
Extracellular Adenosine Generation in the Regulation of Pro-Inflammatory Responses and Pathogen Colonization

{kind=link}
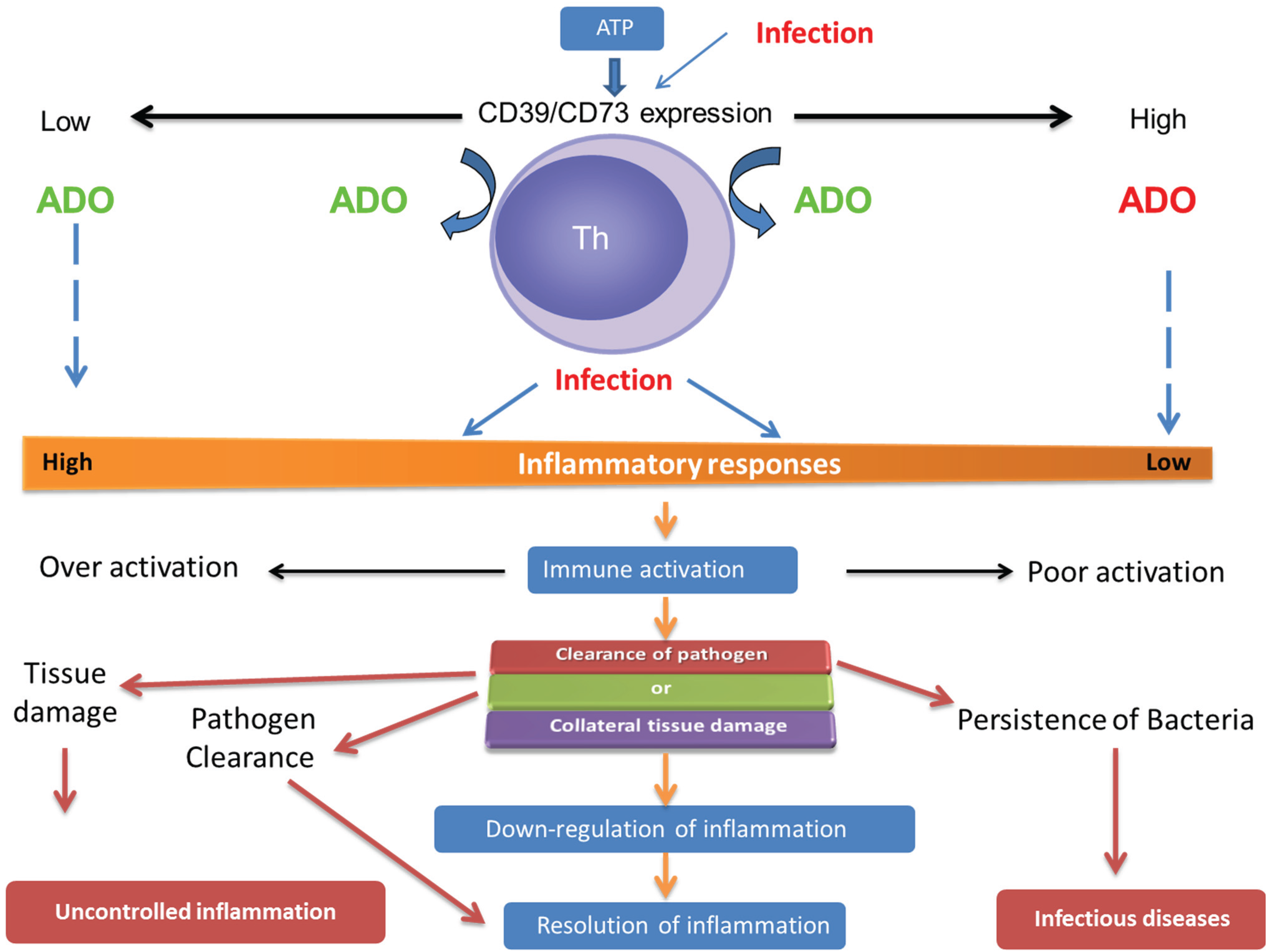
Abstract
:1. Introduction
2. Adenosine Generated by CD39/CD73 Expression and Its Mechanism of Action
3. Adenosine Generated by CD39/CD73 Expression and Impairment of Immunity to Infections
3.1. CD73-Regulated Immune Response during Helicobacter-Induced Gastritis and Persistent Infection
3.2. CD73-Regulated Immune Response during Salmonellosis
3.3. CD39/CD73 Expression in the Control of Other Infections
4. Adenosine Generated by CD39/CD73 Expression in the Innate Host Response to Infection
4.1. Role of Adenosine in Macrophage and Dendritic Cell (DC) Function
4.2. Role of Adenosine in Neutrophil Function
4.3. Role of Adenosine in Natural Killer (NK) Cell Function
4.4. Role of Adenosine in the Inflammasome
5. Conclusions
Acknowledgments
Author Contributions
Conflict of Interest
References
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Hasko, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Crimeen-Irwin, B.; Scalzo, K.; Gloster, S.; Mottram, P.L.; Plebanski, M. Failure of immune homeostasis—The consequences of under and over reactivity. Curr. Drug. Targets Immune Endocr. Metabol. Disord. 2005, 5, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Regateiro, F.S.; Cobbold, S.P.; Waldmann, H. CD73 and adenosine generation in the creation of regulatory microenvironments. Clin. Exp. Immunol. 2013, 171, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Fernandez, P.; Wilder, T.; Yee, H.; Chiriboga, L.; Chan, E.S.; Cronstein, B.N. Ecto-5'-nucleotidase (CD73)-mediated extracellular adenosine production plays a critical role in hepatic fibrosis. FASEB J. 2008, 22, 2263–2272. [Google Scholar] [CrossRef] [PubMed]
- Sitkovsky, M.V.; Lukashev, D.; Apasov, S.; Kojima, H.; Koshiba, M.; Caldwell, C.; Ohta, A.; Thiel, M. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu. Rev. Immunol. 2004, 22, 657–682. [Google Scholar] [CrossRef] [PubMed]
- Hasko, G.; Linden, J.; Cronstein, B.; Pacher, P. Adenosine receptors: Therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug. Discov. 2008, 7, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Sakowicz-Burkiewicz, M.; Pawelczyk, T. Recent advances in understanding the relationship between adenosine metabolism and the function of T and B lymphocytes in diabetes. J. Physiol. Pharmacol. 2011, 62, 505–512. [Google Scholar] [PubMed]
- Hasko, G.; Cronstein, B.N. Adenosine: An endogenous regulator of innate immunity. Trends Immunol. 2004, 25, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Okusa, M.D.; Linden, J.; Huang, L.; Rieger, J.M.; Macdonald, T.L.; Huynh, L.P. A2A adenosine receptor-mediated inhibition of renal injury and neutrophil adhesion. Am. J. Physiol. Renal Physiol. 2000, 279, F809–F818. [Google Scholar] [PubMed]
- Hasko, G.; Nemeth, Z.H.; Vizi, E.S.; Salzman, A.L.; Szabo, C. An agonist of adenosine A3 receptors decreases interleukin-12 and interferon-gamma production and prevents lethality in endotoxemic mice. Eur. J. Pharmacol. 1998, 358, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Koziak, K.; Sevigny, J.; Robson, S.C.; Siegel, J.B.; Kaczmarek, E. Analysis of CD39/ATP diphosphohydrolase (ATPDase) expression in endothelial cells, platelets and leukocytes. Thromb. Haemost. 1999, 82, 1538–1544. [Google Scholar] [PubMed]
- Dwyer, K.M.; Deaglio, S.; Gao, W.; Friedman, D.; Strom, T.B.; Robson, S.C. CD39 and control of cellular immune responses. Purinergic Signal. 2007, 3, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Pulte, E.D.; Broekman, M.J.; Olson, K.E.; Drosopoulos, J.H.; Kizer, J.R.; Islam, N.; Marcus, A.J. CD39/NTPDase-1 activity and expression in normal leukocytes. Thromb. Res. 2007, 121, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Beldi, G.; Banz, Y.; Kroemer, A.; Sun, X.; Wu, Y.; Graubardt, N.; Rellstab, A.; Nowak, M.; Enjyoji, K.; Li, X.; Junger, W.G.; et al. Deletion of CD39 on natural killer cells attenuates hepatic ischemia/reperfusion injury in mice. Hepatology 2010, 51, 1702–1711. [Google Scholar] [CrossRef] [PubMed]
- Beldi, G.; Wu, Y.; Banz, Y.; Nowak, M.; Miller, L.; Enjyoji, K.; Haschemi, A.; Yegutkin, G.G.; Candinas, D.; Exley, M.; et al. Natural killer T cell dysfunction in CD39-null mice protects against concanavalin A-induced hepatitis. Hepatology 2008, 48, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Robson, S.C. Ectonucleotidases as regulators of purinergic signaling in thrombosis, inflammation, and immunity. Adv. Pharmacol. 2011, 61, 301–332. [Google Scholar] [PubMed]
- Eltzschig, H.K.; Kohler, D.; Eckle, T.; Kong, T.; Robson, S.C.; Colgan, S.P. Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood 2009, 113, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Chalmin, F.; Mignot, G.; Bruchard, M.; Chevriaux, A.; Vegran, F.; Hichami, A.; Ladoire, S.; Derangère, V.; Vincent, J.; Masson, D.; et al. Stat3 and Gfi-1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity 2012, 36, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Thomson, L.F.; Ruedi, J.M.; Glass, A.; Moldenhauer, G.; Moller, P.; Low, M.G.; Klemens, M.R.; Massaia, M.; Lucas, A.H. Production and characterization of monoclonal antibodies to the glycosyl phosphatidylinositol-anchored lymphocyte differentiation antigen ecto-5'-nucleotidase (CD73). Tissue Antigens 1990, 35, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.F.; Eltzschig, H.K.; Ibla, J.C.; van de Wiele, C.J.; Resta, R.; Morote-Garcia, J.C.; Colgan, S.P. Crucial role for ecto-5'-nucleotidase (CD73) in vascular leakage during hypoxia. J. Exp. Med. 2004, 200, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Resta, R.; Yamashita, Y.; Thompson, L.F. Ecto-enzyme and signaling functions of lymphocyte CD73. Immunol. Rev. 1998, 161, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Ernst, P.B.; Garrison, J.C.; Thompson, L.F. Much Ado about adenosine: Adenosine synthesis and function in regulatory T cell biology. J. Immunol. 2010, 185, 1993–1998. [Google Scholar] [CrossRef] [PubMed]
- Kobie, J.J.; Shah, P.R.; Yang, L.; Rebhahn, J.A.; Fowell, D.J.; Mosmann, T.R. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5'-adenosine monophosphate to adenosine. J. Immunol. 2006, 177, 6780–6786. [Google Scholar] [CrossRef] [PubMed]
- Synnestvedt, K.; Furuta, G.T.; Comerford, K.M.; Louis, N.; Karhausen, J.; Eltzschig, H.K.; Hansen, K.R.; Thompson, L.F.; Colgan, S.P. Ecto-5'-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J. Clin. Invest. 2002, 110, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Ibla, J.C.; Furuta, G.T.; Leonard, M.O.; Jacobson, K.A.; Enjyoji, K.; Robson, S.C.; Colgan, S.P. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: Role of ectonucleotidases and adenosine A2B receptors. J. Exp. Med. 2003, 198, 783–796. [Google Scholar] [CrossRef] [PubMed]
- Beavis, P.A.; Stagg, J.; Darcy, P.K.; Smyth, M.J. CD73: A potent suppressor of antitumor immune responses. Trends Immunol. 2012, 33, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Regateiro, F.S.; Howie, D.; Nolan, K.F.; Agorogiannis, E.I.; Greaves, D.R.; Cobbold, S.P.; Waldmann, H. Generation of anti-inflammatory adenosine by leukocytes is regulated by TGF-beta. Eur. J. Immunol. 2011, 41, 2955–2965. [Google Scholar] [CrossRef] [PubMed]
- Niemela, J.; Henttinen, T.; Yegutkin, G.G.; Airas, L.; Kujari, A.M.; Rajala, P.; Jalkanen, S. IFN-alpha induced adenosine production on the endothelium: A mechanism mediated by CD73 (ecto-5'-nucleotidase) up-regulation. J. Immunol. 2004, 172, 1646–1653. [Google Scholar] [CrossRef] [PubMed]
- Fausther, M.; Sheung, N.; Saiman, Y.; Bansal, M.B.; Dranoff, J.A. Activated hepatic stellate cells upregulate transcription of ecto-5'-nucleotidase/CD73 via specific SP1 and SMAD promoter elements. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G904–G914. [Google Scholar] [CrossRef] [PubMed]
- Estrela, A.B.; Abraham, W.R. Adenosine in the inflamed gut: A Janus faced compound. Curr. Med. Chem. 2011, 18, 2791–2815. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.S.; Kurtz, C.C.; Wilson, J.M.; Burnette, B.R.; Wiznerowicz, E.B.; Ross, W.G.; Rieger, J.M.; Figler, R.A.; Linden, J.; Crowe, S.E.; et al. A2A adenosine receptor (AR) activation inhibits pro-inflammatory cytokine production by human CD4+ helper T cells and regulates Helicobacter-induced gastritis and bacterial persistence. Mucosal Immunol. 2009, 2, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Francois, V.; Shehade, H.; Acolty, V.; Preyat, N.; Delree, P.; Moser, M.; Oldenhove, G. Intestinal immunopathology is associated with decreased CD73-generated adenosine during lethal infection. Mucosal Immunol. 2014. [Google Scholar] [CrossRef]
- Alam, M.S.; Kuo, J.L.; Ernst, P.B.; Derr-Castillo, V.; Pereira, M.; Gaines, D.; Costales, M.; Bigley, E.; Williams, K. Ecto-5'-nucleotidase (CD73) regulates host inflammatory responses and exacerbates murine salmonellosis. Sci. Rep. 2014. [Google Scholar] [CrossRef]
- Linden, J. Molecular approach to adenosine receptors: Receptor-mediated mechanisms of tissue protection. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Grenz, A.; Zhang, H.; Eckle, T.; Mittelbronn, M.; Wehrmann, M.; Kohle, C.; Kloor, D.; Thompson, L.F.; Osswald, H.; Eltzschig, H.K. Protective role of ecto-5'-nucleotidase (CD73) in renal ischemia. J. Am. Soc. Nephrol. 2007, 18, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Hasko, G.; Pacher, P. Regulation of macrophage function by adenosine. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Mahamed, D.A.; Mills, J.H.; Egan, C.E.; Denkers, E.Y.; Bynoe, M.S. CD73-generated adenosine facilitates Toxoplasma gondii differentiation to long-lived tissue cysts in the central nervous system. Proc. Natl. Acad. Sci. USA 2012, 109, 16312–16317. [Google Scholar] [CrossRef] [PubMed]
- Thammavongsa, V.; Kern, J.W.; Missiakas, D.M.; Schneewind, O. Staphylococcus aureus synthesizes adenosine to escape host immune responses. J. Exp. Med. 2009, 206, 2417–2427. [Google Scholar] [CrossRef] [PubMed]
- Firon, A.; Dinis, M.; Raynal, B.; Poyart, C.; Trieu-Cuot, P.; Kaminski, P.A. Extracellular nucleotide catabolism by the Group B Streptococcus ectonucleotidase NudP increases bacterial survival in blood. J. Biol. Chem. 2014, 289, 5479–5489. [Google Scholar] [CrossRef] [PubMed]
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nature 2014, 509, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Sansom, F.M.; Newton, H.J.; Crikis, S.; Cianciotto, N.P.; Cowan, P.J.; dʼApice, A.J.; Hartland, E.L. A bacterial ecto-triphosphate diphosphohydrolase similar to human CD39 is essential for intracellular multiplication of Legionella pneumophila. Cell Microbiol. 2007, 9, 1922–1935. [Google Scholar] [CrossRef] [PubMed]
- Riedmaier, P.; Sansom, F.M.; Sofian, T.; Beddoe, T.; Schuelein, R.; Newton, H.J.; Hartland, E.L. Multiple ecto-nucleoside triphosphate diphosphohydrolases facilitate intracellular replication of Legionella pneumophila. Biochem. J. 2014, 462, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Hasko, G.; Csoka, B.; Koscso, B.; Chandra, R.; Pacher, P.; Thompson, L.F.; Deitch, E.A.; Spolarics, Z.; Virág, L.; Gergely, P.; et al. Ecto-5'-nucleotidase (CD73) decreases mortality and organ injury in sepsis. J. Immunol. 2011, 187, 4256–4267. [Google Scholar] [CrossRef] [PubMed]
- Theatre, E.; Frederix, K.; Guilmain, W.; Delierneux, C.; Lecut, C.; Bettendorff, L.; Bours, V.; Oury, C. Overexpression of CD39 in mouse airways promotes bacteria-induced inflammation. J. Immunol. 2012, 189, 1966–1974. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.J.; Kunzli, B.M.; Rahim, Y.I.; Sevigny, J.; Berberat, P.O.; Enjyoji, K.; Csizmadia, E.; Friess, H.; Robson, S.C. From the cover: CD39 deletion exacerbates experimental murine colitis and human polymorphisms increase susceptibility to inflammatory bowel disease. Proc. Natl. Acad. Sci. USA 2009, 106, 16788–16793. [Google Scholar] [CrossRef] [PubMed]
- Bynoe, M.S.; Waickman, A.T.; Mahamed, D.A.; Mueller, C.; Mills, J.H.; Czopik, A. CD73 is critical for the resolution of murine colonic inflammation. J. Biomed. Biotechnol. 2012. [Google Scholar] [CrossRef]
- Wang, L.; Fan, J.; Chen, S.; Zhang, Y.; Curiel, T.J.; Zhang, B. Graft-versus-host disease is enhanced by selective CD73 blockade in mice. PLOS ONE 2013, 8, e58397. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Fox, J.G.; Otto, G.; Murphy, J. A small animal model of human Helicobacter pylori active chronic gastritis. Gastroenterology 1990, 99, 1315–1323. [Google Scholar] [PubMed]
- D’Elios, M.M.; Manghetti, M.; de Carli, M.; Costa, F.; Baldari, C.T.; Burroni, D.; Telford, J.L.; Romagnani, S.; del Prete, G. T helper 1 effector cells specific for Helicobacter pylori in the gastric antrum of patients with peptic ulcer disease. J. Immunol. 1997, 158, 962–967. [Google Scholar] [PubMed]
- Bamford, K.B.; Fan, X.; Crowe, S.E.; Leary, J.F.; Gourley, W.K.; Luthra, G.K.; Brooks, E.G.; Graham, D.Y.; Reyes, V.E.; Ernst, P.B. Lymphocytes in the human gastric mucosa during Helicobacter pylori have a T helper cell 1 phenotype. Gastroenterology 1998, 114, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Karttunen, R.; Karttunen, T.; Ekre, H.P.; MacDonald, T.T. Interferon gamma and interleukin 4 secreting cells in the gastric antrum in Helicobacter pylori positive and negative gastritis. Gut 1995, 36, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Goll, R.; Gruber, F.; Olsen, T.; Cui, G.; Raschpichler, G.; Buset, M.; Asfeldt, A.M.; Husebekk, A.; Florholmen, J. Helicobacter pylori stimulates a mixed adaptive immune response with a strong T-regulatory component in human gastric mucosa. Helicobacter 2007, 12, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Enarsson, K.; Lundgren, A.; Kindlund, B.; Hermansson, M.; Roncador, G.; Banham, A.H.; Lundin, B.S.; Quiding-Järbrink, M. Function and recruitment of mucosal regulatory T cells in human chronic Helicobacter pylori infection and gastric adenocarcinoma. Clin. Immunol. 2006, 121, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Stromberg, E.; Edebo, A.; Lundin, B.S.; Bergin, P.; Brisslert, M.; Svennerholm, A.M.; Lindholm, C. Down-regulation of epithelial IL-8 responses in Helicobacter pylori-infected duodenal ulcer patients depends on host factors, rather than bacterial factors. Clin. Exp. Immunol. 2005, 140, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Lundgren, A.; Stromberg, E.; Sjoling, A.; Lindholm, C.; Enarsson, K.; Edebo, A.; Johnsson, E.; Suri-Payer, E.; Larsson, P.; Rudin, A.; et al. Mucosal FOXP3-expressing CD4+ CD25high regulatory T cells in Helicobacter pylori-infected patients. Infect. Immun. 2005, 73, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Rad, R.; Brenner, L.; Bauer, S.; Schwendy, S.; Layland, L.; da Costa, C.P.; Reindl, W.; Dossumbekova, A.; Friedrich, M.; Saur, D.; et al. CD25+/Foxp3+ T cells regulate gastric inflammation and Helicobacter pylori colonization in vivo. Gastroenterology 2006, 131, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.S.; Kurtz, C.C.; Rowlett, R.M.; Reuter, B.K.; Wiznerowicz, E.; Das, S.; Linden, J.; Crowe, S.E.; Ernst, P.B. CD73 is expressed by human regulatory T helper cells and suppresses proinflammatory cytokine production and Helicobacter felis-induced gastritis in mice. J. Infect. Dis. 2009, 199, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Lund, B.M.; OʼBrien, S.J. The occurrence and prevention of foodborne disease in vulnerable people. Foodborne Pathog. Dis. 2011, 8, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Hess, J.; Ladel, C.; Miko, D.; Kaufmann, S.H. Salmonella typhimurium aroA- infection in gene-targeted immunodeficient mice: Major role of CD4+ TCR-alpha beta cells and IFN-gamma in bacterial clearance independent of intracellular location. J. Immunol. 1996, 156, 3321–3326. [Google Scholar] [PubMed]
- Ravindran, R.; Foley, J.; Stoklasek, T.; Glimcher, L.H.; McSorley, S.J. Expression of T-bet by CD4 T cells is essential for resistance to Salmonella infection. J. Immunol. 2005, 175, 4603–4610. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, A.; Nanton, M.; Griffin, A.; McSorley, S.J. Culling of activated CD4 T cells during typhoid is driven by Salmonella virulence genes. J. Immunol. 2009, 182, 7838–7845. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.S.; Akaike, T.; Okamoto, S.; Kubota, T.; Yoshitake, J.; Sawa, T.; Miyamoto, Y.; Tamura, F.; Maeda, H. Role of nitric oxide in host defense in murine salmonellosis as a function of its antibacterial and antiapoptotic activities. Infect. Immun. 2002, 70, 3130–3142. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.S.; Zaki, M.H.; Yoshitake, J.; Akuta, T.; Ezaki, T.; Akaike, T. Involvement of Salmonella enterica serovar Typhi RpoS in resistance to NO-mediated host defense against serovar Typhi infection. Microb. Pathog. 2006, 40, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.S.; Zaki, M.H.; Sawa, T.; Islam, S.; Ahmed, K.A.; Fujii, S.; Okamoto, T.; Akaike, T. Nitric oxide produced in Peyerʼs patches exhibits antiapoptotic activity contributing to an antimicrobial effect in murine salmonellosis. Microbiol. Immunol. 2008, 52, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, M.U.; MacMicking, J.D.; Nicholson, S.; Brause, J.E.; Potter, S.; Marino, M.; Fang, F.; Dinauer, M.; Nathan, C. Phenotype of mice and macrophages deficient in both phagocyte oxidase and inducible nitric oxide synthase. Immunity 1999, 10, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Torres, A.; Jones-Carson, J.; Mastroeni, P.; Ischiropoulos, H.; Fang, F.C. Antimicrobial actions of the NADPH phagocyte oxidase and inducible nitric oxide synthase in experimental salmonellosis. I. Effects on microbial killing by activated peritoneal macrophages in vitro. J. Exp. Med. 2000, 192, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Geddes, K.; Rubino, S.J.; Magalhaes, J.G.; Streutker, C.; Le, B.L.; Cho, J.H.; Robertson, S.J.; Kim, C.J.; Kaul, R.; Philpott, D.J.; et al. Identification of an innate T helper type 17 response to intestinal bacterial pathogens. Nat. Med. 2011, 17, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Ramarathinam, L.; Niesel, D.W.; Klimpel, G.R. Ity influences the production of IFN-gamma by murine splenocytes stimulated in vitro with Salmonella typhimurium. J. Immunol. 1993, 150, 3965–3972. [Google Scholar] [PubMed]
- Alam, M.; Costales, M.; Williams, K. wn-regulation of CD73 expression favors host protection during Intracellular foodborne bacterial infections (IRC8P. 496). In proceedings of AAI Annual Meeting, Pittsburgh, PA, USA, 2–6 May, 2014.
- Mendez, S.; Reckling, S.K.; Piccirillo, C.A.; Sacks, D.; Belkaid, Y. Role for CD4+CD25+ regulatory T cells in reactivation of persistent leishmaniasis and control of concomitant immunity. J. Exp. Med. 2004, 200, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Walther, M.; Tongren, J.E.; Andrews, L.; Korbel, D.; King, E.; Fletcher, H.; Andersen, R.F.; Bejon, P.; Thompson, F.; Dunachie, S.J.; et al. Upregulation of TGF-beta, FOXP3, and CD4+CD25+ regulatory T cells correlates with more rapid parasite growth in human malaria infection. Immunity 2005, 23, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.F.; Takedachi, M.; Ebisuno, Y.; Tanaka, T.; Miyasaka, M.; Mills, J.H.; Bynoe, M.S. Regulation of leukocyte migration across endothelial barriers by ecto-5'-nucleotidase-generated adenosine. Nucleosides Nucleotides Nucleic Acids 2008, 27, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Takedachi, M.; Qu, D.; Ebisuno, Y.; Oohara, H.; Joachims, M.L.; McGee, S.T.; Maeda, E.; McEver, R.P.; Tanaka, T.; Miyasaka, M.; et al. CD73-generated adenosine restricts lymphocyte migration into draining lymph nodes. J. Immunol. 2008, 180, 6288–6296. [Google Scholar] [CrossRef] [PubMed]
- Yegutkin, G.G.; Hytonen, J.; Samburski, S.S.; Yrjanainen, H.; Jalkanen, S.; Viljanen, M.K. Disordered lymphoid purine metabolism contributes to the pathogenesis of persistent Borrelia garinii infection in mice. J. Immunol. 2010, 184, 5112–5120. [Google Scholar] [CrossRef] [PubMed]
- Belikoff, B.G.; Hatfield, S.; Georgiev, P.; Ohta, A.; Lukashev, D.; Buras, J.A.; Remick, D.G.; Sitkovsky, M. A2B adenosine receptor blockade enhances macrophage-mediated bacterial phagocytosis and improves polymicrobial sepsis survival in mice. J. Immunol. 2011, 186, 2444–2453. [Google Scholar] [CrossRef] [PubMed]
- Barletta, K.E.; Cagnina, R.E.; Burdick, M.D.; Linden, J.; Mehrad, B. Adenosine A2B receptor deficiency promotes host defenses against gram-negative bacterial pneumonia. Am. J. Respir. Crit. Care. Med. 2012, 186, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Kolachala, V.L.; Vijay-Kumar, M.; Dalmasso, G.; Yang, D.; Linden, J.; Wang, L.; Gewirtz, A.; Ravid, K.; Merlin, D.; Sitaraman, S.V. A2B adenosine receptor gene deletion attenuates murine colitis. Gastroenterology 2008, 135, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Boer, M.C.; van Meijgaarden, K.E.; Bastid, J.; Ottenhoff, T.H.; Joosten, S.A. CD39 is involved in mediating suppression by Mycobacterium bovis BCG-activated human CD8+CD39+ regulatory T cells. Eur. J. Immunol. 2013, 43, 1925–1932. [Google Scholar] [CrossRef] [PubMed]
- Toth, I.; Le, A.Q.; Hartjen, P.; Thomssen, A.; Matzat, V.; Lehmann, C.; Scheurich, C.; Beisel, C.; Busch, P.; Degen, O.; et al. Decreased frequency of CD73+CD8+ T cells of HIV-infected patients correlates with immune activation and T cell exhaustion. J. Leukoc. Biol. 2013, 94, 551–561. [Google Scholar] [CrossRef] [PubMed]
- MacMicking, J.; Xie, Q.W.; Nathan, C. Nitric oxide and macrophage function. Annu. Rev. Immunol. 1997, 15, 323–350. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Hyman, M.C.; Baek, A.E.; Fukase, K.; Pinsky, D.J. cAMP/CREB-mediated transcriptional regulation of ectonucleoside triphosphate diphosphohydrolase 1 (CD39) expression. J. Biol. Chem. 2010, 285, 14791–14805. [Google Scholar] [CrossRef] [PubMed]
- Levesque, S.A.; Kukulski, F.; Enjyoji, K.; Robson, S.C.; Sevigny, J. NTPDase1 governs P2X7-dependent functions in murine macrophages. Eur. J. Immunol. 2010, 40, 1473–1485. [Google Scholar] [CrossRef] [PubMed]
- Cohen, H.B.; Briggs, K.T.; Marino, J.P.; Ravid, K.; Robson, S.C.; Mosser, D.M. TLR stimulation initiates a CD39-based autoregulatory mechanism that limits macrophage inflammatory responses. Blood 2013, 122, 1935–1945. [Google Scholar] [CrossRef] [PubMed]
- Costales, M.; Alam, M.S.; Williams, K. CD73 regulates nitric oxide production and inflammatory responses in Salmonella infected RAW 264.7 macrophages (P4223). In proceedings of AAI Annual Meeting, Honolulu, HI, USA, 3–7 May, 2013.
- Hasko, G.; Szabo, C.; Nemeth, Z.H.; Kvetan, V.; Pastores, S.M.; Vizi, E.S. Adenosine receptor agonists differentially regulate IL-10, TNF-alpha, and nitric oxide production in RAW 264.7 macrophages and in endotoxemic mice. J. Immunol. 1996, 157, 4634–4640. [Google Scholar] [PubMed]
- Berchtold, S.; Ogilvie, A.L.; Bogdan, C.; Muhl-Zurbes, P.; Ogilvie, A.; Schuler, G.; Steinkasserer, A. Human monocyte derived dendritic cells express functional P2X and P2Y receptors as well as ecto-nucleotidases. FEBS Lett. 1999, 458, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, N.; Kumamoto, T.; Robson, S.C.; Sevigny, J.; Matsue, H.; Enjyoji, K.; Takashima, A. CD39 is the dominant Langerhans cell-associated ecto-NTPDase: Modulatory roles in inflammation and immune responsiveness. Nat. Med. 2002, 8, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Cobbold, S.P.; Adams, E.; Nolan, K.F.; Regateiro, F.S.; Waldmann, H. Connecting the mechanisms of T-cell regulation: Dendritic cells as the missing link. Immunol. Rev. 2010, 236, 203–218. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Colucci, R.; La, M.C.; Tuccori, M.; Awwad, O.; Da, S.F.; Blandizzi, C.; Fornai, M. Adenosine deaminase in the modulation of immune system and its potential as a novel target for treatment of inflammatory disorders. Curr. Drug Targets 2012, 13, 842–862. [Google Scholar] [CrossRef] [PubMed]
- Barletta, K.E.; Ley, K.; Mehrad, B. Regulation of neutrophil function by adenosine. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Flogel, U.; Burghoff, S.; van Lent, P.L.; Temme, S.; Galbarz, L.; Ding, Z.; El-Tayeb, A.; Huels, S.; Bönner, F.; Borg, N.; et al. Selective activation of adenosine A2A receptors on immune cells by a CD73-dependent prodrug suppresses joint inflammation in experimental rheumatoid arthritis. Sci. Transl. Med. 2012. [Google Scholar] [CrossRef]
- Linden, J. Cell biology. Purinergic chemotaxis. Science 2006, 314, 1689–1690. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Thompson, L.F.; Karhausen, J.; Cotta, R.J.; Ibla, J.C.; Robson, S.C.; Colgan, S.P. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: Coordination by extracellular nucleotide metabolism. Blood 2004, 104, 3986–3992. [Google Scholar] [CrossRef] [PubMed]
- Kukulski, F.; Bahrami, F.; Ben, Y.F.; Lecka, J.; Martin-Satue, M.; Levesque, S.A.; Sévigny, J. NTPDase1 controls IL-8 production by human neutrophils. J. Immunol. 2011, 187, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Corriden, R.; Chen, Y.; Inoue, Y.; Beldi, G.; Robson, S.C.; Insel, P.A.; Junger, W.G. Ecto-nucleoside triphosphate diphosphohydrolase 1 (E-NTPDase1/CD39) regulates neutrophil chemotaxis by hydrolyzing released ATP to adenosine. J. Biol. Chem. 2008, 283, 28480–28486. [Google Scholar] [CrossRef] [PubMed]
- Reutershan, J.; Vollmer, I.; Stark, S.; Wagner, R.; Ngamsri, K.C.; Eltzschig, H.K. Adenosine and inflammation: CD39 and CD73 are critical mediators in LPS-induced PMN trafficking into the lungs. FASEB J. 2009, 23, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Weissmuller, T.; Mager, A.; Eckle, T. Nucleotide metabolism and cell-cell interactions. Methods Mol. Biol. 2006, 341, 73–87. [Google Scholar] [PubMed]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Sharma, A. Adenosine: An endogenous modulator of innate immune system with therapeutic potential. Eur. J. Pharmacol. 2009, 616, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Stutz, A.; Golenbock, D.T.; Latz, E. Inflammasomes: Too big to miss. J. Clin. Invest. 2009, 119, 3502–3511. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Munoz-Planillo, R.; Reimer, T.; Eigenbrod, T.; Nunez, G. Inflammasomes as microbial sensors. Eur. J. Immunol. 2010, 40, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Zhou, R.; Tschopp, J. The NLRP3 inflammasome: A sensor for metabolic danger? Science 2010, 327, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Sater, A.A.; Said-Sadier, N.; Ojcius, D.M.; Yilmaz, O.; Kelly, K.A. Inflammasomes bridge signaling between pathogen identification and the immune response. Drugs Today 2009, 45, 105–112. [Google Scholar] [PubMed]
- Yilmaz, O.; Sater, A.A.; Yao, L.; Koutouzis, T.; Pettengill, M.; Ojcius, D.M. ATP-dependent activation of an inflammasome in primary gingival epithelial cells infected by Porphyromonas gingivalis. Cell Microbiol. 2010, 12, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Brosnan, C.F. Regulation of immune response by P2X7 receptor. Crit. Rev. Immunol. 2006, 26, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Kahlenberg, J.M.; Lundberg, K.C.; Kertesy, S.B.; Qu, Y.; Dubyak, G.R. Potentiation of caspase-1 activation by the P2X7 receptor is dependent on TLR signals and requires NF-κB-driven protein synthesis. J. Immunol. 2005, 175, 7611–7622. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, D.; Pizzirani, C.; Adinolfi, E.; Lemoli, R.M.; Curti, A.; Idzko, M.; Panther, E.; di Virgilio, F. The P2X7 receptor: A key player in IL-1 processing and release. J. Immunol. 2006, 176, 3877–3883. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Wang, X.; Yan, C.; Gao, Q.; Li, S.A.; Liu, J.; Zhou, K.; Guo, X.; Lee, W.; Zhang, Y. Adenosine-5'-triphosphate (ATP) protects mice against bacterial infection by activation of the NLRP3 inflammasome. PLOS ONE 2013, 8, e63759. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Ghani, A.; Malik, A.; Wilder, T.; Colegio, O.R.; Flavell, R.A.; Cronstein, B.N.; Mehal, W.Z. Adenosine is required for sustained inflammasome activation via the A2A receptor and the HIF-1alpha pathway. Nat. Commun. 2013, 4, 2909. [Google Scholar] [PubMed]
- Martinon, F. Signaling by ROS drives inflammasome activation. Eur. J. Immunol. 2010, 40, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Coutinho-Silva, R.; Correa, G.; Sater, A.A.; Ojcius, D.M. The P2X7 receptor and intracellular pathogens: A continuing struggle. Purinergic Signal. 2009, 5, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Spooner, R.; Yilmaz, O. The role of reactive-oxygen-species in microbial persistence and inflammation. Int. J. Mol. Sci. 2011, 12, 334–352. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, A. Extracellular ATP in the immune system: More than just a “danger signal”. Sci. Signal. 2009. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alam, M.S.; Costales, M.G.; Cavanaugh, C.; Williams, K. Extracellular Adenosine Generation in the Regulation of Pro-Inflammatory Responses and Pathogen Colonization. Biomolecules 2015, 5, 775-792. https://doi.org/10.3390/biom5020775
Alam MS, Costales MG, Cavanaugh C, Williams K. Extracellular Adenosine Generation in the Regulation of Pro-Inflammatory Responses and Pathogen Colonization. Biomolecules. 2015; 5(2):775-792. https://doi.org/10.3390/biom5020775
Chicago/Turabian StyleAlam, M. Samiul, Matthew G. Costales, Christopher Cavanaugh, and Kristina Williams. 2015. "Extracellular Adenosine Generation in the Regulation of Pro-Inflammatory Responses and Pathogen Colonization" Biomolecules 5, no. 2: 775-792. https://doi.org/10.3390/biom5020775
APA StyleAlam, M. S., Costales, M. G., Cavanaugh, C., & Williams, K. (2015). Extracellular Adenosine Generation in the Regulation of Pro-Inflammatory Responses and Pathogen Colonization. Biomolecules, 5(2), 775-792. https://doi.org/10.3390/biom5020775