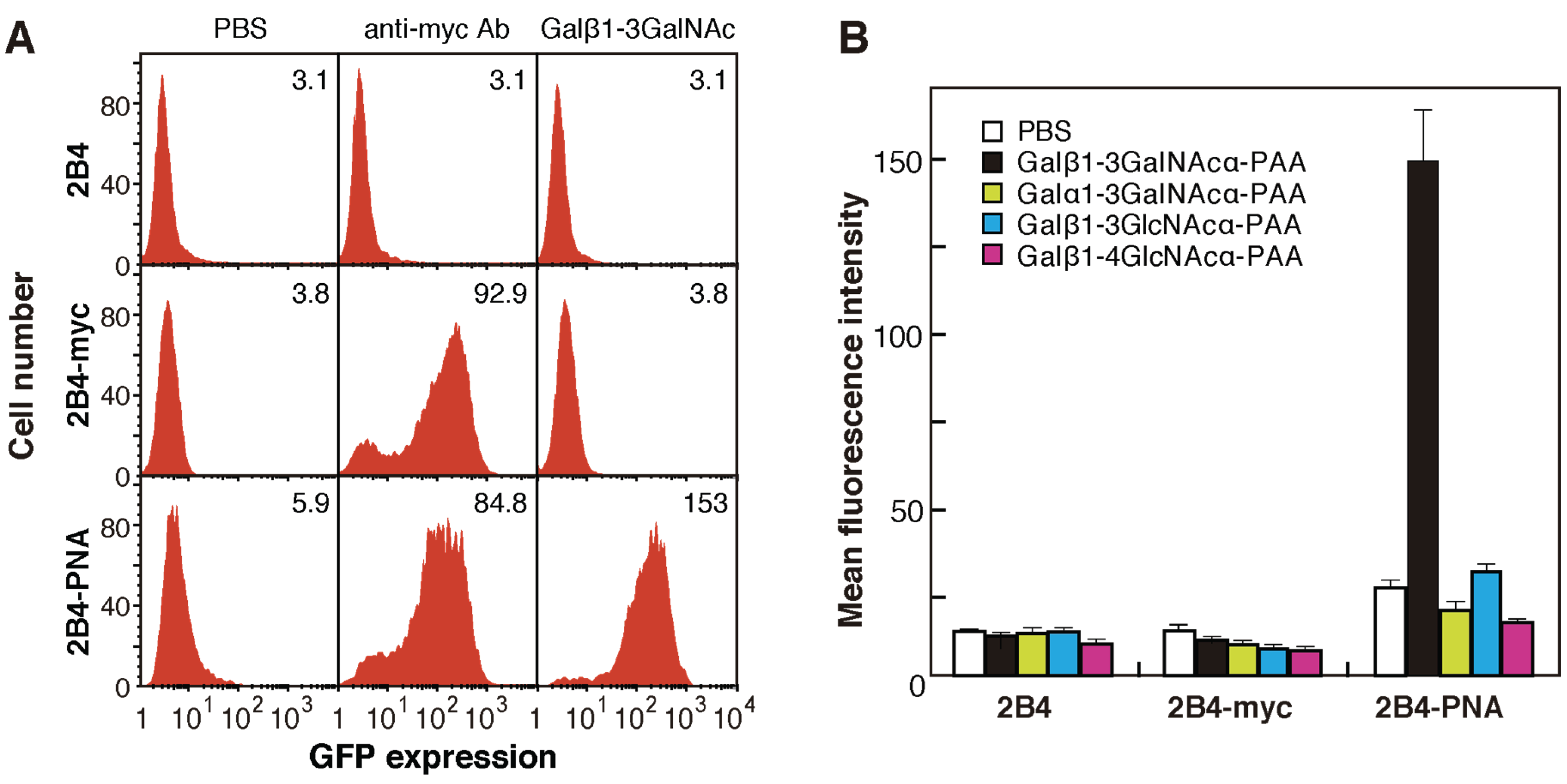
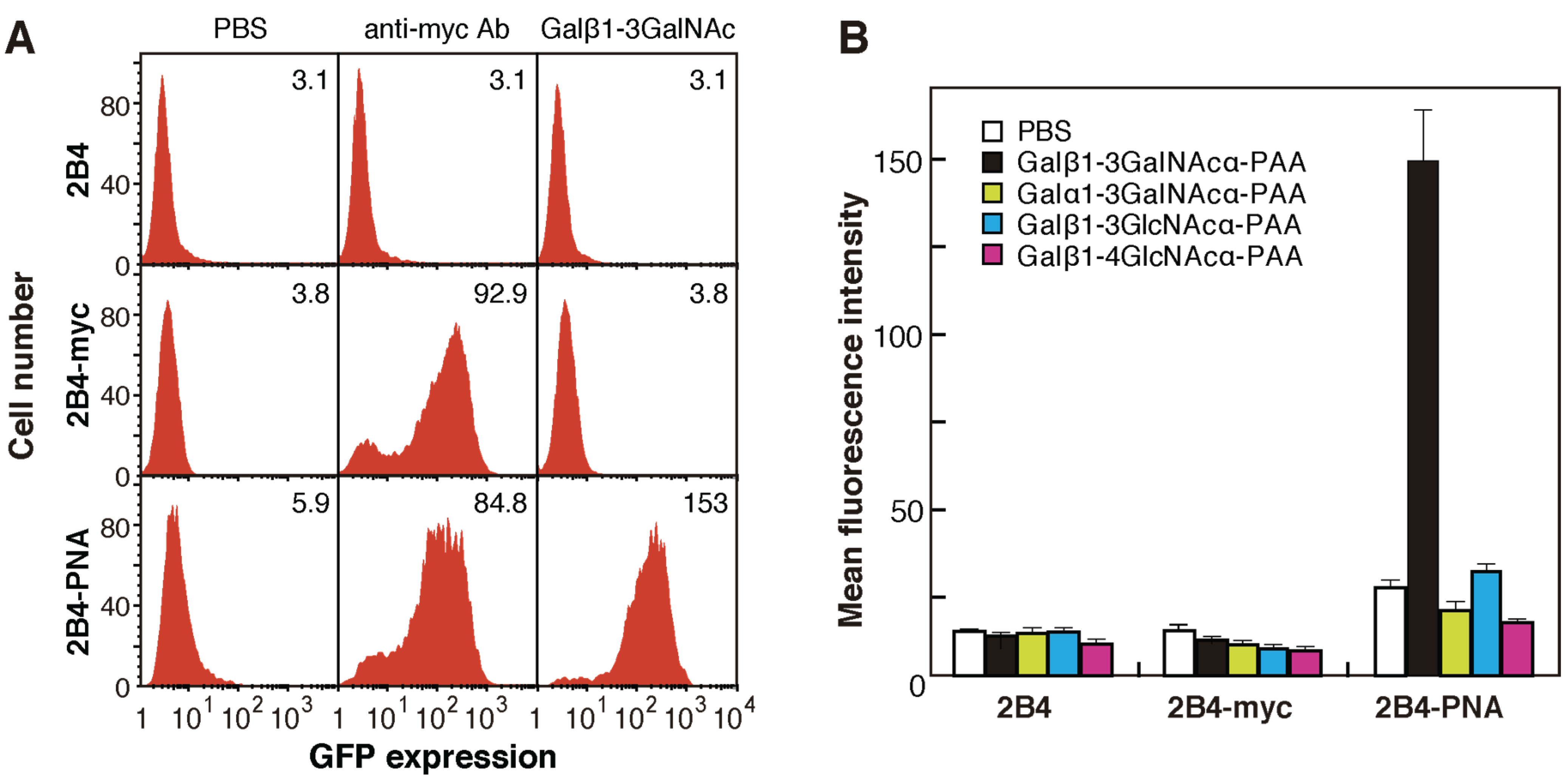
2.2. Generation of 2B4 Reporter Cells Displaying Mutated PNAs on the Cell Surface
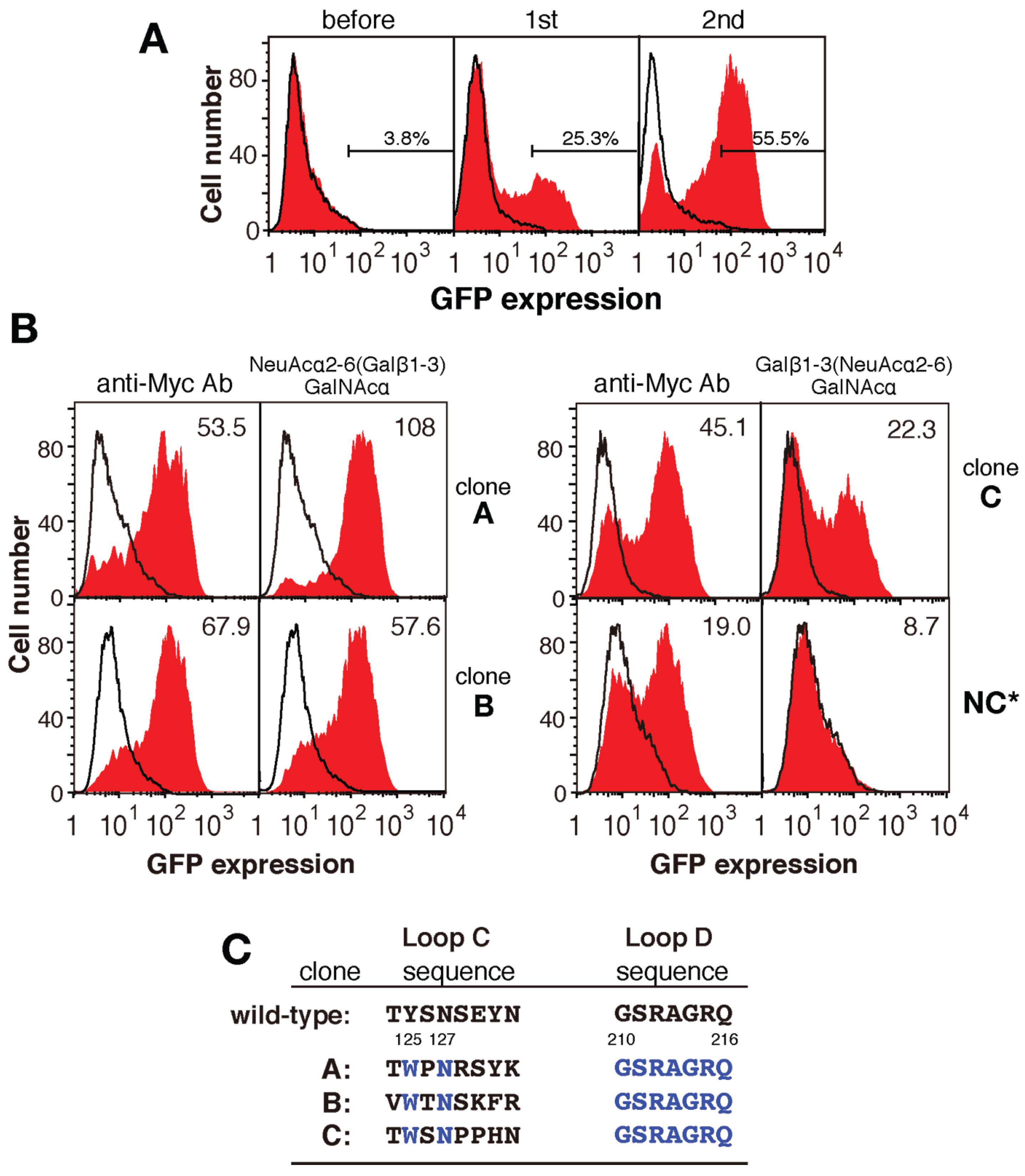
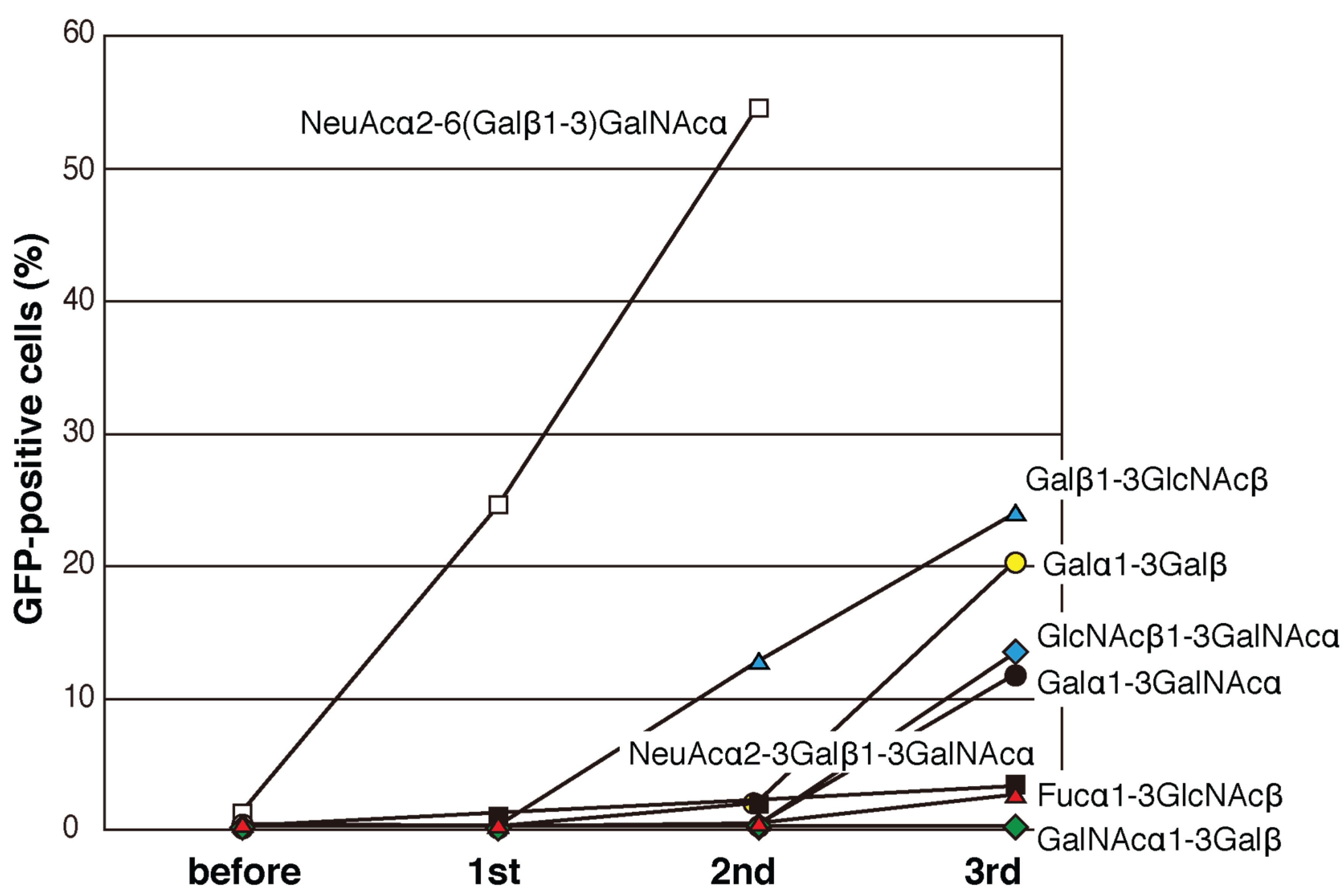
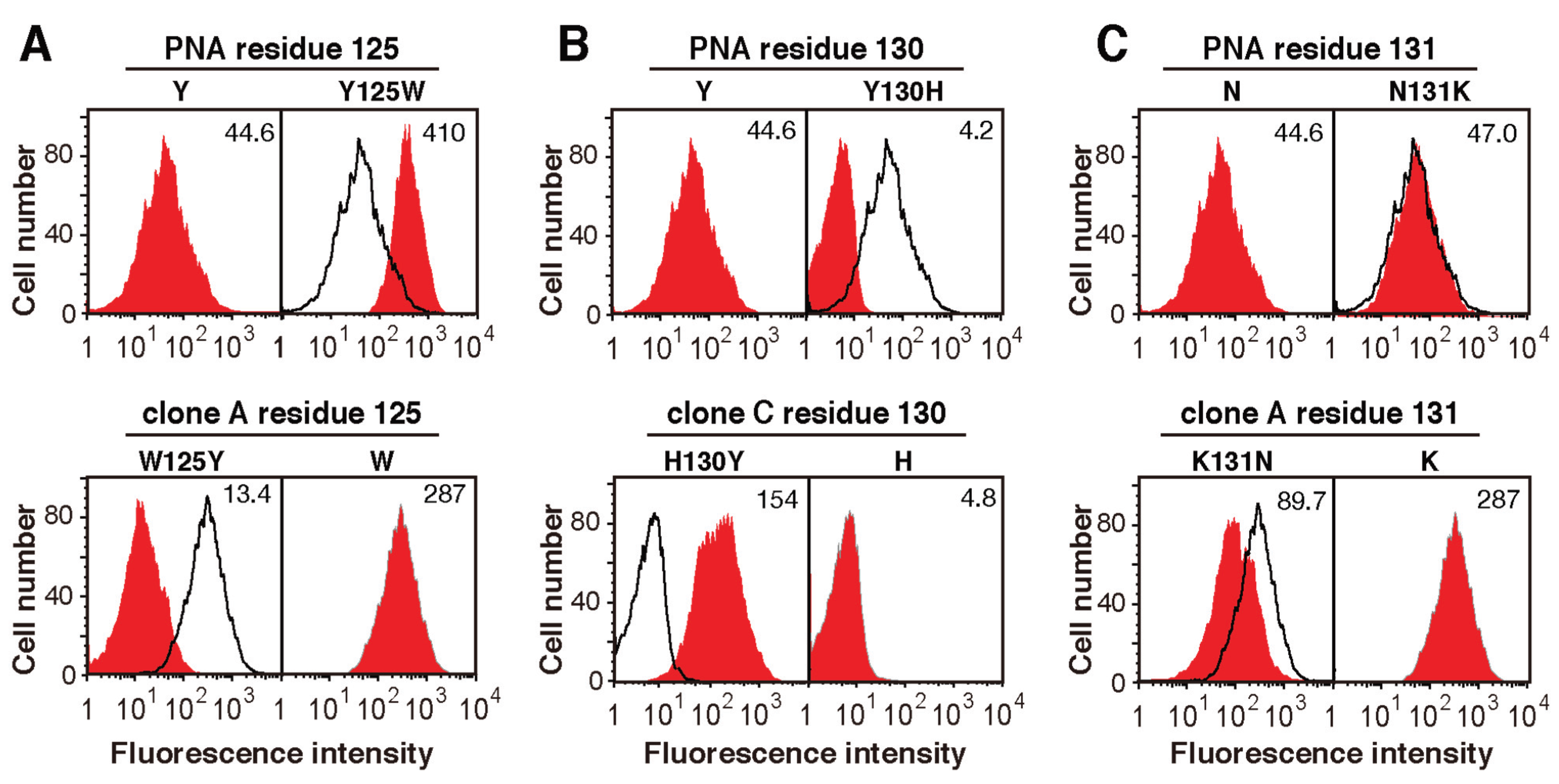
2.3. Screening for Mutated Lectins with Novel Sugar-Binding Specificities
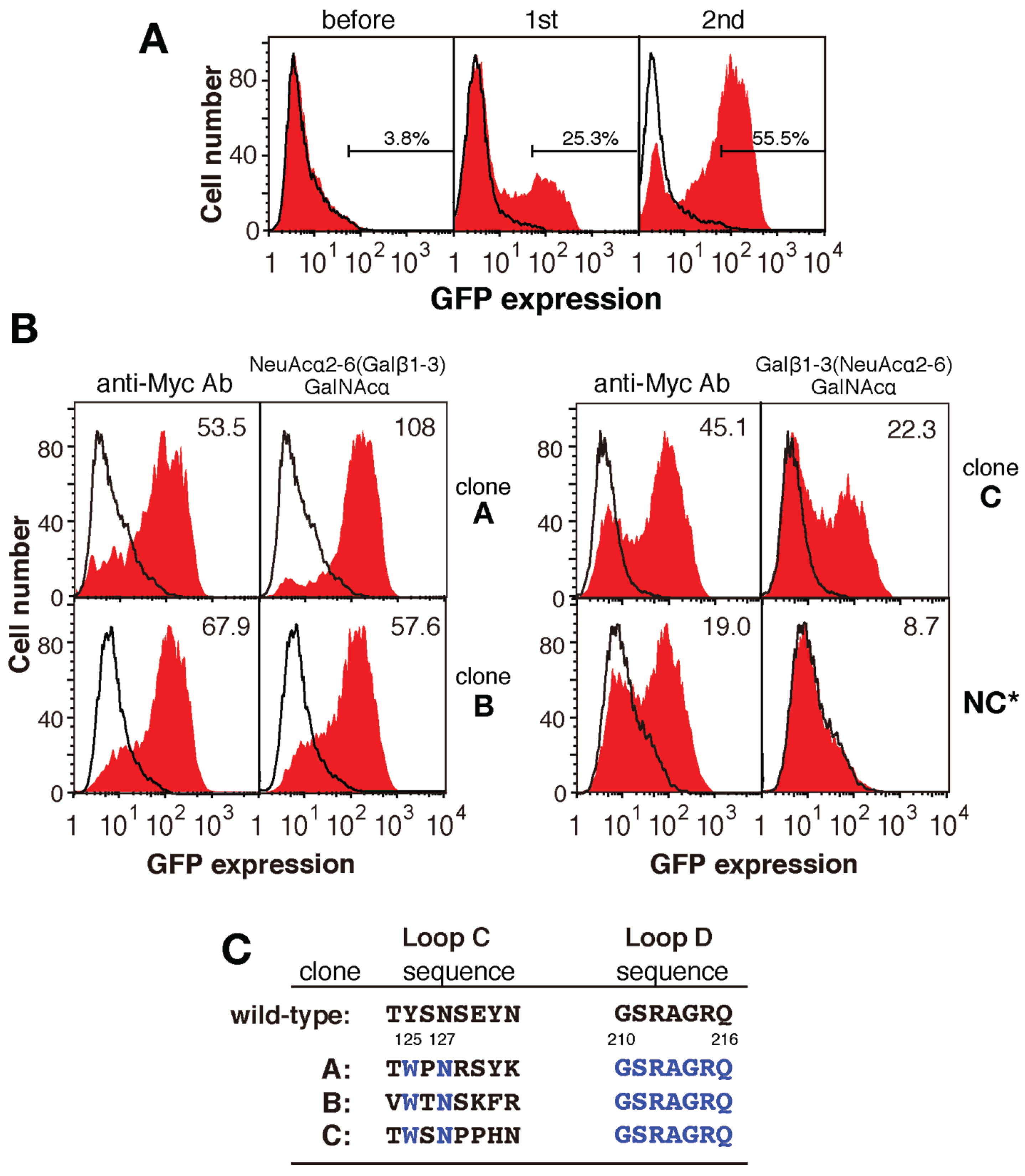

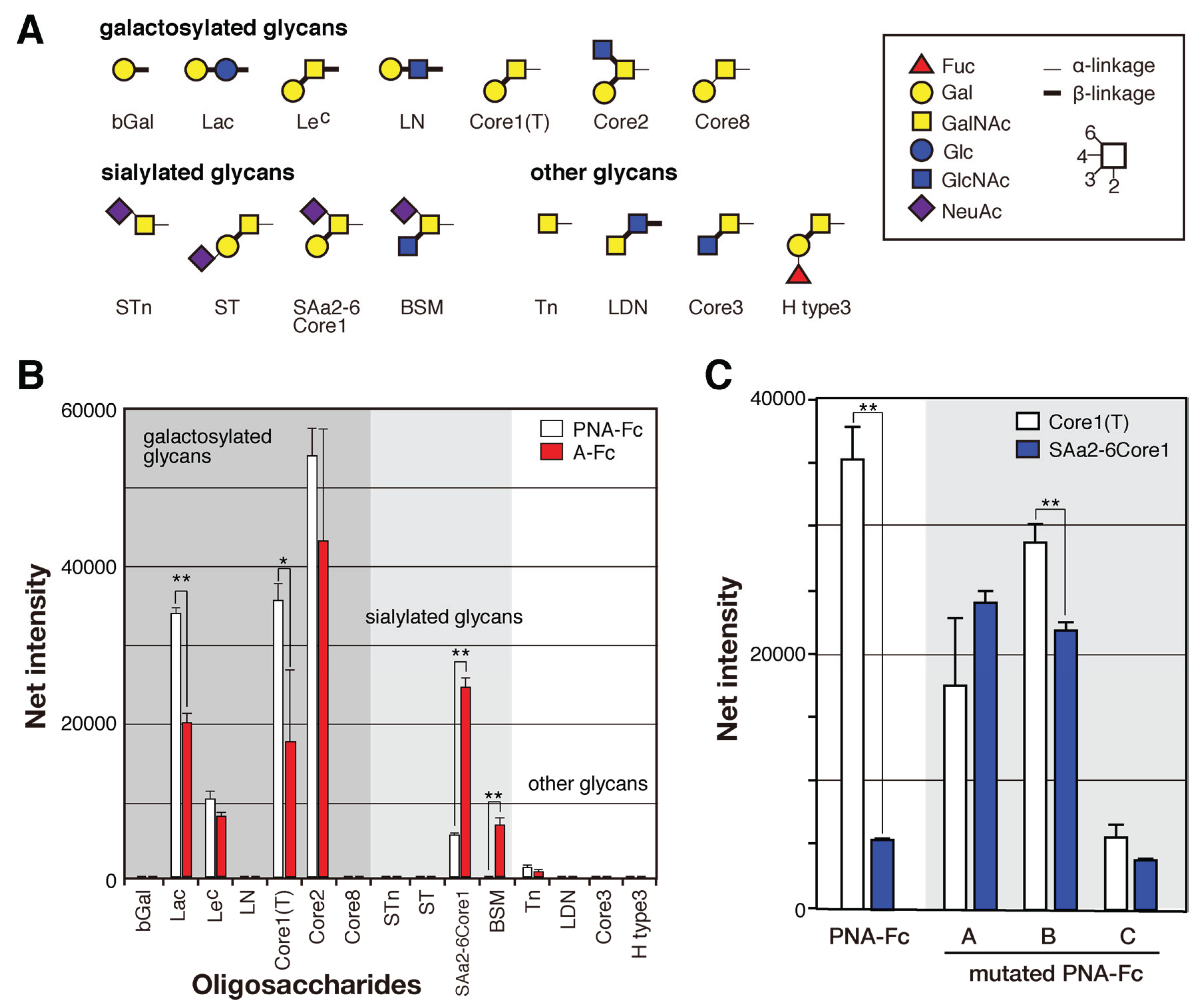
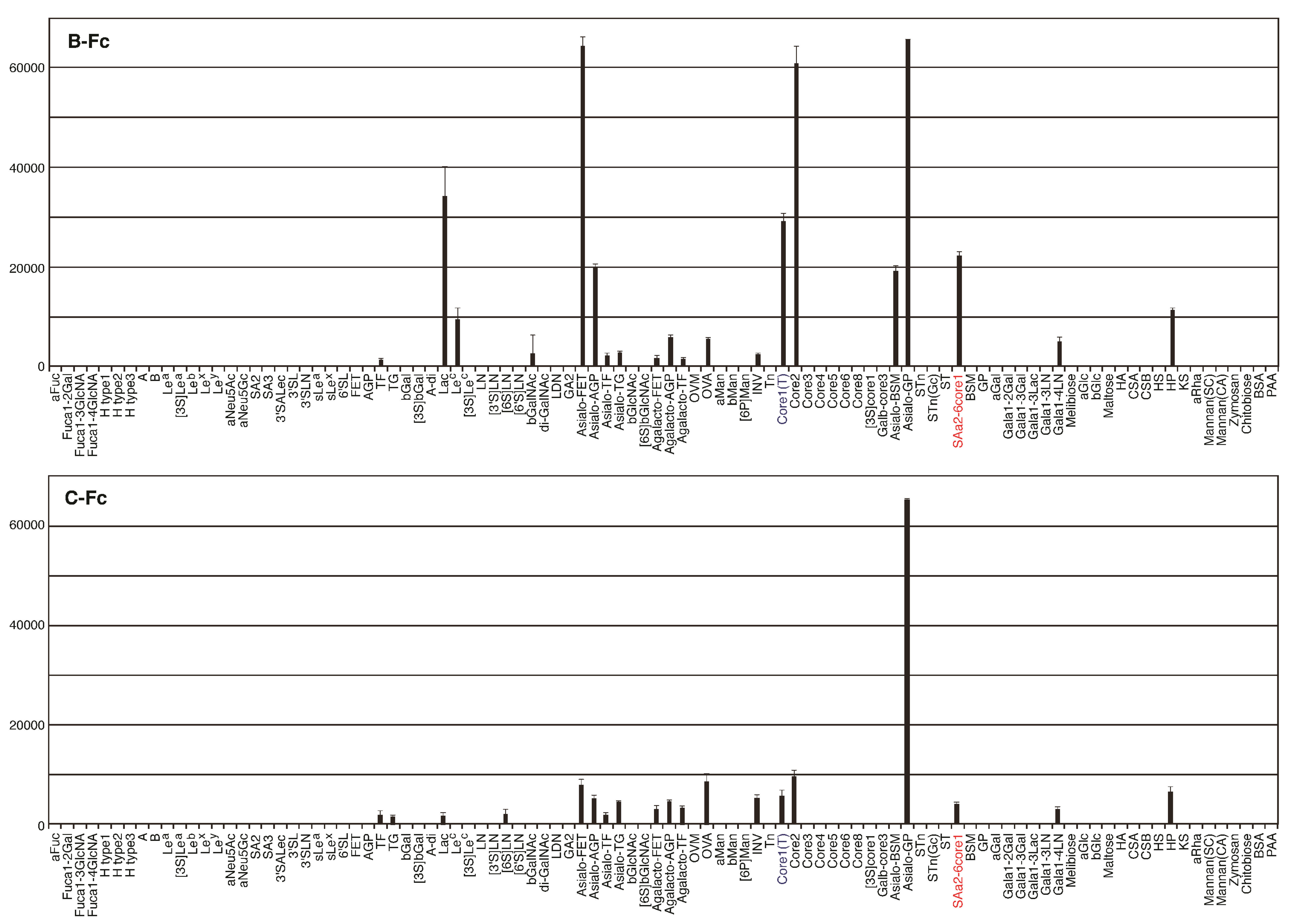
2.4. Identification of the Sugar-Binding Specificities of Mutated PNA-IgG Fc Fusion Proteins Using a Glycan Microarray
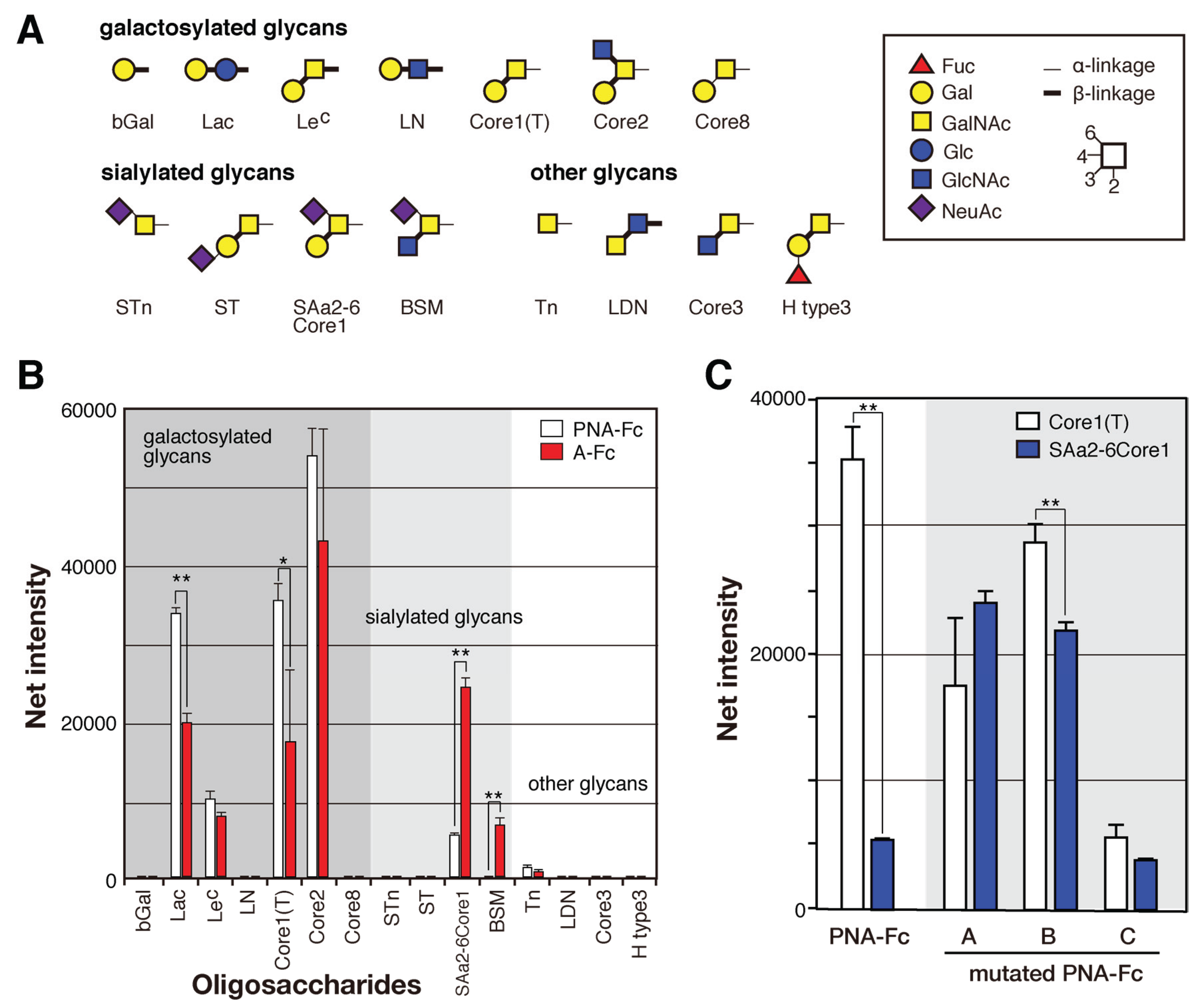
2.5. The Amino Acid Sequences of Loops C and D from the Mutated PNA Proteins Isolated Using NeuAcα2-6(Galβ1-3)GalNAc
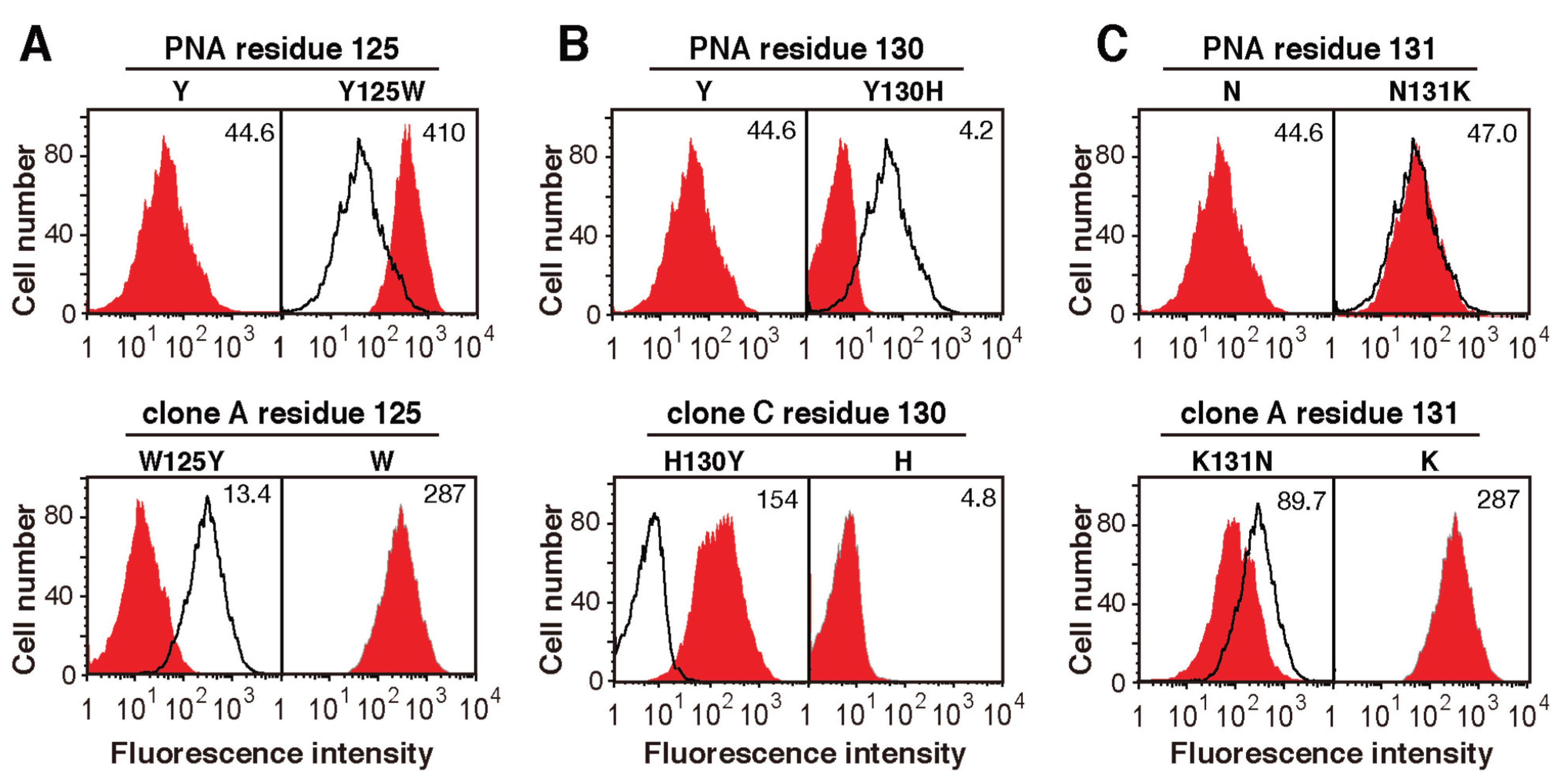
3. Discussion
4. Experimental Section
4.1. Cells and Reagents
4.2. Establishment of PNA and Mutated PNA Library-Expressing Reporter Cells
4.3. Screening of Engineered Lectins from Mutated PNA Library-Expressing Cells by a GFP-Reporter Assay
4.4. Isolation and Sequencing of Cloned Mutated PNA cDNAs
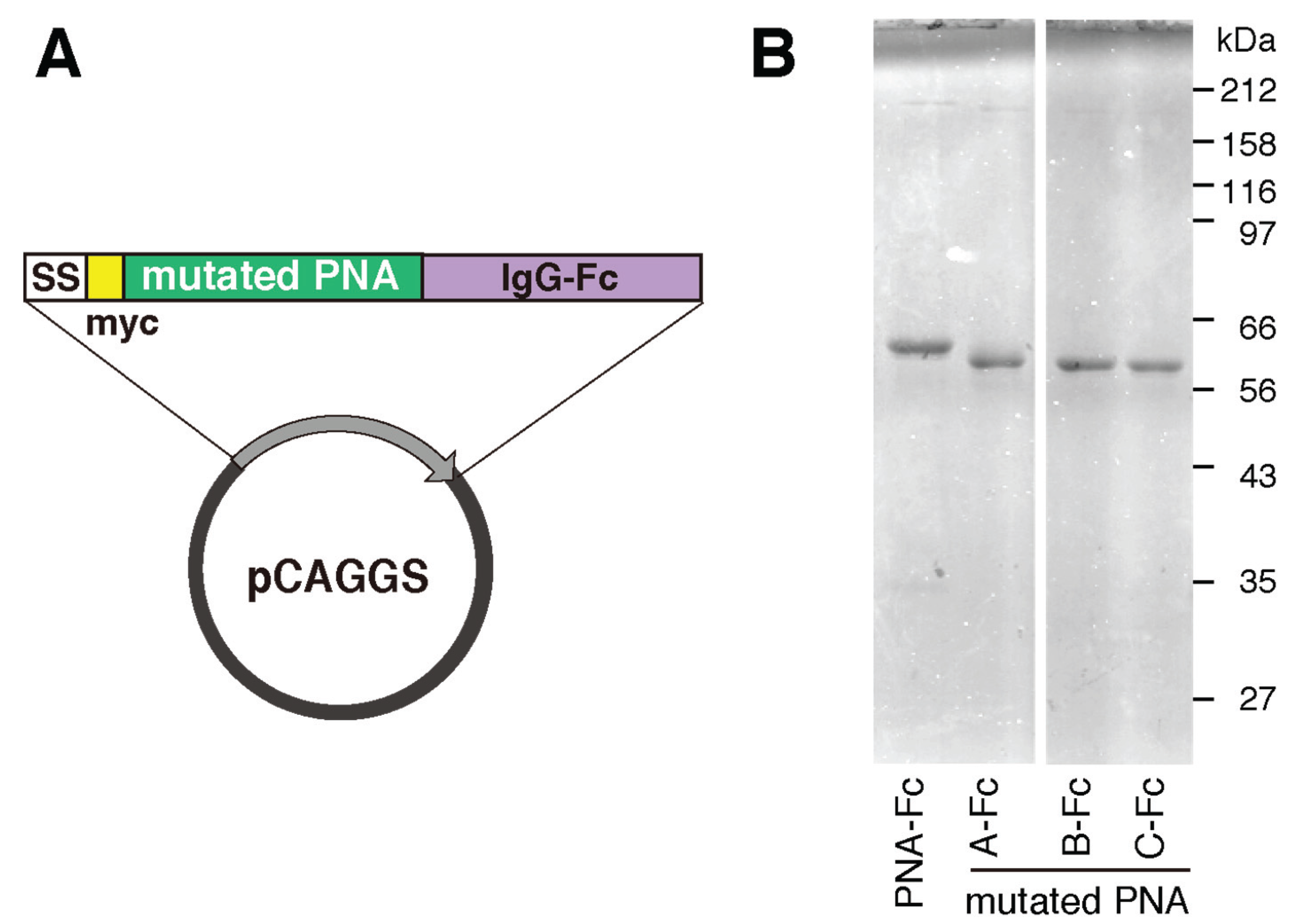
4.5. Preparation of Mutated PNA-IgG Fc Fusion Proteins
4.6. Glycan Microarray
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix



References
- Jang-Lee, J.; North, S.J.; Sutton-Smith, M.; Goldberg, D.; Panico, M.; Morris, H.; Haslam, S.; Dell, A. Glycomic profiling of cells and tissues by mass spectrometry: Fingerprinting and sequencing methodologies. Methods Enzymol. 2006, 415, 59–86. [Google Scholar] [PubMed]
- Pilobello, K.T.; Slawek, D.E.; Mahal, L.K. A ratiometric lectin microarray approach to analysis of the dynamic mammalian glycome. Proc. Natl. Acad. Sci. USA 2007, 104, 11534–11539. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, J.; Yamada, M.; Kuno, A.; Tateno, H. Lectin microarrays: Concept, principle and applications. Chem. Soc. Rev. 2013, 42, 4443–4458. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.; Surolia, A.; Sampathkumar, S.G. Lectin microarrays for glycomic analysis. Omics J. Integr. Biol. 2010, 14, 419–436. [Google Scholar] [CrossRef] [PubMed]
- Katrlík, J.; Svitel, J.; Gemeiner, P.; Kozár, T.; Tkac, J. Glycan and lectin microarrays for glycomics and medicinal applications. Med. Res. Rev. 2010, 30, 394–418. [Google Scholar] [PubMed]
- Sharon, N.; Lis, H. Lectins, 2nd ed.; Kluwer: Dordrecht, The Netherlands, 1993; pp. 105–117. [Google Scholar]
- Yamamoto, K.; Konami, Y.; Osawa, T. Purification and characterization of a carbohydrate-binding peptide from Bauhinia purpurea lectin. FEBS Lett. 1991, 281, 258–262. [Google Scholar] [CrossRef]
- Konami, Y.; Yamamoto, K.; Osawa, T.; Irimura, T. Correlation between carbohydrate-binding specificity and amino acid sequence of carbohydrate-binding regions of Cytisus-type anti-H(O) lectins. FEBS Lett. 1992, 304, 129–135. [Google Scholar] [CrossRef]
- Yamamoto, K.; Konami, Y.; Osawa, T.; Irimura, T. Carbohydrate-binding peptides from several anti-H(O) lectins. J. Biochem. 1992, 111, 436–439. [Google Scholar] [PubMed]
- Yamamoto, K.; Konami, Y.; Osawa, T. A chimeric lectin formed from Bauhinia purpurea lectin and Lens culinaris lectin recognizes a unique carbohydrate structure. J. Biochem. 2000, 127, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Tateno, H.; Hirabayashi, J. Lectin engineering, a molecular evolutionary approach to expanding the lectin utilities. Molecules 2015, 20, 7637–7656. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Dan, X.; Ng, C.C.W.; Ng, T.B. Lectins with potential for anti-cancer therapy. Molecules 2015, 20, 3791–3810. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Zhang, C.Z.Y.; Ng, T.B.; Wong, J.H.; Pan, W.L.; Ye, X.J.; Chan, Y.S.; Fong, W.P. Momordica charantia lectin, a type II ribosome inactivating protein, exhibits antitumor activity toward human nasopharyngeal carcinoma cells in vitro and in vivo. Cancer Prev. Res. 2012, 5, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Sharon, N.; Lis, H. Legume lectins: A large family of homologous proteins. FASEB J. 1990, 4, 3198–3208. [Google Scholar] [PubMed]
- Yamamoto, K.; Konami, Y.; Osawa, T.; Irimura, T. Alteration of the carbohydrate-binding specificity of the Bauhinia purpurea lectin through the preparation of a chimeric lectin. J. Biochem. 1992, 111, 87–90. [Google Scholar] [PubMed]
- Loris, R.; Hamelryck, T.; Bouckaert, J.; Wyns, L. Legume lectin structure. Biochim. Biophys. Acta 1998, 1383, 9–36. [Google Scholar] [CrossRef]
- Sharma, V.; Surolia, A. Analyses of carbohydrate recognition by legume lectins: Size of the combining site loops and their primary specificity. J. Mol. Biol. 1997, 267, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.E.; Kabat, E.A.; Lotan, R.; Sharon, N. Immunochemical studies on the specificity of the peanut (Arachis hypogaea) agglutinin. Carbohydr. Res. 1976, 51, 107–118. [Google Scholar] [CrossRef]
- Lotan, R.; Skutelsky, E.; Danon, D.; Sharon, N. The purification, composition, and specificity of the anti-T lectin from peanut (Arachis hypogaea). J. Biol. Chem. 1975, 250, 8518–8523. [Google Scholar] [PubMed]
- Reddi, A.L.; Sankaranarayanan, K.; Arulraj, H.S.; Devaraj, N.; Devaraj, H. Enzyme-linked PNA lectin-binding assay of serum T-antigen in patients with SCC of the uterine cervix. Cancer Lett. 2000, 149, 207–211. [Google Scholar] [CrossRef]
- Zebda, N.; Bailly, M.; Brown, S.; Dore, J.F.; Berthier-Vergnes, O. Expression of PNA-binding sites on specific glycoproteins by human melanoma cells is associated with a high metastatic potential. J. Cell. Biochem. 1994, 54, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Priatel, J.J.; Chui, D.; Hiraoka, N.; Simmons, C.J.; Richardson, K.B.; Page, D.M.; Fukuda, M.; Varki, N.M.; Marth, J.D. The ST3Gal-I sialyltransferase controls CD8+ T lymphocyte homeostasis by modulating O-glycan biosynthesis. Immunity 2000, 12, 273–283. [Google Scholar] [CrossRef]
- Valenzuela, H.F.; Pace, K.E.; Cabrera, P.V.; White, R.; Porvari, K.; Kaija, H.; Vihko, P.; Baum, L.G. O-glycosylation regulates LNCaP prostate cancer cell susceptibility to apoptosis induced by galectin-1. Cancer Res. 2007, 67, 6155–6162. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Yamamoto, K.; Konami, Y.; Irimura, T.; Osawa, T. Separation of acidic oligosaccharides by liquid chromatography: Application to analysis of sugar chains of glycoproteins. Carbohydr. Res. 1982, 109, 259–269. [Google Scholar] [CrossRef]
- Zhu, K.; Bressan, R.A.; Hasegawa, P.M.; Murdock, L.L. Identification of N-acetylglucosamine binding residues in Griffonia simplicifolia lectin II. FEBS Lett. 1996, 390, 271–274. [Google Scholar] [CrossRef]
- Adar, R.; Sharon, N. Mutational studies of the amino acid residues in the combining site of Erythrina corallodendron lectin. Eur. J. Biochem. 1996, 239, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Van Eijsden, R.R.; Hoedemaeker, F.J.; Diaz, C.L.; Lugtenberg, B.J.; de Pater, B.S.; Kijne, J.W. Mutational analysis of pea lectin. Substitution of Asn125 for Asp in the monosaccharide-binding site eliminates mannose/glucose-binding activity. Plant. Mol. Biol. 1992, 20, 1049–1058. [Google Scholar]
- Samyn-Petit, B.; Krzewinski-Recchi, M.A.; Steelant, W.F.; Delannoy, P.; Harduin-Lepers, A. Molecular cloning and functional expression of human ST6GalNAc II. Molecular expression in various human cultured cells. Biochim. Biophys. Acta 2000, 1474, 201–211. [Google Scholar] [CrossRef]
- Natchiar, S.K.; Srinivas, O.; Mitra, N.; Surolia, A.; Jayaraman, N.; Vijayan, M. Structural studies on peanut lectin complexed with disaccharides involving different linkages: Further insights into the structure and interactions of the lectin. Acta Cryst. 2006, D62, 1413–1421. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Maruyama, I.N.; Osawa, T. Cyborg lectins: Novel leguminous lectins with unique specificities. J. Biochem. 2000, 127, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Yabe, R.; Suzuki, R.; Kuno, A.; Fujimoto, Z.; Jigami, Y.; Hirabayashi, J. Tailoring a novel sialic acid-binding lectin from a ricin-B chain-like galactose-binding protein by natural evolution-mimicry. J. Biochem. 2007, 141, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Tateno, H.; Kuno, A.; Yabe, R.; Hirabayashi, J. Directed evolution of lectins with sugar-binding specificity for 6-sulfo-galactose. J. Biol. Chem. 2012, 287, 20313–20320. [Google Scholar] [CrossRef] [PubMed]
- Deantonio, C.; Cotella, D.; Macor, P.; Santoro, C.; Sblattero, D. Phage display technology for human monoclonal antibodies. Methods Mol. Biol. 2014, 1060, 277–295. [Google Scholar] [PubMed]
- Hu, D.; Tateno, H.; Hirabayashi, J. Directed evolution of lectins by an improved error-prone PCR and ribosome display method. Methods Mol. Biol. 2014, 1200, 527–538. [Google Scholar] [PubMed]
- Edwards, B.M.; He, M. Evolution of antibodies in vitro by ribosome display. Methods Mol. Biol. 2012, 907, 281–292. [Google Scholar] [PubMed]
- Umiel, T.; Daley, J.F.; Bhan, A.K.; Levey, R.H.; Schlossman, S.F.; Reinherz, E.L. Acquisition of immune competence by a subset of human cortical thymocytes expressing mature T cell antigens. J. Immunol. 1982, 129, 1054–1060. [Google Scholar] [PubMed]
- Fukuda, M. Leukosialin, a major O-glycan-containing sialoglycoprotein defining leukocyte differentiation and malignancy. Glycobiology 1991, 1, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Konami, Y.; Yamamoto, K.; Osawa, T.; Irimura, T. Strong affinity of Maackia amurensis hemagglutinin (MAH) for sialic acid-containing Ser/Thr-linked carbohydrate chains of N-terminal octapeptides from human glycophorin A. FEBS Lett. 1994, 342, 334–338. [Google Scholar] [CrossRef]
- Konami, Y.; Ishida, C.; Yamamoto, K.; Osawa, T.; Irimura, T. A unique amino acid sequence involved in the putative carbohydrate-binding domain of a legume lectin specific for sialylated carbohydrate chains: Primary sequence determination of Maackia amurensis hemagglutinin (MAH). J. Biochem. 1994, 115, 767–777. [Google Scholar] [PubMed]
- Yamamoto, K.; Konami, Y.; Irimura, T. Sialic acid-binding motif of Maackia amurensis lectins. J. Biochem. 1997, 121, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Imberty, A.; Gautier, C.; Lescar, J.; Perez, S.; Wyns, L.; Loris, R. An unusual carbohydrate binding site revealed by the structures of two Maackia amurensis lectins complexed with sialic acid-containing oligosaccharides. J. Biol. Chem. 2000, 275, 17541–17548. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, M.; Arase, H.; Takeuchi, A.; Yamasaki, S.; Shiina, R.; Suenaga, T.; Sakurai, D.; Yokosuka, T.; Arase, N.; Iwashima, M.; et al. NFAM1, an immunoreceptor tyrosine-based activation motif-bearing molecule that regulates B cell development and signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 8126–8131. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.; Kojima, T.; Kitamura, T. Plat-E: An efficient and stable system for transient packaging of retroviruses. Gene Therapy 2000, 7, 1063–1066. [Google Scholar] [CrossRef] [PubMed]
- Young, N.M.; Johnston, R.A.; Watson, D.C. The amino acid sequence of peanut agglutinin. Eur. J. Biochem. 1991, 196, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Niwa, H.; Yamamura, K.; Miyazaki, J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 1991, 108, 193–199. [Google Scholar] [PubMed]
- Tateno, H.; Mori, A.; Uchiyama, N.; Yabe, R.; Iwaki, J.; Shikanai, T.; Angata, T.; Narimatsu, H.; Hirabayashi, J. Glycoconjugate microarray based on an evanescent-field fluorescence-assisted detection principle for investigation of glycan-binding proteins. Glycobiology 2008, 18, 789–798. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soga, K.; Abo, H.; Qin, S.-Y.; Kyoutou, T.; Hiemori, K.; Tateno, H.; Matsumoto, N.; Hirabayashi, J.; Yamamoto, K. Mammalian Cell Surface Display as a Novel Method for Developing Engineered Lectins with Novel Characteristics. Biomolecules 2015, 5, 1540-1562. https://doi.org/10.3390/biom5031540
Soga K, Abo H, Qin S-Y, Kyoutou T, Hiemori K, Tateno H, Matsumoto N, Hirabayashi J, Yamamoto K. Mammalian Cell Surface Display as a Novel Method for Developing Engineered Lectins with Novel Characteristics. Biomolecules. 2015; 5(3):1540-1562. https://doi.org/10.3390/biom5031540
Chicago/Turabian StyleSoga, Keisuke, Hirohito Abo, Sheng-Ying Qin, Takuya Kyoutou, Keiko Hiemori, Hiroaki Tateno, Naoki Matsumoto, Jun Hirabayashi, and Kazuo Yamamoto. 2015. "Mammalian Cell Surface Display as a Novel Method for Developing Engineered Lectins with Novel Characteristics" Biomolecules 5, no. 3: 1540-1562. https://doi.org/10.3390/biom5031540
APA StyleSoga, K., Abo, H., Qin, S.-Y., Kyoutou, T., Hiemori, K., Tateno, H., Matsumoto, N., Hirabayashi, J., & Yamamoto, K. (2015). Mammalian Cell Surface Display as a Novel Method for Developing Engineered Lectins with Novel Characteristics. Biomolecules, 5(3), 1540-1562. https://doi.org/10.3390/biom5031540