
NF-kappaB Signaling in Chronic Inflammatory Airway Disease

Abstract
:1. Introduction
2. Asthma and COPD
2.1. Overview of Asthma and COPD
2.2. Evidence of NF-κB in Asthma
2.3. Evidence of NF-κB in COPD
3. Role of NF-κB in Airway Cells in Disease

{kind=link}
| Cell Type | Genes |
|---|---|
| Lymphocytes (Th1/Th2) | Eotaxin-1, regulated and activation normal T cell expressed and secreted (RANTES), Th1 [interferon (IFN)-gamma and interleukin (IL)-2], Th2 [IL-4, IL-5 and IL-13] [46] |
| Eosinophils | TNF-α, IL-8, intercellular adhesion molecule (ICAM)-1 and leukocyte function-associated antigen-1 (LFA-1) [47,48] |
| Neutrophils | IL-8, granulocyte macrophage-colony-stimulating factor (GM-CSF), IL-1Ra [49] |
| Macrophages | Monocyte chemotactic protein-1 (MCP-1), IL-8 and growth-regulated oncogene-α (GROα) [50,51] |
| Epithelial cells | Thymic stromal lymphopoietin (TSLP), ICAM-1, vascular adhesion molecule (VCAM)-1, IL-8, IL-6, GM-CSF, chemokine (C-X-C motif) ligand (CXCL)1, RANTES, GROα, MCP-1, eotaxin-1 and MUC5AC [52,53,5455] |
| Smooth muscle | TSLP, CD38, VCAM-1, ICAM-1, cyclooxygenase-2, IL-6, IL-8, CXCL10 (a chemoattractant for mast cells), GM-CSF, RANTES, MCP-1, GROα, neutrophil-activating protein-2 (NAP-2) and epithelial neutrophil activating peptide 78 (ENA-78) [56,57,58,59,60,61,6263] |
3.1. Lymphocytes
3.2. Eosinophils
3.3. Neutrophils
3.4. Macrophages
3.5. Epithelial Cells
3.6. Smooth Muscle Cell
4. GC-Insensitivity in Severe Asthma and COPD
5. The Effect of GC-Transactivation on NF-κB Signaling
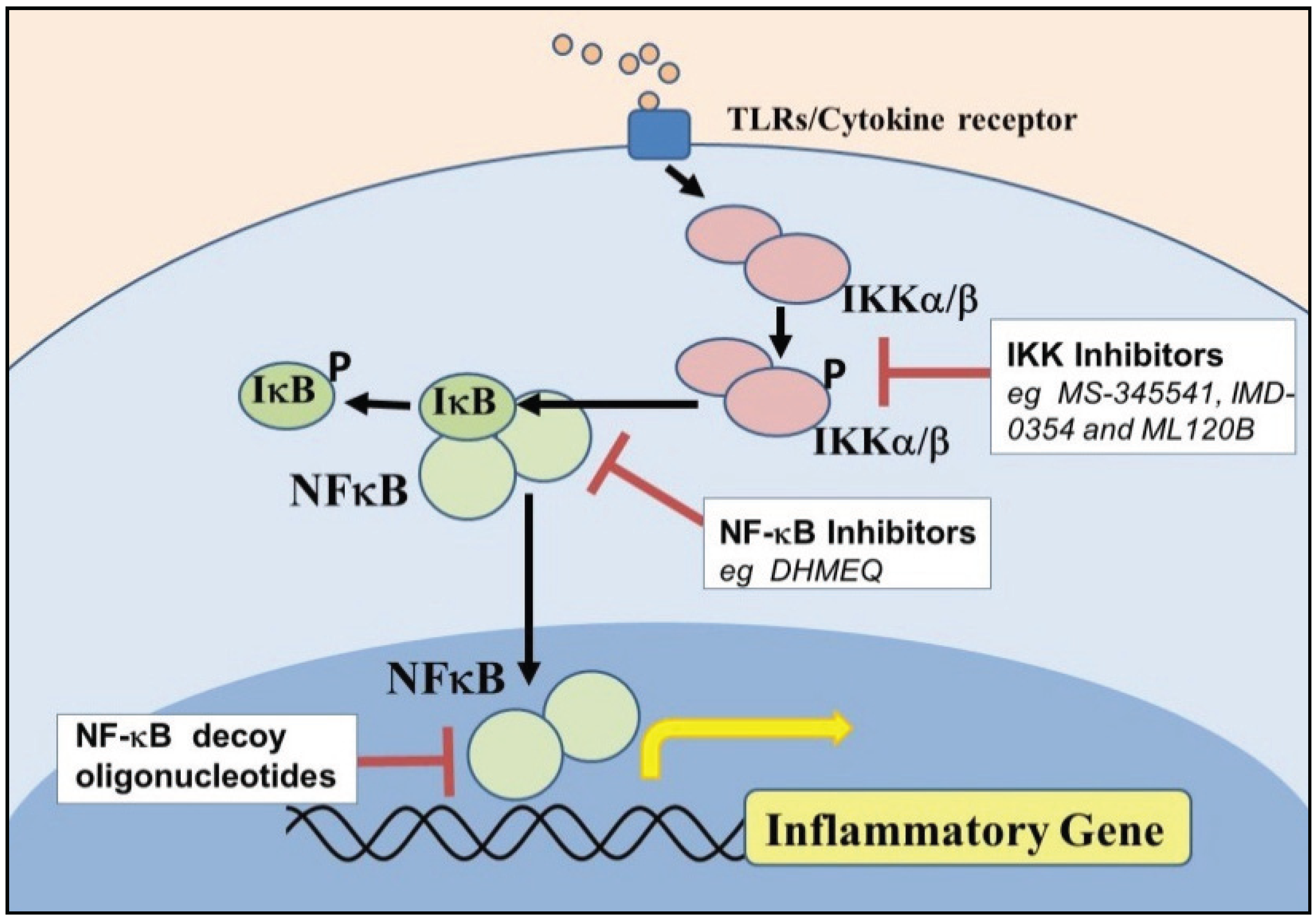
6. Targeting NF-κB Signaling in Airway Obstructive Diseases
6.1. Inhibitors of IKK and NF-κB
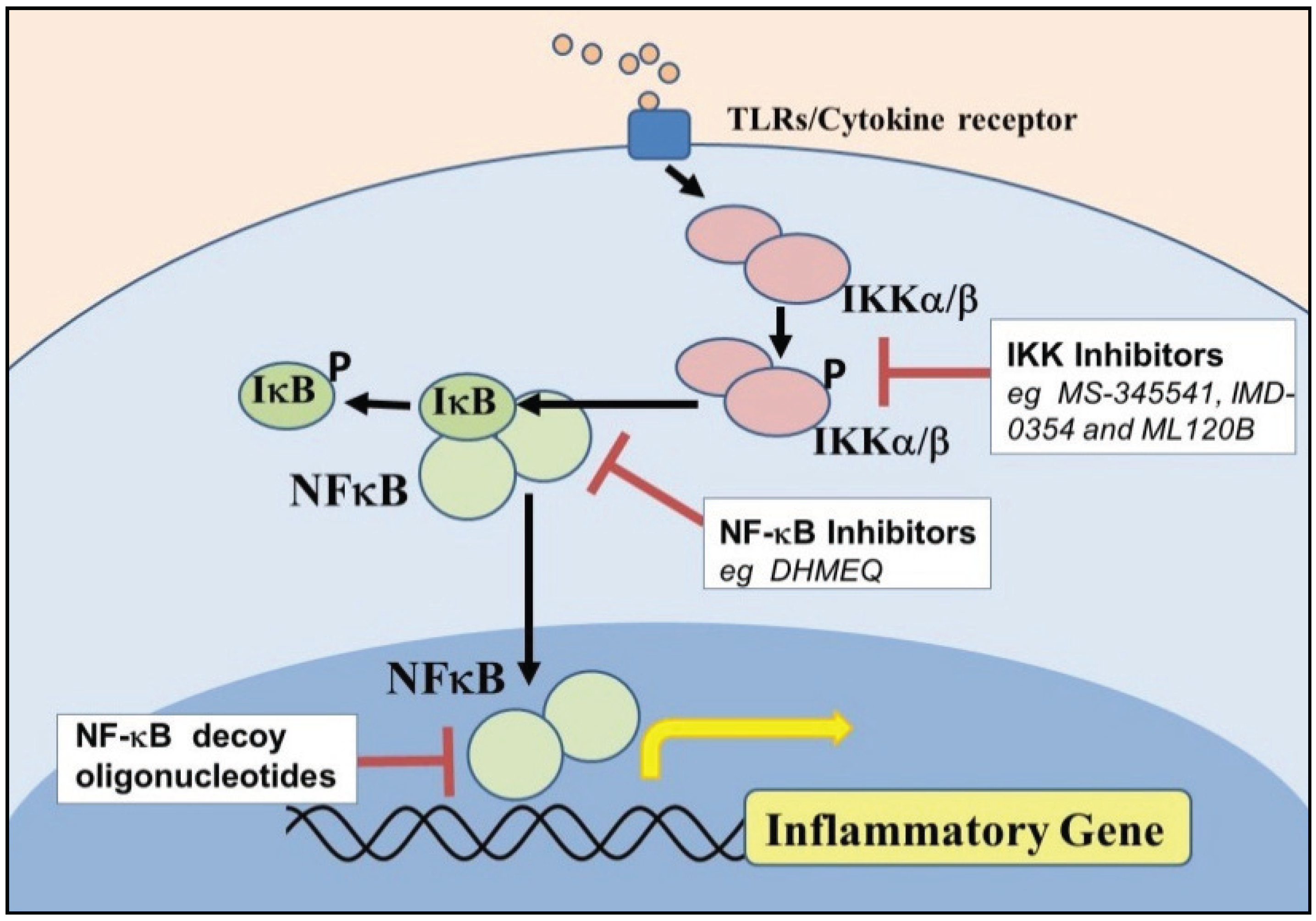
| Inhibitor | Target | Model (Animal and Challenge) | Outcome (as Compared to Vehicle Control Mice Following Challenge) |
|---|---|---|---|
| Compound A | IKKβ | Mouse, allergen (cockroach extract) | Reduced airway inflammation and reactivity [99] |
| Compound A | IKKβ | Rat, allergen (OVA) | Reduced airway inflammation and reactivity [99] |
| DHMEQ | NF-κB | Mouse, allergen (OVA) | Reduced Th2 eosinophilic airway inflammation and AWR [54] |
| GSK 657311A | IKKβ | Mouse, LPS and CS extract | Attenuated LPS-stimulated airway inflammation but had no effect on CS-evoked airway inflammation (despite decreasing NF-κB:DNA binding) [44] |
| IMD-0354 [N-(3,5-bis-trifluoromethyl-phenyl)-5-chloro-2-hydroxy-benzamide] | IKKβ | Mouse, allergen (OVA) | Reduced Th2 eosinophilic airway inflammation [100] |
| IMD-0354 | IKKβ | Mouse, allergen (HDM) | Reduced airway IL-13 production and AWR [101] |
| NF-κB decoy oligonucleotides | NF-κB | Mouse, CS extract | Decreased lung macrophage number but had no effect on other parameters examined (e.g., TNF-α levels) [42] |
| PHA-408 [8-(5-chloro-2-(4-methylpiperazin-1-yl)isonicotinamido)-1-(4-fluorophenyl)-4,5-dihydro-1H-benzo[gamma]indazole-3-carboxamide] | IKKβ | Rat, CS extract and LPS | Reduced airway levels of IL-6, TNF-α, IL-1β and GM-CSF [43] |
| PF-184 [8-(2-(3,4-bis(hydroxymethyl)-3,4-dimethylpyrrolidin-1-yl)-5-chloroisonicotinamido)-1-(4-fluorophenyl)-4,5-dihydro-1H-benzo-[g]indazole-3-carboxamide)] | IKKβ | Rat, LPS | Intratracheal administered PF-184 reduced airway inflammatory cell infiltration and cytokine levels [102] |
| TPCA-1 | IKKβ | Mouse, LPS and CS extract | Attenuated LPS-stimulated airway inflammation but had no effect on CS-evoked airway inflammation (despite decreasing NF-κB:DNA binding) [44] |
6.2. Indirect Modulators of NF-κB Signaling
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- WHO. Global Surveillance, Prevention and Control of Chronic Respiratory Diseases: A Comprehensive Approach; WHO: Geneva, Switzerland, 2007; p. 155. [Google Scholar]
- Decramer, M.; Janssens, W.; Miravitlles, M. Chronic obstructive pulmonary disease. Lancet 2012, 379, 1341–1351. [Google Scholar] [CrossRef]
- Ambrosino, N.; Paggiaro, P. The management of asthma and chronic obstructive pulmonary disease: Current status and future perspectives. Expert Rev. Respir. Med. 2012, 6, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Tetley, T.D. Inflammatory cells and chronic obstructive pulmonary disease. Curr. Drug Targets Inflamm. Allergy 2005, 4, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Tubby, C.; Harrison, T.; Todd, I.; Fairclough, L. Immunological basis of reversible and fixed airways disease. Clin. Sci. 2011, 121, 285–296. [Google Scholar] [PubMed]
- Papiris, S.A.; Kollintza, A.; Karatza, M.; Manali, E.D.; Sotiropoulou, C.; Milic-Emili, J.; Roussos, C.; Daniil, Z. CD8+ T lymphocytes in bronchoalveolar lavage in idiopathic pulmonary fibrosis. J. Inflamm. 2007. [Google Scholar] [CrossRef] [PubMed]
- Durham, A.; Adcock, I.M.; Tliba, O. Steroid resistance in severe asthma: Current mechanisms and future treatment. Curr. Pharm. Des. 2011, 17, 674–684. [Google Scholar] [CrossRef] [PubMed]
- Salter, M.; Biggadike, K.; Matthews, J.L.; West, M.R.; Haase, M.V.; Farrow, S.N.; Uings, I.J.; Gray, D.W. Pharmacological properties of the enhanced-affinity glucocorticoid fluticasone furoate in vitro and in an in vivo model of respiratory inflammatory disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L660–L667. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-κB and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef] [PubMed]
- Hinz, M.; Scheidereit, C. The IκB kinase complex in NF-κB regulation and beyond. EMBO Rep. 2014, 15, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Koziol-White, C.J.; Panettieri, R.A., Jr. Airway smooth muscle and immunomodulation in acute exacerbations of airway disease. Immunol. Rev. 2011, 242, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.E.; Harris, T.; Bamford, T.; Mast, A.; Pain, M.C.; Robertson, C.; Smallwood, D.; Tran, T.; Wilson, J.; Stewart, A.G. Proliferation is not increased in airway myofibroblasts isolated from asthmatics. Eur. Respir. J. 2008, 32, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Flavell, S.J.; Hou, T.Z.; Lax, S.; Filer, A.D.; Salmon, M.; Buckley, C.D. Fibroblasts as novel therapeutic targets in chronic inflammation. Br. J. Pharmacol. 2008, 153, S241–S246. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Riddle, S.R.; Frid, M.G.; el Kasmi, K.C.; McKinsey, T.A.; Sokol, R.J.; Strassheim, D.; Meyrick, B.; Yeager, M.E.; Flockton, A.R.; et al. Emergence of fibroblasts with a proinflammatory epigenetically altered phenotype in severe hypoxic pulmonary hypertension. J. Immunol. 2011, 187, 2711–2722. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.R.; Bartlett, N.W.; Clarke, D.; Birrell, M.; Belvisi, M.; Johnston, S.L. Targeting the NF-κB pathway in asthma and chronic obstructive pulmonary disease. Pharmacol. Ther. 2009, 121, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rajendrasozhan, S.; Yang, S.R.; Edirisinghe, I.; Yao, H.; Adenuga, D.; Rahman, I. Deacetylases and NF-κB in redox regulation of cigarette smoke-induced lung inflammation: Epigenetics in pathogenesis of COPD. Antioxid. Redox Signal. 2008, 10, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Anti-inflammatory actions of glucocorticoids: Molecular mechanisms. Clin. Sci. 1998, 94, 557–572. [Google Scholar] [PubMed]
- Kagoshima, M.; Ito, K.; Cosio, B.; Adcock, I.M. Glucocorticoid suppression of nuclear factor-κB: A role for histone modifications. Biochem. Soc. Trans. 2003, 31, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Adcock, I.M.; Ito, K.; Barnes, P.J. Histone deacetylation: An important mechanism in inflammatory lung diseases. COPD 2005, 2, 445–455. [Google Scholar] [CrossRef] [PubMed]
- To, T.; Stanojevic, S.; Moores, G.; Gershon, A.S.; Bateman, E.D.; Cruz, A.A.; Boulet, L.P. Global asthma prevalence in adults: Findings from the cross-sectional world health survey. BMC Public Health 2012. [Google Scholar] [CrossRef] [PubMed]
- Buist, A.S.; McBurnie, M.A.; Vollmer, W.M.; Gillespie, S.; Burney, P.; Mannino, D.M.; Menezes, A.M.; Sullivan, S.D.; Lee, T.A.; Weiss, K.B.; et al. International variation in the prevalence of COPD (the BOLD Study): A population-based prevalence study. Lancet 2007, 370, 741–750. [Google Scholar] [CrossRef]
- Brusselle, G.; Bracke, K. Targeting immune pathways for therapy in asthma and chronic obstructive pulmonary disease. Ann. Am. Thorac. Soc. 2014, 11, S322–S328. [Google Scholar] [CrossRef] [PubMed]
- Hart, L.A.; Krishnan, V.L.; Adcock, I.M.; Barnes, P.J.; Chung, K.F. Activation and localization of transcription factor, nuclear factor-κB, in asthma. Am. J. Respir. Crit. Care Med. 1998, 158, 1585–1592. [Google Scholar] [CrossRef] [PubMed]
- Gagliardo, R.; Chanez, P.; Mathieu, M.; Bruno, A.; Costanzo, G.; Gougat, C.; Vachier, I.; Bousquet, J.; Bonsignore, G.; Vignola, A.M. Persistent activation of nuclear factor-κB signaling pathway in severe uncontrolled asthma. Am. J. Respir. Crit. Care Med. 2003, 168, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Tully, J.E.; Hoffman, S.M.; Lahue, K.G.; Nolin, J.D.; Anathy, V.; Lundblad, L.K.; Daphtary, N.; Aliyeva, M.; Black, K.E.; Dixon, A.E.; et al. Epithelial NF-κB orchestrates house dust mite-induced airway inflammation, hyperresponsiveness, and fibrotic remodeling. J. Immunol. 2013, 191, 5811–5821. [Google Scholar] [CrossRef] [PubMed]
- Poynter, M.E.; Cloots, R.; van Woerkom, T.; Butnor, K.J.; Vacek, P.; Taatjes, D.J.; Irvin, C.G.; Janssen-Heininger, Y.M. NF-κB activation in airways modulates allergic inflammation but not hyperresponsiveness. J. Immunol. 2004, 173, 7003–7009. [Google Scholar] [CrossRef] [PubMed]
- Poynter, M.E.; Irvin, C.G.; Janssen-Heininger, Y.M. Rapid activation of nuclear factor-κB in airway epithelium in a murine model of allergic airway inflammation. Am. J. Pathol. 2002, 160, 1325–1334. [Google Scholar] [CrossRef]
- Poynter, M.E.; Irvin, C.G.; Janssen-Heininger, Y.M. A prominent role for airway epithelial NF-κB activation in lipopolysaccharide-induced airway inflammation. J. Immunol. 2003, 170, 6257–6265. [Google Scholar] [CrossRef] [PubMed]
- Quinton, L.J.; Jones, M.R.; Simms, B.T.; Kogan, M.S.; Robson, B.E.; Skerrett, S.J.; Mizgerd, J.P. Functions and regulation of NF-κB RelA during pneumococcal pneumonia. J. Immunol. 2007, 178, 1896–1903. [Google Scholar] [CrossRef] [PubMed]
- Donovan, C.E.; Mark, D.A.; He, H.Z.; Liou, H.C.; Kobzik, L.; Wang, Y.; de Sanctis, G.T.; Perkins, D.L.; Finn, P.W. NF-κB/Rel transcription factors: c-Rel Promotes airway hyperresponsiveness and allergic pulmonary inflammation. J. Immunol. 1999, 163, 6827–3683. [Google Scholar] [PubMed]
- Li, X.; Chen, Q.; Chu, C.; You, H.; Jin, M.; Zhao, X.; Zhu, X.; Zhou, W.; Ji, W. Ovalbumin-induced experimental allergic asthma is Toll-like receptor 2 dependent. Allergy Asthma Proc. 2014, 35, e15–e20. [Google Scholar] [CrossRef] [PubMed]
- Lam, D.; Ng, N.; Lee, S.; Batzer, G.; Horner, A.A. Airway house dust extract exposures modify allergen-induced airway hypersensitivity responses by TLR4-dependent and independent pathways. J. Immunol. 2008, 181, 2925–2932. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.J.; Wallin, A.; Della-Cioppa, G.; Sandstrom, T.; Holgate, S.T. Effects of budesonide and formoterol on NF-κB, adhesion molecules, and cytokines in asthma. Am. J. Respir. Crit. Care Med. 2001, 164, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Hancox, R.J.; Stevens, D.A.; Adcock, I.M.; Barnes, P.J.; Taylor, D.R. Effects of inhaled beta agonist and corticosteroid treatment on nuclear transcription factors in bronchial mucosa in asthma. Thorax 1999, 54, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Hart, L.; Lim, S.; Adcock, I.; Barnes, P.J.; Chung, K.F. Effects of inhaled corticosteroid therapy on expression and DNA-binding activity of nuclear factor kappaB in asthma. Am. J. Respir. Crit. Care Med. 2000, 161, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, A.; Caramori, G.; Oates, T.; Capelli, A.; Lusuardi, M.; Gnemmi, I.; Ioli, F.; Chung, K.F.; Donner, C.F.; Barnes, P.J.; et al. Increased expression of nuclear factor-κB in bronchial biopsies from smokers and patients with COPD. Eur. Respir. J. 2002, 20, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Caramori, G.; Romagnoli, M.; Casolari, P.; Bellettato, C.; Casoni, G.; Boschetto, P.; Chung, K.F.; Barnes, P.J.; Adcock, I.M.; Ciaccia, A.; et al. Nuclear localisation of p65 in sputum macrophages but not in sputum neutrophils during COPD exacerbations. Thorax 2003, 58, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Szulakowski, P.; Crowther, A.J.; Jimenez, L.A.; Donaldson, K.; Mayer, R.; Leonard, T.B.; MacNee, W.; Drost, E.M. The effect of smoking on the transcriptional regulation of lung inflammation in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2006, 174, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Gagliardo, R.; Chanez, P.; Profita, M.; Bonanno, A.; Albano, G.D.; Montalbano, A.M.; Pompeo, F.; Gagliardo, C.; Merendino, A.M.; Gjomarkaj, M. IκB kinase-driven nuclear factor-κB activation in patients with asthma and chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2011, 128, 635.e2–645.e2. [Google Scholar] [CrossRef] [PubMed]
- Brown, V.; Elborn, J.S.; Bradley, J.; Ennis, M. Dysregulated apoptosis and NFκB expression in COPD subjects. Respir. Res. 2009. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.R.; Yao, H.; Rajendrasozhan, S.; Chung, S.; Edirisinghe, I.; Valvo, S.; Fromm, G.; McCabe, M.J., Jr.; Sime, P.J.; Phipps, R.P.; et al. RelB is differentially regulated by IκB Kinase-α in B cells and mouse lung by cigarette smoke. Am. J. Respir. Cell Mol. Biol. 2009, 40, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.T.; He, B.; Wang, Y.Z.; Wang, J. Effects of intratracheal administration of nuclear factor-κB decoy oligodeoxynucleotides on long-term cigarette smoke-induced lung inflammation and pathology in mice. Respir. Res. 2009. [Google Scholar] [CrossRef] [PubMed]
- Rajendrasozhan, S.; Hwang, J.W.; Yao, H.; Kishore, N.; Rahman, I. Anti-inflammatory effect of a selective IκB kinase-beta inhibitor in rat lung in response to LPS and cigarette smoke. Pulm. Pharmacol. Ther. 2010, 23, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Rastrick, J.M.; Stevenson, C.S.; Eltom, S.; Grace, M.; Davies, M.; Kilty, I.; Evans, S.M.; Pasparakis, M.; Catley, M.C.; Lawrence, T.; et al. Cigarette smoke induced airway inflammation is independent of NF-κB signalling. PLoS ONE 2013, 8, e54128. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Edirisinghe, I.; Rajendrasozhan, S.; Yang, S.R.; Caito, S.; Adenuga, D.; Rahman, I. Cigarette smoke-mediated inflammatory and oxidative responses are strain-dependent in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L1174–L1186. [Google Scholar] [CrossRef] [PubMed]
- Das, J.; Chen, C.H.; Yang, L.; Cohn, L.; Ray, P.; Ray, A. A critical role for NF-κB in GATA3 expression and TH2 differentiation in allergic airway inflammation. Nat. Immunol. 2001, 2, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Coward, W.R.; Sagara, H.; Wilson, S.J.; Holgate, S.T.; Church, M.K. Allergen activates peripheral blood eosinophil nuclear factor-κB to generate granulocyte macrophage-colony stimulating factor, tumour necrosis factor-α and interleukin-8. Clin. Exp. Allergy 2004, 34, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.K.; Wang, C.B.; Li, M.L.; Ip, W.K.; Tian, Y.P.; Lam, C.W. Induction of adhesion molecules upon the interaction between eosinophils and bronchial epithelial cells: Involvement of p38 MAPK and NF-κB. Int. Immunopharmacol. 2006, 6, 1859–1871. [Google Scholar] [CrossRef] [PubMed]
- Langereis, J.D.; Raaijmakers, H.A.; Ulfman, L.H.; Koenderman, L. Abrogation of NF-κB signaling in human neutrophils induces neutrophil survival through sustained p38-MAPK activation. J. Leukoc. Biol. 2010, 88, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Murugan, V.; Peck, M.J. Signal transduction pathways linking the activation of alveolar macrophages with the recruitment of neutrophils to lungs in chronic obstructive pulmonary disease. Exp. Lung Res. 2009, 35, 439–485. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.R.; Chida, A.S.; Bauter, M.R.; Shafiq, N.; Seweryniak, K.; Maggirwar, S.B.; Kilty, I.; Rahman, I. Cigarette smoke induces proinflammatory cytokine release by activation of NF-κB and posttranslational modifications of histone deacetylase in macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L46–L57. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Ziegler, S.F. Inducible expression of the proallergic cytokine thymic stromal lymphopoietin in airway epithelial cells is controlled by NFκB. Proc. Natl. Acad. Sci. USA 2007, 104, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Newton, R.; Holden, N.S.; Catley, M.C.; Oyelusi, W.; Leigh, R.; Proud, D.; Barnes, P.J. Repression of inflammatory gene expression in human pulmonary epithelial cells by small-molecule IκB kinase inhibitors. J. Pharmacol. Exp. Ther. 2007, 321, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Konno, S.; Ozaki, M.; Umezawa, K.; Yamashita, K.; Todo, S.; Nishimura, M. Dehydroxymethylepoxyquinomicin (DHMEQ), a novel NF-κB inhibitor, inhibits allergic inflammation and airway remodelling in murine models of asthma. Clin. Exp. Allergy 2012, 42, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Ren, G.; Gong, Y.; Dong, S.; Yin, Y.; Zhang, L. Bronchial epithelial cells release IL-6, CXCL1 and CXCL8 upon mast cell interaction. Cytokine 2011, 56, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Redhu, N.S.; Saleh, A.; Halayko, A.J.; Ali, A.S.; Gounni, A.S. Essential role of NF-κB and AP-1 transcription factors in TNF-α-induced TSLP expression in human airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L479–L485. [Google Scholar] [CrossRef] [PubMed]
- Tirumurugaan, K.G.; Kang, B.N.; Panettieri, R.A.; Foster, D.N.; Walseth, T.F.; Kannan, M.S. Regulation of the CD38 promoter in human airway smooth muscle cells by TNF-α and dexamethasone. Respir. Res. 2008. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Lin, W.N.; Lin, C.C.; Luo, S.F.; Wang, J.S.; Pouyssegur, J.; Yang, C.M. Transcriptional regulation of VCAM-1 expression by tumor necrosis factor-alpha in human tracheal smooth muscle cells: Involvement of MAPKs, NF-κB, p300, and histone acetylation. J. Cell. Physiol. 2006, 207, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Catley, M.C.; Sukkar, M.B.; Chung, K.F.; Jaffee, B.; Liao, S.M.; Coyle, A.J.; Haddad, El-B.; Barnes, P.J.; Newton, R. Validation of the anti-inflammatory properties of small-molecule IκB Kinase (IKK)-2 inhibitors by comparison with adenoviral-mediated delivery of dominant-negative IKK1 and IKK2 in human airways smooth muscle. Mol. Pharmacol. 2006, 70, 697–705. [Google Scholar] [CrossRef] [PubMed]
- John, A.E.; Zhu, Y.M.; Brightling, C.E.; Pang, L.; Knox, A.J. Human airway smooth muscle cells from asthmatic individuals have CXCL8 hypersecretion due to increased NF-κB p65, C/EBP β, and RNA polymerase II binding to the CXCL8 promoter. J. Immunol. 2009, 183, 4682–4692. [Google Scholar] [CrossRef] [PubMed]
- Alrashdan, Y.A.; Alkhouri, H.; Chen, E.; Lalor, D.J.; Poniris, M.; Henness, S.; Brightling, C.E.; Burgess, J.K.; Armour, C.L.; Ammit, A.J.; et al. Asthmatic airway smooth muscle CXCL10 production: Mitogen-activated protein kinase JNK involvement. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L1118–L1127. [Google Scholar] [CrossRef] [PubMed]
- Gras, D.; Chanez, P.; Vachier, I.; Petit, A.; Bourdin, A. Bronchial epithelium as a target for innovative treatments in asthma. Pharmacol. Ther. 2013, 140, 290–305. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.L.; Clifford, R.L.; Jindarat, S.; Proud, D.; Pang, L.; Belvisi, M.; Knox, A.J. TNFα and IFNγ synergistically enhance transcriptional activation of CXCL10 in human airway smooth muscle cells via STAT-1, NF-κB, and the transcriptional coactivator CREB-binding protein. J. Biol. Chem. 2010, 285, 29101–29110. [Google Scholar] [CrossRef] [PubMed]
- Pelaia, G.; Vatrella, A.; Busceti, M.T.; Gallelli, L.; Calabrese, C.; Terracciano, R.; Maselli, R. Cellular mechanisms underlying eosinophilic and neutrophilic airway inflammation in asthma. Mediat. Inflamm. 2015. [Google Scholar] [CrossRef] [PubMed]
- Brusselle, G.G.; Maes, T.; Bracke, K.R. Eosinophils in the spotlight: Eosinophilic airway inflammation in nonallergic asthma. Nat. Med. 2013, 19, 977–979. [Google Scholar] [CrossRef] [PubMed]
- Chien, J.W.; Lin, C.Y.; Yang, K.D.; Lin, C.H.; Kao, J.K.; Tsai, Y.G. Increased IL-17A secreting CD4+ T cells, serum IL-17 levels and exhaled nitric oxide are correlated with childhood asthma severity. Clin. Exp. Allergy 2013, 43, 1018–1026. [Google Scholar] [CrossRef] [PubMed]
- Chesne, J.; Braza, F.; Mahay, G.; Brouard, S.; Aronica, M.; Magnan, A. IL-17 in severe asthma. Where do we stand? Am. J. Respir. Crit. Care Med. 2014, 190, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Chang, M.M.; Velichko, S.; Thai, P.; Hung, L.Y.; Huang, F.; Phuong, N.; Chen, Y.; Wu, R. NF-κB mediates IL-1β- and IL-17A-induced MUC5B expression in airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2011, 45, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Dragon, S.; Hirst, S.J.; Lee, T.H.; Gounni, A.S. IL-17A mediates a selective gene expression profile in asthmatic human airway smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Al-Alwan, L.; Risse, P.A.; Halayko, A.J.; Martin, J.G.; Baglole, C.J.; Eidelman, D.H.; Hamid, Q. Th17-associated cytokines promote human airway smooth muscle cell proliferation. FASEB J. 2012, 26, 5152–5160. [Google Scholar] [CrossRef] [PubMed]
- Kankaanranta, H.; Ilmarinen, P.; Zhang, X.; Adcock, I.M.; Lahti, A.; Barnes, P.J.; Giembycz, M.A.; Lindsay, M.A.; Moilanen, E. Tumour necrosis factor-α regulates human eosinophil apoptosis via ligation of TNF-receptor 1 and balance between NF-κB and AP-1. PLoS ONE 2014, 9, e90298. [Google Scholar] [CrossRef] [PubMed]
- Ordonez, C.L.; Shaughnessy, T.E.; Matthay, M.A.; Fahy, J.V. Increased neutrophil numbers and IL-8 levels in airway secretions in acute severe asthma: Clinical and biologic significance. Am. J. Respir. Crit. Care Med. 2000, 161, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Nakagome, K.; Matsushita, S.; Nagata, M. Neutrophilic inflammation in severe asthma. Int. Arch. Allergy Immunol. 2012, 158, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Pappas, K.; Papaioannou, A.I.; Kostikas, K.; Tzanakis, N. The role of macrophages in obstructive airways disease: Chronic obstructive pulmonary disease and asthma. Cytokine 2013, 64, 613–625. [Google Scholar] [CrossRef] [PubMed]
- Cundall, M.; Sun, Y.; Miranda, C.; Trudeau, J.B.; Barnes, S.; Wenzel, S.E. Neutrophil-derived matrix metalloproteinase-9 is increased in severe asthma and poorly inhibited by glucocorticoids. J. Allergy Clin. Immunol. 2003, 112, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.B.; Wong, C.K.; Ip, W.K.; Li, M.L.; Tian, Y.P.; Lam, C.W. Induction of IL-6 in co-culture of bronchial epithelial cells and eosinophils is regulated by p38 MAPK and NF-κB. Allergy 2005, 60, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Hwang, S.M.; Chung, I.Y. S100A8, S100A9 and S100A12 activate airway epithelial cells to produce MUC5AC via extracellular signal-regulated kinase and nuclear factor-κB pathways. Immunology 2015, 144, 79–90. [Google Scholar] [CrossRef] [PubMed]
- James, A.L.; Wenzel, S. Clinical relevance of airway remodelling in airway diseases. Eur. Respir. J. 2007, 30, 134–155. [Google Scholar] [CrossRef] [PubMed]
- Amin, K.; Ludviksdottir, D.; Janson, C.; Nettelbladt, O.; Bjornsson, E.; Roomans, G.M.; Boman, G.; Seveus, L.; Venge, P. Inflammation and structural changes in the airways of patients with atopic and nonatopic asthma. BHR Group. Am. J. Respir. Crit. Care Med. 2000, 162, 2295–2301. [Google Scholar] [CrossRef] [PubMed]
- Gosselink, J.V.; Hayashi, S.; Elliott, W.M.; Xing, L.; Chan, B.; Yang, L.; Wright, C.; Sin, D.; Pare, P.D.; Pierce, J.A.; et al. Differential expression of tissue repair genes in the pathogenesis of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 181, 1329–1335. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.C.; Redhu, N.S.; Moir, L.M.; Koziol-White, C.; Ammit, A.J.; Al-Alwan, L.; Camoretti-Mercado, B.; Clifford, R.L. Pro-inflammatory and immunomodulatory functions of airway smooth muscle: Emerging concepts. Pulm. Pharmacol. Ther. 2013, 26, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.; Fernandes, D.J.; Schuliga, M.; Harris, T.; Landells, L.; Stewart, A.G. Stimulus-dependent glucocorticoid-resistance of GM-CSF production in human cultured airway smooth muscle. Br. J. Pharmacol. 2005, 145, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Keenan, C.R.; Salem, S.; Fietz, E.R.; Gualano, R.C.; Stewart, A.G. Glucocorticoid-resistant asthma and novel anti-inflammatory drugs. Drug Discov. Today 2012, 17, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Salem, S.; Harris, T.; Mok, J.S.; Li, M.Y.; Keenan, C.R.; Schuliga, M.J.; Stewart, A.G. Transforming growth factor-β impairs glucocorticoid activity in the A549 lung adenocarcinoma cell line. Br. J. Pharmacol. 2012, 166, 2036–2048. [Google Scholar] [CrossRef] [PubMed]
- Langenbach, S.Y.; Wheaton, B.J.; Fernandes, D.J.; Jones, C.; Sutherland, T.E.; Wraith, B.C.; Harris, T.; Schuliga, M.J.; McLean, C.; Stewart, A.G. Resistance of fibrogenic responses to glucocorticoid and 2-methoxyestradiol in bleomycin-induced lung fibrosis in mice. Can. J. Physiol. Pharmacol. 2007, 85, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Bonacci, J.V.; Schuliga, M.; Harris, T.; Stewart, A.G. Collagen impairs glucocorticoid actions in airway smooth muscle through integrin signalling. Br. J. Pharmacol. 2006, 149, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2013, 131, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Adcock, I.M.; Cosio, B.; Tsaprouni, L.; Barnes, P.J.; Ito, K. Redox regulation of histone deacetylases and glucocorticoid-mediated inhibition of the inflammatory response. Antioxid. Redox Signal. 2005, 7, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Kagoshima, M.; Wilcke, T.; Ito, K.; Tsaprouni, L.; Barnes, P.J.; Punchard, N.; Adcock, I.M. Glucocorticoid-mediated transrepression is regulated by histone acetylation and DNA methylation. Eur. J. Pharmacol. 2001, 429, 327–334. [Google Scholar] [CrossRef]
- Ito, K.; Yamamura, S.; Essilfie-Quaye, S.; Cosio, B.; Ito, M.; Barnes, P.J.; Adcock, I.M. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-κB suppression. J. Exp. Med. 2006, 203, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Rajendrasozhan, S.; Yang, S.R.; Kinnula, V.L.; Rahman, I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2008, 177, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, H.; Luo, G.; Dai, X. SIRT4 inhibits cigarette smoke extracts-induced mononuclear cell adhesion to human pulmonary microvascular endothelial cells via regulating NF-κB activity. Toxicol. Lett. 2014, 226, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Keenan, C.R.; Schuliga, M.J.; Stewart, A.G. Pro-inflammatory mediators increase levels of the noncoding RNA GAS5 in airway smooth muscle and epithelial cells. Can. J. Physiol. Pharmacol. 2015, 93, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Eddleston, J.; Herschbach, J.; Wagelie-Steffen, A.L.; Christiansen, S.C.; Zuraw, B.L. The anti-inflammatory effect of glucocorticoids is mediated by glucocorticoid-induced leucine zipper in epithelial cells. J. Allergy Clin. Immunol. 2007, 119, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Ayroldi, E.; Migliorati, G.; Bruscoli, S.; Marchetti, C.; Zollo, O.; Cannarile, L.; D’Adamio, F.; Riccardi, C. Modulation of T-cell activation by the glucocorticoid-induced leucine zipper factor via inhibition of nuclear factor κB. Blood 2001, 98, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Che, W.; Manetsch, M.; Quante, T.; Rahman, M.M.; Patel, B.S.; Ge, Q.; Ammit, A.J. Sphingosine 1-phosphate induces MKP-1 expression via p38 MAPK- and CREB-mediated pathways in airway smooth muscle cells. Biochim. Biophys. Acta 2012, 1823, 1658–1665. [Google Scholar] [CrossRef] [PubMed]
- Manetsch, M.; Che, W.; Seidel, P.; Chen, Y.; Ammit, A.J. MKP-1: A negative feedback effector that represses MAPK-mediated pro-inflammatory signaling pathways and cytokine secretion in human airway smooth muscle cells. Cell Signal. 2012, 24, 907–913. [Google Scholar] [CrossRef] [PubMed]
- King, E.M.; Holden, N.S.; Gong, W.; Rider, C.F.; Newton, R. Inhibition of NF-κB-dependent transcription by MKP-1: Transcriptional repression by glucocorticoids occurring via p38 MAPK. J. Biol. Chem. 2009, 284, 26803–26815. [Google Scholar] [CrossRef] [PubMed]
- Ziegelbauer, K.; Gantner, F.; Lukacs, N.W.; Berlin, A.; Fuchikami, K.; Niki, T.; Sakai, K.; Inbe, H.; Takeshita, K.; Ishimori, M.; et al. A selective novel low-molecular-weight inhibitor of IκB kinase-β (IKK-β) prevents pulmonary inflammation and shows broad anti-inflammatory activity. Br. J. Pharmacol. 2005, 145, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Sugita, A.; Ogawa, H.; Azuma, M.; Muto, S.; Honjo, A.; Yanagawa, H.; Nishioka, Y.; Tani, K.; Itai, A.; Sone, S. Antiallergic and anti-inflammatory effects of a novel IκB kinase beta inhibitor, IMD-0354, in a mouse model of allergic inflammation. Int. Arch. Allergy Immunol. 2009, 148, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, H.; Azuma, M.; Muto, S.; Nishioka, Y.; Honjo, A.; Tezuka, T.; Uehara, H.; Izumi, K.; Itai, A.; Sone, S. IκB kinase beta inhibitor IMD-0354 suppresses airway remodelling in a Dermatophagoides pteronyssinus-sensitized mouse model of chronic asthma. Clin. Exp. Allergy 2011, 41, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Sommers, C.D.; Thompson, J.M.; Guzova, J.A.; Bonar, S.L.; Rader, R.K.; Mathialagan, S.; Venkatraman, N.; Holway, V.W.; Kahn, L.E.; Hu, G.; et al. Novel tight-binding inhibitory factor-κB kinase (IKK-2) inhibitors demonstrate target-specific anti-inflammatory activities in cellular assays and following oral and local delivery in an in vivo model of airway inflammation. J. Pharmacol. Exp. Ther. 2009, 330, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Wada, H.; Rossios, C.; Takagi, D.; Charron, C.; Barnes, P.J.; Ito, K. A novel macrolide/fluoroketolide, solithromycin (CEM-101), reverses corticosteroid insensitivity via phosphoinositide 3-kinase pathway inhibition. Br. J. Pharmacol. 2013, 169, 1024–1034. [Google Scholar] [CrossRef] [PubMed]
- To, Y.; Ito, K.; Kizawa, Y.; Failla, M.; Ito, M.; Kusama, T.; Elliott, W.M.; Hogg, J.C.; Adcock, I.M.; Barnes, P.J. Targeting phosphoinositide-3-kinase-delta with theophylline reverses corticosteroid insensitivity in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 182, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Mercado, N.; To, Y.; Ito, K.; Barnes, P.J. Nortriptyline reverses corticosteroid insensitivity by inhibition of phosphoinositide-3-kinase-delta. J. Pharmacol. Exp. Ther. 2011, 337, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Rossios, C.; To, Y.; Osoata, G.; Ito, M.; Barnes, P.J.; Ito, K. Corticosteroid insensitivity is reversed by formoterol via phosphoinositide-3-kinase inhibition. Br. J. Pharmacol. 2012, 167, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Wada, H.; Rossios, C.; Takagi, D.; Higaki, M.; Mikura, S.; Goto, H.; Barnes, P.J.; Ito, K. A novel macrolide solithromycin exerts superior anti-inflammatory effect via NF-κB inhibition. J. Pharmacol. Exp. Ther. 2013, 345, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhong, X.; He, Z.; Wen, M.; Li, J.; Peng, X.; Liu, G.; Deng, J.; Zhang, J.; Bai, J. Effect of erythromycin on cigarette-induced histone deacetylase protein expression and nuclear factor-κB activity in human macrophages in vitro. Int. Immunopharmacol. 2012, 12, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Park, S.J.; Kim, S.R.; Min, K.H.; Jin, S.M.; Puri, K.D.; Lee, Y.C. Phosphoinositide 3-kinase-delta inhibitor reduces vascular permeability in a murine model of asthma. J. Allergy Clin. Immunol. 2006, 118, 403–409. [Google Scholar] [CrossRef] [PubMed]
- De Souza Alves, C.C.; Collison, A.; Hatchwell, L.; Plank, M.; Morten, M.; Foster, P.S.; Johnston, S.L.; da Costa, C.F.; de Almeida, M.V.; Couto Teixeira, H.; et al. Inhibiting AKT phosphorylation employing non-cytotoxic anthraquinones ameliorates TH2 mediated allergic airways disease and rhinovirus exacerbation. PLoS ONE 2013, 8, e79565. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Jin, G.Y.; Li, L.C.; Yan, G.H. Inhibition of protein kinase C delta attenuates allergic airway inflammation through suppression of PI3K/Akt/mTOR/HIF-1 alpha/VEGF pathway. PLoS ONE 2013, 8, e81773. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Lee, K.S.; Park, S.J.; Min, K.H.; Choe, Y.H.; Moon, H.; Yoo, W.H.; Chae, H.J.; Han, M.K.; Lee, Y.C. Involvement of sirtuin 1 in airway inflammation and hyperresponsiveness of allergic airway disease. J. Allergy Clin. Immunol. 2010, 125, 449.e14–460.e14. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhou, H.; Wang, J.; Zhang, B.; Liu, F.; Huang, J.; Li, J.; Lin, J.; Bai, J.; Liu, R. Therapeutic effects of resveratrol in a mouse model of HDM-induced allergic asthma. Int. Immunopharmacol. 2015, 25, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, T.; Hayashi, R.; Suzuki, K.; Imanishi, S.; Kambara, K.; Okazawa, S.; Inomata, M.; Yamada, T.; Yamazaki, Y.; Koshimizu, Y.; et al. Sirtuin 1 activator SRT1720 suppresses inflammation in an ovalbumin-induced mouse model of asthma. Respirology 2013, 18, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Royce, S.G.; Dang, W.; Yuan, G.; Tran, J.; el Osta, A.; Karagiannis, T.C.; Tang, M.L. Resveratrol has protective effects against airway remodeling and airway hyperreactivity in a murine model of allergic airways disease. Pathobiol. Aging Age Relat. Dis. 2011. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Kim, S.; Kwon, O.K.; Oh, S.R.; Lee, H.K.; Ahn, K. Anti-inflammatory and anti-asthmatic effects of resveratrol, a polyphenolic stilbene, in a mouse model of allergic asthma. Int. Immunopharmacol. 2009, 9, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Chung, S.; Hwang, J.W.; Rajendrasozhan, S.; Sundar, I.K.; Dean, D.A.; McBurney, M.W.; Guarente, L.; Gu, W.; Ronty, M.; et al. SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. J. Clin. Invest. 2012, 122, 2032–2045. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Sundar, I.K.; Ahmad, T.; Lerner, C.; Gerloff, J.; Friedman, A.E.; Phipps, R.P.; Sime, P.J.; McBurney, M.W.; Guarente, L.; et al. SIRT1 protects against cigarette smoke-induced lung oxidative stress via a FOXO3-dependent mechanism. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L816–L828. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schuliga, M. NF-kappaB Signaling in Chronic Inflammatory Airway Disease. Biomolecules 2015, 5, 1266-1283. https://doi.org/10.3390/biom5031266
Schuliga M. NF-kappaB Signaling in Chronic Inflammatory Airway Disease. Biomolecules. 2015; 5(3):1266-1283. https://doi.org/10.3390/biom5031266
Chicago/Turabian StyleSchuliga, Michael. 2015. "NF-kappaB Signaling in Chronic Inflammatory Airway Disease" Biomolecules 5, no. 3: 1266-1283. https://doi.org/10.3390/biom5031266
APA StyleSchuliga, M. (2015). NF-kappaB Signaling in Chronic Inflammatory Airway Disease. Biomolecules, 5(3), 1266-1283. https://doi.org/10.3390/biom5031266