
Sirtuins and Proteolytic Systems: Implications for Pathogenesis of Synucleinopathies

{kind=link}
Abstract
:1. Introduction
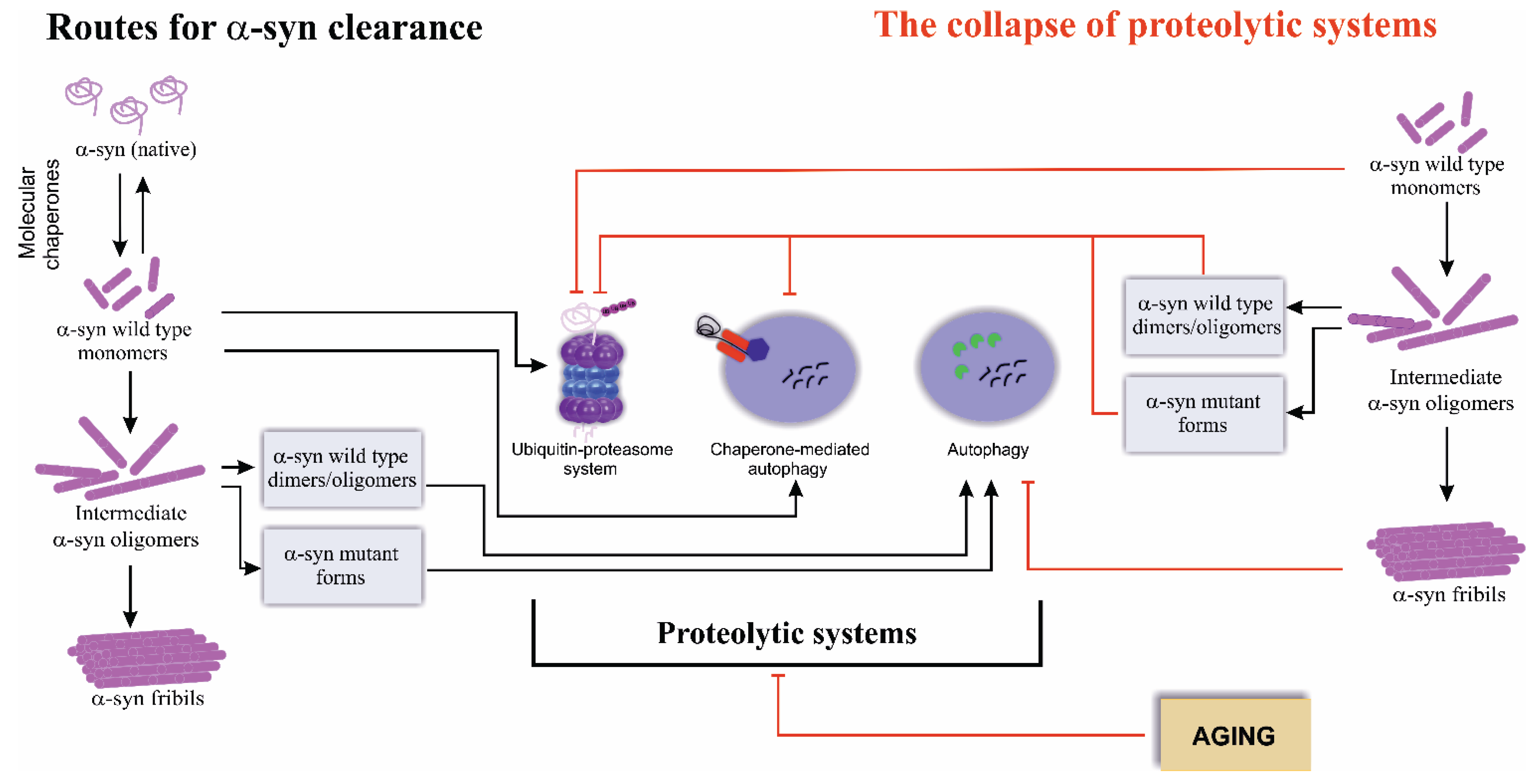
2. Routes for Alpha-Synuclein Clearance
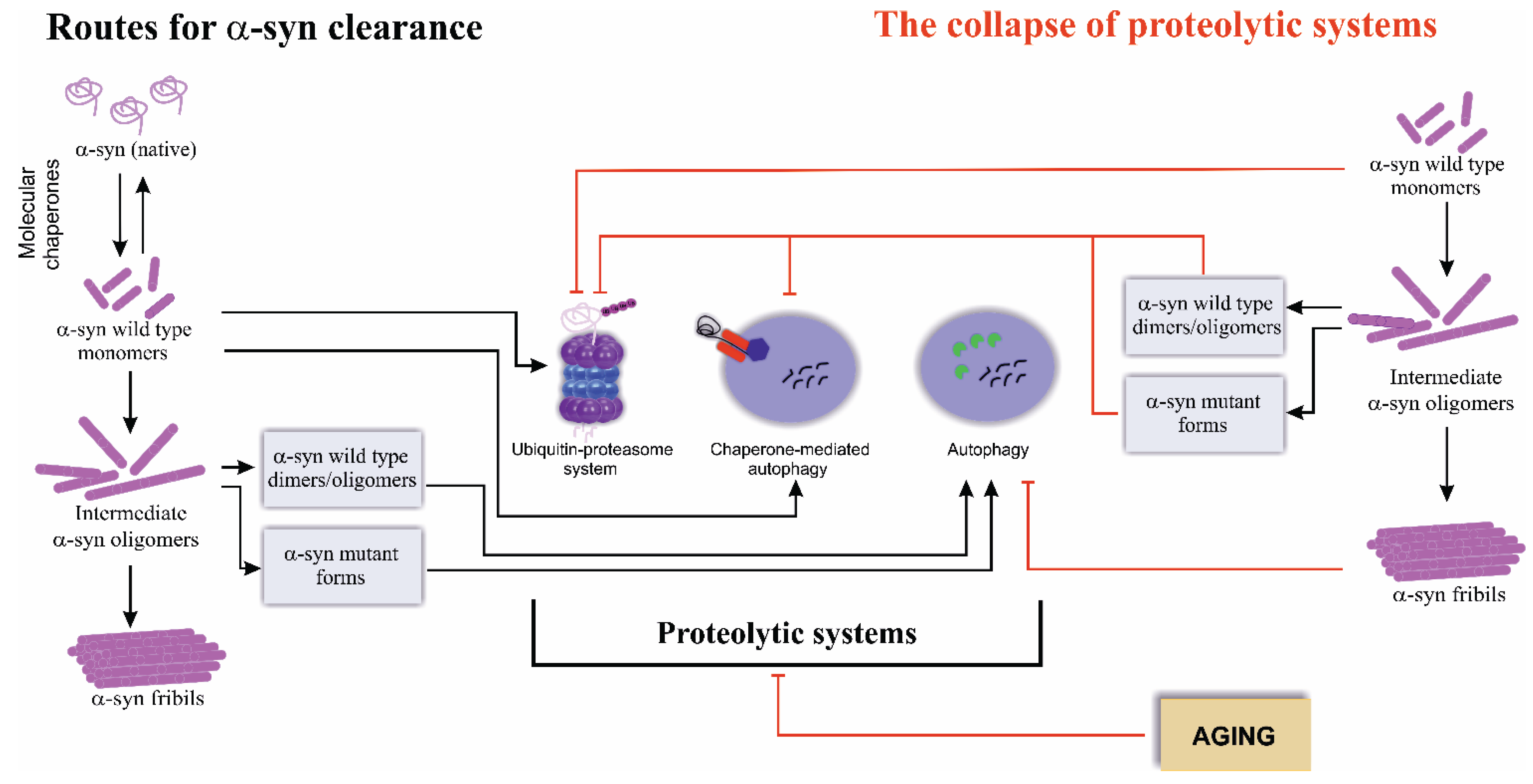
3. The Collapse of Proteolytic Systems
4. The Role of Sirtuins on the Proteostasis Control
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [PubMed]
- Marques, O.; Outeiro, T.F. Alpha-synuclein: From secretion to dysfunction and death. Cell Death Dis. 2012. [Google Scholar] [CrossRef]
- Bendor, J.T.; Logan, T.P.; Edwards, R.H. The function of alpha-synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. Alpha-synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [PubMed]
- Kruger, R.; Kuhn, W.; Muller, T.; Woitalla, D.; Graeber, M.; Kosel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atares, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honore, A.; Rozas, N.; Pieri, L.; Madiona, K.; Durr, A.; Melki, R.; et al. G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Proukakis, C.; Dudzik, C.G.; Brier, T.; MacKay, D.S.; Cooper, J.M.; Millhauser, G.L.; Houlden, H.; Schapira, A.H. A novel alpha-synuclein missense mutation in Parkinson disease. Neurology 2013, 80, 1062–1064. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Lewis, P.; Revesz, T.; Lees, A.; Paisan-Ruiz, C. The genetics of Parkinson’s syndromes: A critical review. Curr. Opin. Genet. Dev. 2009, 19, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. Alpha-synuclein locus triplication causes Parkinson’s disease. Science 2003. [Google Scholar] [CrossRef]
- Karpinar, D.P.; Balija, M.B.; Kugler, S.; Opazo, F.; Rezaei-Ghaleh, N.; Wender, N.; Kim, H.Y.; Taschenberger, G.; Falkenburger, B.H.; Heise, H.; et al. Pre-fibrillar alpha-synuclein variants with impaired beta-structure increase neurotoxicity in Parkinson’s disease models. EMBO J. 2009, 28, 3256–3268. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.; Angot, E.; Bergstrom, A.L.; Steiner, J.A.; Pieri, L.; Paul, G.; Outeiro, T.F.; Melki, R.; Kallunki, P.; Fog, K.; et al. Alpha-synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Investig. 2011, 121, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Bjorklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Patel, S.; Lee, S.J. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 2005, 25, 6016–6024. [Google Scholar] [CrossRef] [PubMed]
- Amm, I.; Sommer, T.; Wolf, D.H. Protein quality control and elimination of protein waste: The role of the ubiquitin-proteasome system. Biochim. Biophys. Acta 2014, 1843, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Nijholt, D.A.; de Graaf, T.R.; van Haastert, E.S.; Oliveira, A.O.; Berkers, C.R.; Zwart, R.; Ovaa, H.; Baas, F.; Hoozemans, J.J.; Scheper, W. Endoplasmic reticulum stress activates autophagy but not the proteasome in neuronal cells: Implications for alzheimer’s disease. Cell Death Differ. 2011, 18, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Akerfelt, M.; Morimoto, R.I.; Sistonen, L. Heat shock factors: Integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 2010, 11, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Fredrickson, E.K.; Gardner, R.G. Selective destruction of abnormal proteins by ubiquitin-mediated protein quality control degradation. Semin. Cell Dev. Biol. 2012, 23, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Ann. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A role for ubiquitin in selective autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Stolz, A.; Wolf, D.H. Endoplasmic reticulum associated protein degradation: A chaperone assisted journey to hell. Biochim. Biophys. Acta 2010, 1803, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Tyedmers, J.; Mogk, A.; Bukau, B. Cellular strategies for controlling protein aggregation. Nat. Rev. Mol. Cell Biol. 2010, 11, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Voisine, C.; Pedersen, J.S.; Morimoto, R.I. Chaperone networks: Tipping the balance in protein folding diseases. Neurobiol. Dis. 2010, 40, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Anelli, T.; Sitia, R. Protein quality control in the early secretory pathway. EMBO J. 2008, 27, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. Alpha-synuclein and protein degradation systems: A reciprocal relationship. Mol. Neurobiol. 2013, 47, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Sampaio-Marques, B.; Burhans, W.C.; Ludovico, P. Longevity pathways and maintenance of the proteome: The role of autophagy and mitophagy during yeast ageing. Microb. Cell 2014, 1, 118–127. [Google Scholar] [CrossRef]
- Gallastegui, N.; Groll, M. The 26S proteasome: Assembly and function of a destructive machine. Trends Biochem. Sci. 2010, 35, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Cuervo, A.M. Integration of clearance mechanisms: The proteasome and autophagy. Cold Spring Harb. Perspect. Biol. 2010. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy. Methods Mol. Biol. 2008, 445, 227–244. [Google Scholar] [PubMed]
- Liu, C.W.; Corboy, M.J.; DeMartino, G.N.; Thomas, P.J. Endoproteolytic activity of the proteasome. Science 2003, 299, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G. Lewy body diseases and multiple system atrophy as α-synucleinopathies. Mol. Psychiatry 1998, 3, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Tortosa, E.; Newell, K.; Irizarry, M.C.; Sanders, J.L.; Hyman, B.T. Alpha-synuclein immunoreactivity in dementia with Lewy bodies: Morphological staging and comparison with ubiquitin immunostaining. Acta Neuropathol. 2000, 99, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Lowe, J.; Blanchard, A.; Morrell, K.; Lennox, G.; Reynolds, L.; Billett, M.; Landon, M.; Mayer, R.J. Ubiquitin is a common factor in intermediate filament inclusion bodies of diverse type in man, including those of Parkinson’s disease, pick’s disease, and alzheimer’s disease, as well as rosenthal fibres in cerebellar astrocytomas, cytoplasmic bodies in muscle, and mallory bodies in alcoholic liver disease. J. Pathol. 1988, 155, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Tofaris, G.K.; Razzaq, A.; Ghetti, B.; Lilley, K.S.; Spillantini, M.G. Ubiquitination of alpha-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J. Biol. Chem. 2003, 278, 44405–44411. [Google Scholar] [CrossRef] [PubMed]
- Kuusisto, E.; Parkkinen, L.; Alafuzoff, I. Morphogenesis of Lewy bodies: Dissimilar incorporation of alpha-synuclein, ubiquitin, and p62. J. Neuropathol. Exp. Neurol. 2003, 62, 1241–1253. [Google Scholar] [PubMed]
- Kuusisto, E.; Salminen, A.; Alafuzoff, I. Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport 2001, 12, 2085–2090. [Google Scholar] [CrossRef] [PubMed]
- Zatloukal, K.; Stumptner, C.; Fuchsbichler, A.; Heid, H.; Schnoelzer, M.; Kenner, L.; Kleinert, R.; Prinz, M.; Aguzzi, A.; Denk, H. P62 is a common component of cytoplasmic inclusions in protein aggregation diseases. Am. J. Pathol. 2002, 160, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Bove, J.; Rodriguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Dodiya, H.; Aebischer, P.; Olanow, C.W.; Kordower, J.H. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: Relationship to alpha-synuclein inclusions. Neurobiol. Dis. 2009, 35, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Fergusson, J.; Landon, M.; Lowe, J.; Dawson, S.P.; Layfield, R.; Hanger, D.P.; Mayer, R.J. Pathological lesions of alzheimer’s disease and dementia with Lewy bodies brains exhibit immunoreactivity to an atpase that is a regulatory subunit of the 26S proteasome. Neurosci. Lett. 1996, 219, 167–170. [Google Scholar] [CrossRef] [PubMed]
- McNaught, K.S.; Bjorklund, L.M.; Belizaire, R.; Isacson, O.; Jenner, P.; Olanow, C.W. Proteasome inhibition causes nigral degeneration with inclusion bodies in rats. Neuroreport 2002, 13, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- McNaught, K.S.; Mytilineou, C.; Jnobaptiste, R.; Yabut, J.; Shashidharan, P.; Jennert, P.; Olanow, C.W. Impairment of the ubiquitin-proteasome system causes dopaminergic cell death and inclusion body formation in ventral mesencephalic cultures. J. Neurochem. 2002, 81, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Nuytemans, K.; Theuns, J.; Cruts, M.; van Broeckhoven, C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: A mutation update. Hum. Mutat. 2010, 31, 763–780. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.F.; Tang, B.S.; Zhang, Y.H.; Liu, H.J.; Yan, X.X.; Chen, T.; Shen, L.; Jiang, H.; Xia, K.; Cai, F.; et al. Genotype and phenotype analyses of three families with autosomal recessive juvenile parkinsonism. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2006, 23, 70–73. [Google Scholar] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Shimura, H.; Hattori, N.; Kubo, S.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nature Genet. 2000, 25, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Cantuti-Castelvetri, I.; Fan, Z.; Rockenstein, E.; Masliah, E.; Hyman, B.T.; McLean, P.J.; Unni, V.K. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of alpha-synuclein. J. Neurosci. 2011, 31, 14508–14520. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Wahlster, L.; McLean, P.J. Protein degradation pathways in Parkinson’s disease: Curse or blessing. Acta Neuropathol. 2012, 124, 153–172. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Wheeler, T.C.; Li, L.; Chin, L.S. Ubiquitination of alpha-synuclein by SIAH-1 promotes alpha-synuclein aggregation and apoptotic cell death. Human Mol. Genet. 2008, 17, 906–917. [Google Scholar] [CrossRef]
- Liani, E.; Eyal, A.; Avraham, E.; Shemer, R.; Szargel, R.; Berg, D.; Bornemann, A.; Riess, O.; Ross, C.A.; Rott, R.; et al. Ubiquitylation of synphilin-1 and alpha-synuclein by SIAH and its presence in cellular inclusions and Lewy bodies imply a role in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 5500–5505. [Google Scholar] [CrossRef] [PubMed]
- Rott, R.; Szargel, R.; Haskin, J.; Shani, V.; Shainskaya, A.; Manov, I.; Liani, E.; Avraham, E.; Engelender, S. Monoubiquitylation of alpha-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J. Biol. Chem. 2008, 283, 3316–3328. [Google Scholar] [CrossRef] [PubMed]
- Rott, R.; Szargel, R.; Haskin, J.; Bandopadhyay, R.; Lees, A.J.; Shani, V.; Engelender, S. α-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc. Natl. Acad. Sci. USA 2011, 108, 18666–18671. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Klucken, J.; Patterson, C.; Hyman, B.T.; McLean, P.J. The co-chaperone carboxyl terminus of hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J. Biol. Chem. 2005, 280, 23727–23734. [Google Scholar] [CrossRef] [PubMed]
- Tetzlaff, J.E.; Putcha, P.; Outeiro, T.F.; Ivanov, A.; Berezovska, O.; Hyman, B.T.; McLean, P.J. CHIP targets toxic alpha-synuclein oligomers for degradation. J. Biol. Chem. 2008, 283, 17962–17968. [Google Scholar] [CrossRef] [PubMed]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef] [PubMed]
- Mak, S.K.; McCormack, A.L.; Manning-Bog, A.B.; Cuervo, A.M.; di Monte, D.A. Lysosomal degradation of alpha-synuclein in vivo. J. Biol. Chem. 2010, 285, 13621–13629. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Rodriguez-Oroz, M.C.; Obeso, J.A.; Cooper, J.M. Influence of microrna deregulation on chaperone-mediated autophagy and alpha-synuclein pathology in Parkinson’s disease. Cell Death Dis. 2013. [Google Scholar] [CrossRef]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [PubMed]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar] [PubMed]
- Lan, D.; Wang, W.; Zhuang, J.; Zhao, Z. Proteasome inhibitor-induced autophagy in PC12 cells overexpressing A53T mutant alpha-synuclein. Mol. Med. Rep. 2015, 11, 1655–1660. [Google Scholar] [PubMed]
- Xilouri, M.; Vogiatzi, T.; Vekrellis, K.; Park, D.; Stefanis, L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLOS ONE 2009, 4, e5515. [Google Scholar] [CrossRef] [PubMed]
- Hoozemans, J.J.; van Haastert, E.S.; Eikelenboom, P.; de Vos, R.A.; Rozemuller, J.M.; Scheper, W. Activation of the unfolded protein response in Parkinson’s disease. Biochem. Biophys. Res. Commun. 2007, 354, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Makioka, K.; Yamazaki, T.; Fujita, Y.; Takatama, M.; Nakazato, Y.; Okamoto, K. Involvement of endoplasmic reticulum stress defined by activated unfolded protein response in multiple system atrophy. J. Neurol. Sci. 2010, 297, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Sampaio-Marques, B.; Felgueiras, C.; Silva, A.; Rodrigues, M.; Tenreiro, S.; Franssens, V.; Reichert, A.S.; Outeiro, T.F.; Winderickx, J.; Ludovico, P. SNCA (α-synuclein)-induced toxicity in yeast cells is dependent on sirtuin 2 (SIR2)-mediated mitophagy. Autophagy 2012, 8, 1494–1509. [Google Scholar] [CrossRef] [PubMed]
- Bertoncini, C.W.; Fernandez, C.O.; Griesinger, C.; Jovin, T.M.; Zweckstetter, M. Familial mutants of alpha-synuclein with increased neurotoxicity have a destabilized conformation. J. Biol. Chem. 2005, 280, 30649–30652. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T., Jr. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, E.A.; Graves, C.L.; Mishizen-Eberz, A.J.; Lupoli, M.A.; Lynch, D.R.; Englander, S.W.; Axelsen, P.H.; Giasson, B.I. The E46K mutation in alpha-synuclein increases amyloid fibril formation. J. Biol. Chem. 2005, 280, 7800–7807. [Google Scholar] [CrossRef] [PubMed]
- Schildknecht, S.; Gerding, H.R.; Karreman, C.; Drescher, M.; Lashuel, H.A.; Outeiro, T.F.; di Monte, D.A.; Leist, M. Oxidative and nitrative alpha-synuclein modifications and proteostatic stress: Implications for disease mechanisms and interventions in synucleinopathies. J. Neurochem. 2013, 125, 491–511. [Google Scholar] [CrossRef] [PubMed]
- Hodara, R.; Norris, E.H.; Giasson, B.I.; Mishizen-Eberz, A.J.; Lynch, D.R.; Lee, V.M.; Ischiropoulos, H. Functional consequences of alpha-synuclein tyrosine nitration: Diminished binding to lipid vesicles and increased fibril formation. J. Biol. Chem. 2004, 279, 47746–47753. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Talloczy, Z.; Kaushik, S.; Massey, A.C.; Mazzulli, J.; Mosharov, E.V.; Hodara, R.; Fredenburg, R.; Wu, D.C.; Follenzi, A.; et al. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J. Clin. Invest. 2008, 118, 777–788. [Google Scholar] [PubMed]
- Tenreiro, S.; Reimao-Pinto, M.M.; Antas, P.; Rino, J.; Wawrzycka, D.; Macedo, D.; Rosado-Ramos, R.; Amen, T.; Waiss, M.; Magalhaes, F.; et al. Phosphorylation modulates clearance of α-synuclein inclusions in a yeast model of Parkinson’s disease. PLOS Genet. 2014, 10, e1004302. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, D.; Dybdal, N.; Donaldson, M.T.; Miller, L.; Cribb, A.E. Nitration and increased alpha-synuclein expression associated with dopaminergic neurodegeneration in equine pituitary pars intermedia dysfunction. J. Neuroendocrinol. 2005, 17, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Paxinou, E.; Chen, Q.; Weisse, M.; Giasson, B.I.; Norris, E.H.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M.; Ischiropoulos, H. Induction of α-synuclein aggregation by intracellular nitrative insult. J. Neurosci. 2001, 21, 8053–8061. [Google Scholar] [PubMed]
- Smith, W.W.; Margolis, R.L.; Li, X.; Troncoso, J.C.; Lee, M.K.; Dawson, V.L.; Dawson, T.M.; Iwatsubo, T.; Ross, C.A. Alpha-synuclein phosphorylation enhances eosinophilic cytoplasmic inclusion formation in SH-SY5Y cells. J. Neurosci. 2005, 25, 5544–5552. [Google Scholar] [CrossRef] [PubMed]
- Volles, M.J.; Lansbury, P.T., Jr. Zeroing in on the pathogenic form of alpha-synuclein and its mechanism of neurotoxicity in Parkinson’s disease. Biochemistry 2003, 42, 7871–7878. [Google Scholar] [CrossRef] [PubMed]
- Chau, K.Y.; Ching, H.L.; Schapira, A.H.; Cooper, J.M. Relationship between α synuclein phosphorylation, proteasomal inhibition and cell death: Relevance to Parkinson’s disease pathogenesis. J. Neurochem. 2009, 110, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Khoshaghideh, F.; Patel, S.; Lee, S.J. Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J. Neurosci. 2004, 24, 1888–1896. [Google Scholar] [CrossRef] [PubMed]
- Malkus, K.A.; Tsika, E.; Ischiropoulos, H. Oxidative modifications, mitochondrial dysfunction, and impaired protein degradation in Parkinson’s disease: How neurons are lost in the bermuda triangle. Mol. Neurodegener. 2009. [Google Scholar] [CrossRef]
- Ebrahimi-Fakhari, D.; McLean, P.J.; Unni, V.K. α-Synuclein’s degradation in vivo: Opening a new (cranial) window on the roles of degradation pathways in Parkinson disease. Autophagy 2012, 8, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Dantuma, N.P.; Bott, L.C. The ubiquitin-proteasome system in neurodegenerative diseases: Precipitating factor, yet part of the solution. Front. Mol. Neurosci. 2014. [Google Scholar] [CrossRef]
- Stefanis, L.; Larsen, K.E.; Rideout, H.J.; Sulzer, D.; Greene, L.A. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J. Neurosci. 2001, 21, 9549–9560. [Google Scholar] [PubMed]
- Caneda-Ferron, B.; de Girolamo, L.A.; Costa, T.; Beck, K.E.; Layfield, R.; Billett, E.E. Assessment of the direct and indirect effects of MPP+ and dopamine on the human proteasome: Implications for Parkinson’s disease aetiology. J. Neurochem. 2008, 105, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Fornai, F.; Schluter, O.M.; Lenzi, P.; Gesi, M.; Ruffoli, R.; Ferrucci, M.; Lazzeri, G.; Busceti, C.L.; Pontarelli, F.; Battaglia, G.; et al. Parkinson-like syndrome induced by continuous MPTP infusion: Convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 3413–3418. [Google Scholar] [CrossRef] [PubMed]
- Snyder, H.; Mensah, K.; Theisler, C.; Lee, J.; Matouschek, A.; Wolozin, B. Aggregated and monomeric alpha-synuclein bind to the S6' proteasomal protein and inhibit proteasomal function. J. Biol. Chem. 2003, 278, 11753–11759. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Engelender, S.; Igarashi, S.; Rao, R.K.; Wanner, T.; Tanzi, R.E.; Sawa, A.; Valina, L.D.; Dawson, T.M.; Ross, C.A. Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum. Mol. Genet. 2001, 10, 919–926. [Google Scholar] [CrossRef] [PubMed]
- McNaught, K.S.; Belizaire, R.; Isacson, O.; Jenner, P.; Olanow, C.W. Altered proteasomal function in sporadic Parkinson’s disease. Exp. Neurol. 2003, 179, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Vigouroux, S.; Wong, H.; Guttman, M.; Rajput, A.H.; Ang, L.; Briand, M.; Kish, S.J.; Briand, Y. Brain proteasomal function in sporadic Parkinson’s disease and related disorders. Ann. Neurol. 2002, 51, 779–782. [Google Scholar] [CrossRef] [PubMed]
- McNaught, K.S.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Ghee, M.; Fournier, A.; Mallet, J. Rat alpha-synuclein interacts with tat binding protein 1, a component of the 26S proteasomal complex. J. Neurochem. 2000, 75, 2221–2224. [Google Scholar] [CrossRef] [PubMed]
- Lindersson, E.; Beedholm, R.; Hojrup, P.; Moos, T.; Gai, W.; Hendil, K.B.; Jensen, P.H. Proteasomal inhibition by alpha-synuclein filaments and oligomers. J. Biol. Chem. 2004, 279, 12924–12934. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Stefanis, L. Chaperone mediated autophagy to the rescue: A new-fangled target for the treatment of neurodegenerative diseases. Mol. Cell. Neurosci. 2015. [Google Scholar] [CrossRef]
- Sala, G.; Arosio, A.; Stefanoni, G.; Melchionda, L.; Riva, C.; Marinig, D.; Brighina, L.; Ferrarese, C. Rotenone upregulates α-synuclein and myocyte enhancer factor 2D independently from lysosomal degradation inhibition. BioMed Res. Int. 2013. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Landeck, N.; Pitychoutis, P.M.; Papasilekas, T.; Papadopoulou-Daifoti, Z.; Kirik, D.; Stefanis, L. Boosting chaperone-mediated autophagy in vivo mitigates α-synuclein-induced neurodegeneration. Brain 2013, 136, 2130–2146. [Google Scholar] [CrossRef] [PubMed]
- Decressac, M.; Bjorklund, A. TFEB: Pathogenic role and therapeutic target in Parkinson disease. Autophagy 2013, 9, 1244–1246. [Google Scholar] [CrossRef] [PubMed]
- Decressac, M.; Mattsson, B.; Weikop, P.; Lundblad, M.; Jakobsson, J.; Bjorklund, A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc. Natl. Acad. Sci. USA 2013, 110, E1817–E1826. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Stefanis, L. Autophagy in the central nervous system: Implications for neurodegenerative disorders. CNS neurol. Disord. Drug Targets 2010, 9, 701–719. [Google Scholar] [CrossRef] [PubMed]
- Massey, A.C.; Kaushik, S.; Sovak, G.; Kiffin, R.; Cuervo, A.M. Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2006, 103, 5805–5810. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Massey, A.C.; Mizushima, N.; Cuervo, A.M. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol. Biol. Cell 2008, 19, 2179–2192. [Google Scholar] [CrossRef] [PubMed]
- Castino, R.; Lazzeri, G.; Lenzi, P.; Bellio, N.; Follo, C.; Ferrucci, M.; Fornai, F.; Isidoro, C. Suppression of autophagy precipitates neuronal cell death following low doses of methamphetamine. J. Neurochem. 2008, 106, 1426–1439. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Santos, C.; Ferrer, I.; Santidrian, A.F.; Barrachina, M.; Gil, J.; Ambrosio, S. Dopamine induces autophagic cell death and alpha-synuclein increase in human neuroblastoma SH-SY5Y cells. J. Neurosci. Res. 2003, 73, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Sampaio-Marques, B.; Felgueiras, C.; Silva, A.; Rodrigues, F.; Ludovico, P. Yeast chronological lifespan and proteotoxic stress: Is autophagy good or bad? Biochem. Soc. Trans. 2011, 39, 1466–1470. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.H.; Dorado, B.; Figueroa, H.Y.; Wang, L.; Planel, E.; Cookson, M.R.; Clark, L.N.; Duff, K.E. Metabolic activity determines efficacy of macroautophagic clearance of pathological oligomeric alpha-synuclein. Am. J. Pathol. 2009, 175, 736–747. [Google Scholar] [CrossRef] [PubMed]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. Alpha-synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Bevis, B.J.; Shorter, J.; Strathearn, K.E.; Hamamichi, S.; Su, L.J.; Caldwell, K.A.; Caldwell, G.A.; Rochet, J.C.; McCaffery, J.M.; et al. The Parkinson’s disease protein α-synuclein disrupts cellular rab homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Cho, E.D.; Lee, K.W.; Kim, J.H.; Cho, S.G.; Lee, S.J. Autophagic failure promotes the exocytosis and intercellular transfer of alpha-synuclein. Exp. Mol. Med. 2013. [Google Scholar] [CrossRef]
- Neumann, J.; Bras, J.; Deas, E.; O’Sullivan, S.S.; Parkkinen, L.; Lachmann, R.H.; Li, A.; Holton, J.; Guerreiro, R.; Paudel, R.; et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 2009, 132, 1783–1794. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [PubMed]
- Kinghorn, K.J. Pathological looping in the synucleinopathies: Investigating the link between Parkinson’s disease and gaucher disease. Dis. Models Mech. 2011, 4, 713–715. [Google Scholar] [CrossRef]
- Osellame, L.D.; Duchen, M.R. Defective quality control mechanisms and accumulation of damaged mitochondria link gaucher and Parkinson diseases. Autophagy 2013, 9, 1633–1635. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson’s disease. Brain 2014, 137, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Spiro, A.S.; Furuta, A.; Cooper, A.; Garner, B.; Kabuta, T.; Halliday, G.M. Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson’s disease. Mov. Disord. 2015. [Google Scholar] [CrossRef]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008, 22, 1427–1438. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Klopp, R.G.; Weindruch, R.; Prolla, T.A. Gene expression profile of aging and its retardation by caloric restriction. Science 1999, 285, 1390–1393. [Google Scholar] [CrossRef] [PubMed]
- Ly, D.H.; Lockhart, D.J.; Lerner, R.A.; Schultz, P.G. Mitotic misregulation and human aging. Science 2000, 287, 2486–2492. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Dice, J.F. Age-related decline in chaperone-mediated autophagy. J. Biol. Chem. 2000, 275, 31505–31513. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lesuisse, C.; Xu, Y.; Troncoso, J.C.; Price, D.L.; Lee, M.K. Stabilization of alpha-synuclein protein with aging and familial Parkinson’s disease-linked A53T mutation. J. Neurosci. 2004, 24, 7400–7409. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Kordower, J.H. Age-associated increases of alpha-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: Is this the target for Parkinson’s disease? Neurobiol. Dis. 2007, 25, 134–149. [Google Scholar] [CrossRef] [PubMed]
- Xuan, Q.; Xu, S.L.; Lu, D.H.; Yu, S.; Zhou, M.; Ueda, K.; Cui, Y.Q.; Zhang, B.Y.; Chan, P. Increased expression of alpha-synuclein in aged human brain associated with neuromelanin accumulation. J. Neural Transm. 2011, 118, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- McCormack, A.L.; Mak, S.K.; di Monte, D.A. Increased alpha-synuclein phosphorylation and nitration in the aging primate substantia nigra. Cell Death Dis. 2012, 3, e315. [Google Scholar] [CrossRef] [PubMed]
- Choubey, V.; Safiulina, D.; Vaarmann, A.; Cagalinec, M.; Wareski, P.; Kuum, M.; Zharkovsky, A.; Kaasik, A. Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J. Biol. Chem. 2011, 286, 10814–10824. [Google Scholar] [CrossRef] [PubMed]
- Hollenbeck, P.J.; Saxton, W.M. The axonal transport of mitochondria. J. Cell Sci. 2005, 118, 5411–5419. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Chung, K.K. α-Synuclein impairs normal dynamics of mitochondria in cell and animal models of Parkinson’s disease. J. Neurochem. 2012, 122, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Youle, R.J. PINK1- and parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.K.; Cookson, M.R. Mitochondrial quality control and dynamics in Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 869–882. [Google Scholar] [CrossRef] [PubMed]
- Vives-Bauza, C.; Przedborski, S. Mitophagy: The latest problem for Parkinson’s disease. Trends Mol. Med. 2011, 17, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Ivatt, R.M.; Whitworth, A.J. The many faces of mitophagy. EMBO Rep. 2014, 15, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Mallajosyula, J.K.; Rane, A.; Andersen, J.K. Mitochondrial alpha-synuclein accumulation impairs complex i function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 2010, 486, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Fournier, M.; Vitte, J.; Garrigue, J.; Langui, D.; Dullin, J.P.; Saurini, F.; Hanoun, N.; Perez-Diaz, F.; Cornilleau, F.; Joubert, C.; et al. Parkin deficiency delays motor decline and disease manifestation in a mouse model of synucleinopathy. PLOS ONE 2009, 4, e6629. [Google Scholar] [CrossRef] [PubMed]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Dali-Youcef, N.; Lagouge, M.; Froelich, S.; Koehl, C.; Schoonjans, K.; Auwerx, J. Sirtuins: The ‘magnificent seven’, function, metabolism and longevity. Ann. Med. 2007, 39, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Ramadori, G.; Lee, C.E.; Bookout, A.L.; Lee, S.; Williams, K.W.; Anderson, J.; Elmquist, J.K.; Coppari, R. Brain SIRT1: Anatomical distribution and regulation by energy availability. J. Neurosci. 2008, 28, 9989–9996. [Google Scholar] [CrossRef] [PubMed]
- Donmez, G.; Arun, A.; Chung, C.Y.; McLean, P.J.; Lindquist, S.; Guarente, L. SIRT1 protects against α-synuclein aggregation by activating molecular chaperones. J. Neurosci. 2012, 32, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Wang, H.; Qin, X.; Pang, S.; Yan, B. Genetic analysis of SIRT1 gene promoter in sporadic Parkinson’s disease. Biochem. Biophys. Res. Commun. 2012, 422, 693–696. [Google Scholar] [CrossRef]
- Min, S.W.; Cho, S.H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Martinez-Vicente, M.; Kruger, U.; Kaushik, S.; Wong, E.; Mandelkow, E.M.; Cuervo, A.M.; Mandelkow, E. Tau fragmentation, aggregation and clearance: The dual role of lysosomal processing. Hum. Mol. Genet. 2009, 18, 4153–4170. [Google Scholar] [CrossRef] [PubMed]
- Westerheide, S.D.; Anckar, J.; Stevens, S.M., Jr.; Sistonen, L.; Morimoto, R.I. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 2009, 323, 1063–1066. [Google Scholar] [CrossRef] [PubMed]
- Van Ham, T.J.; Thijssen, K.L.; Breitling, R.; Hofstra, R.M.; Plasterk, R.H.; Nollen, E.A.C. Elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLOS Genet. 2008, 4, e1000027. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Maiuri, M.C.; Markaki, M.; Megalou, E.; Pasparaki, A.; Palikaras, K.; Criollo, A.; Galluzzi, L.; Malik, S.A.; Vitale, I.; et al. The life span-prolonging effect of sirtuin-1 is mediated by autophagy. Autophagy 2010, 6, 186–188. [Google Scholar] [CrossRef] [PubMed]
- Kakefuda, K.; Fujita, Y.; Oyagi, A.; Hyakkoku, K.; Kojima, T.; Umemura, K.; Tsuruma, K.; Shimazawa, M.; Ito, M.; Nozawa, Y.; et al. Sirtuin 1 overexpression mice show a reference memory deficit, but not neuroprotection. Biochem. Biophys. Res. Commun. 2009, 387, 784–788. [Google Scholar] [CrossRef] [PubMed]
- Buttner, S.; Delay, C.; Franssens, V.; Bammens, T.; Ruli, D.; Zaunschirm, S.; de Oliveira, R.M.; Outeiro, T.F.; Madeo, F.; Buee, L.; et al. Synphilin-1 enhances alpha-synuclein aggregation in yeast and contributes to cellular stress and cell death in a SIR2-dependent manner. PLOS ONE 2010, 5, e13700. [Google Scholar] [CrossRef] [PubMed]
- Albani, D.; Polito, L.; Batelli, S.; de Mauro, S.; Fracasso, C.; Martelli, G.; Colombo, L.; Manzoni, C.; Salmona, M.; Caccia, S.; et al. The SIRT1 activator resveratrol protects SK-N-BE cells from oxidative stress and against toxicity caused by alpha-synuclein or amyloid-beta (1–42) peptide. J. Neurochem. 2009, 110, 1445–1456. [Google Scholar] [CrossRef]
- Blanchet, J.; Longpre, F.; Bureau, G.; Morissette, M.; DiPaolo, T.; Bronchti, G.; Martinoli, M.G. Resveratrol, a red wine polyphenol, protects dopaminergic neurons in MPTP-treated mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, X.; Zhu, J.X.; Xie, W.; Le, W.; Fan, Z.; Jankovic, J.; Pan, T. Resveratrol-activated AMPK/SIRT1/autophagy in cellular models of Parkinson’s disease. Neurosignals 2011, 19, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Wareski, P.; Vaarmann, A.; Choubey, V.; Safiulina, D.; Liiv, J.; Kuum, M.; Kaasik, A. PGC-1α and PGC-1β regulate mitochondrial density in neurons. J. Biol. Chem. 2009, 284, 21379–21385. [Google Scholar] [CrossRef] [PubMed]
- Mudo, G.; Makela, J.; di Liberto, V.; Tselykh, T.V.; Olivieri, M.; Piepponen, P.; Eriksson, O.; Malkia, A.; Bonomo, A.; Kairisalo, M.; et al. Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinson’s disease. Cell. Mol. Life Sci. 2012, 69, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.C.; McLean, P.J.; et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 2007, 317, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Gal, J.; Bang, Y.; Choi, H.J. SIRT2 interferes with autophagy-mediated degradation of protein aggregates in neuronal cells under proteasome inhibition. Neurochem. Int. 2012, 61, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Calnan, D.R.; Brunet, A. The foxo code. Oncogene 2008, 27, 2276–2288. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Nguyen, M.; Qin, F.X.; Tong, Q. SIRT2 deacetylates Foxo3a in response to oxidative stress and caloric restriction. Aging Cell 2007, 6, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Arun, A.; Ellis, L.; Peritore, C.; Donmez, G. Sirtuin 2 (SIRT2) enhances 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced nigrostriatal damage via deacetylating forkhead box o3a (Foxo3a) and activating bim protein. J. Biol. Chem. 2012, 287, 32307–32311. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of foxo transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Auburger, G.; Gispert, S.; Jendrach, M. Mitochondrial acetylation and genetic models of Parkinson’s disease. Prog. Mol. Biol. Transl. Sci. 2014, 127, 155–182. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sampaio-Marques, B.; Ludovico, P. Sirtuins and Proteolytic Systems: Implications for Pathogenesis of Synucleinopathies. Biomolecules 2015, 5, 735-757. https://doi.org/10.3390/biom5020735
Sampaio-Marques B, Ludovico P. Sirtuins and Proteolytic Systems: Implications for Pathogenesis of Synucleinopathies. Biomolecules. 2015; 5(2):735-757. https://doi.org/10.3390/biom5020735
Chicago/Turabian StyleSampaio-Marques, Belém, and Paula Ludovico. 2015. "Sirtuins and Proteolytic Systems: Implications for Pathogenesis of Synucleinopathies" Biomolecules 5, no. 2: 735-757. https://doi.org/10.3390/biom5020735
APA StyleSampaio-Marques, B., & Ludovico, P. (2015). Sirtuins and Proteolytic Systems: Implications for Pathogenesis of Synucleinopathies. Biomolecules, 5(2), 735-757. https://doi.org/10.3390/biom5020735