
Role of α- and β-Synucleins in the Axonal Pathology of Parkinson’s Disease and Related Synucleinopathies

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
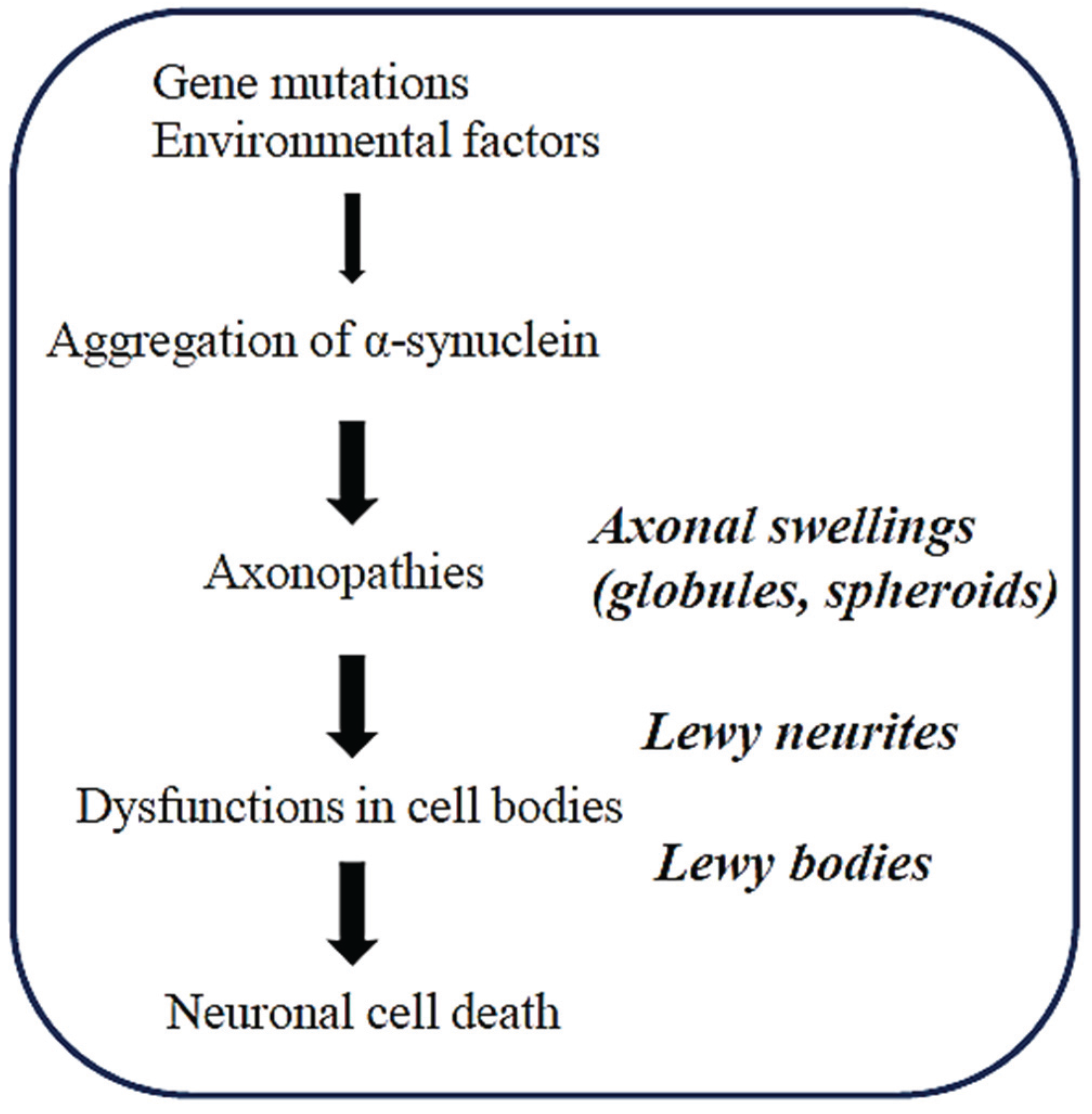
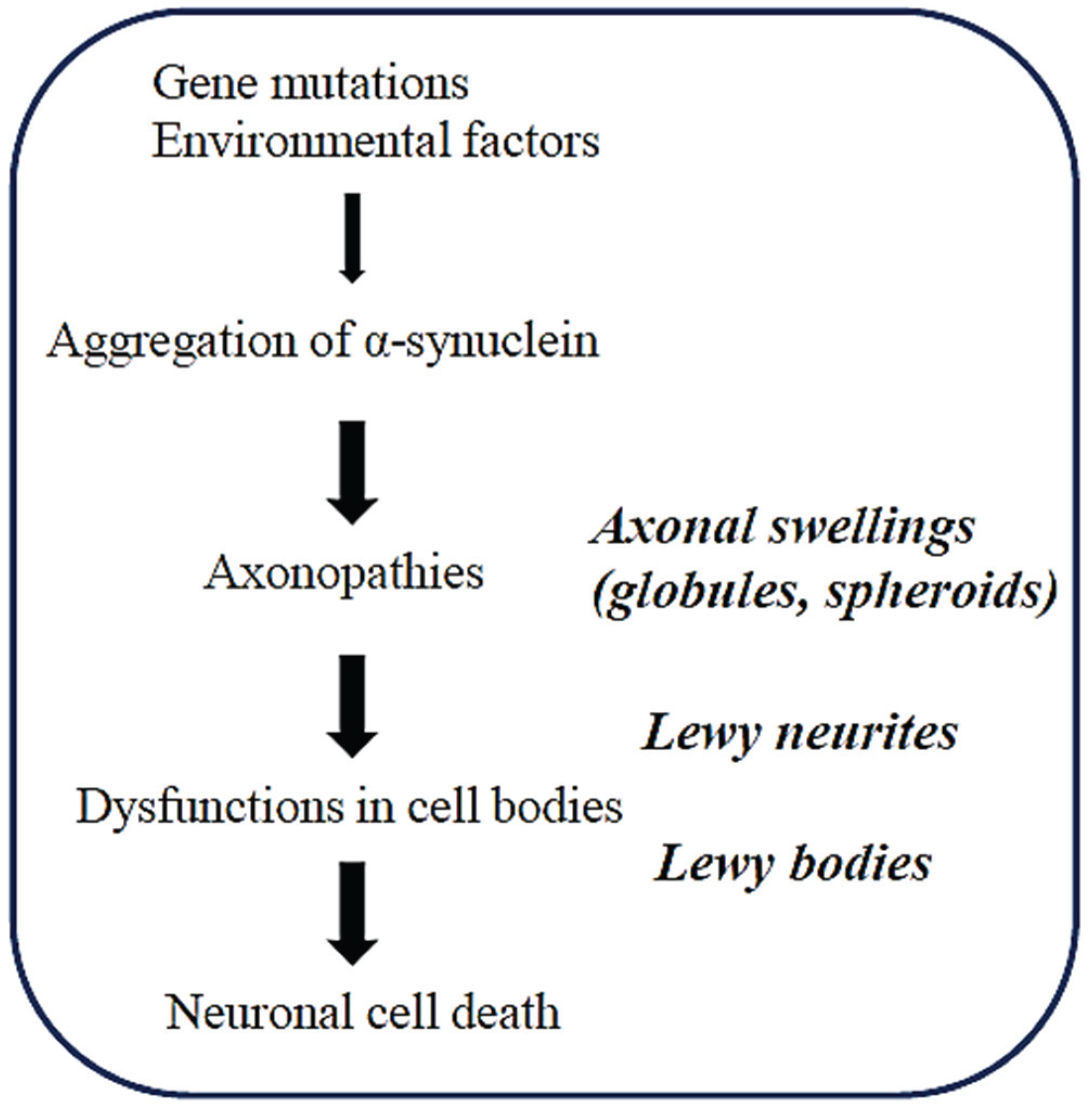
2. Axonopathy is an Early Event in the Pathogenesis of Synucleinopathies
3. Axonal Swellings in the Brains of a Tg Mouse Model with Synucleinopathies
3.1. Age-Dependent Formation of Globules in the Brains of a Tg Mouse Model of Synucleinopathies
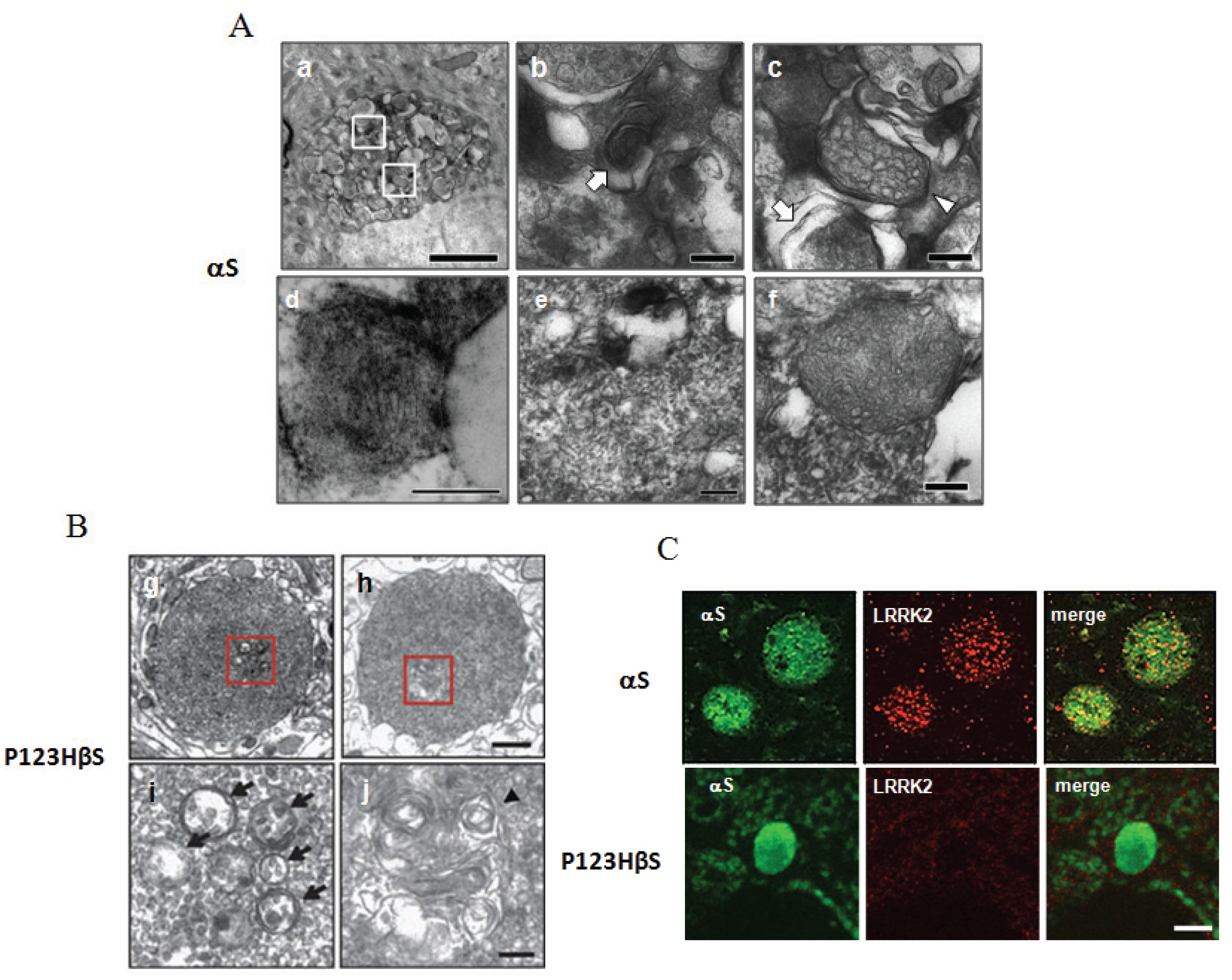
3.2. Lysosomal Pathology is Commonly Observed in Both αS- and P123H βS-Globules
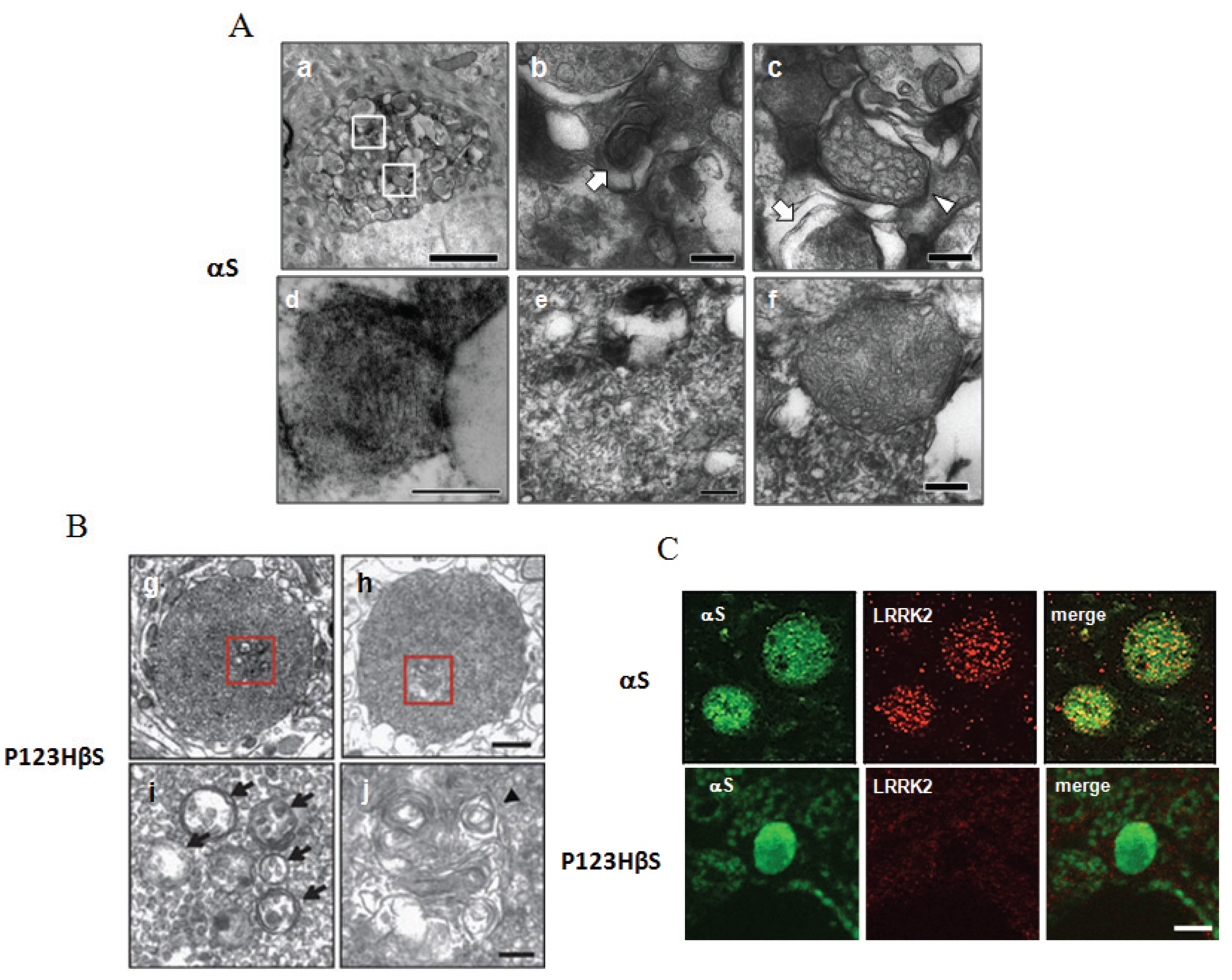
3.3. Alterations of Mitochondria in the αS-Globules but Not in the P123H βS-Globules
3.4. Oxidative Stress Conditions are Enhanced in the αS-Globules and to a Lesser Extent in the P123H βS-Globules
3.5. Accumulation of LRRK2 in the αS-Globules but Not in the P123H βS-Globules
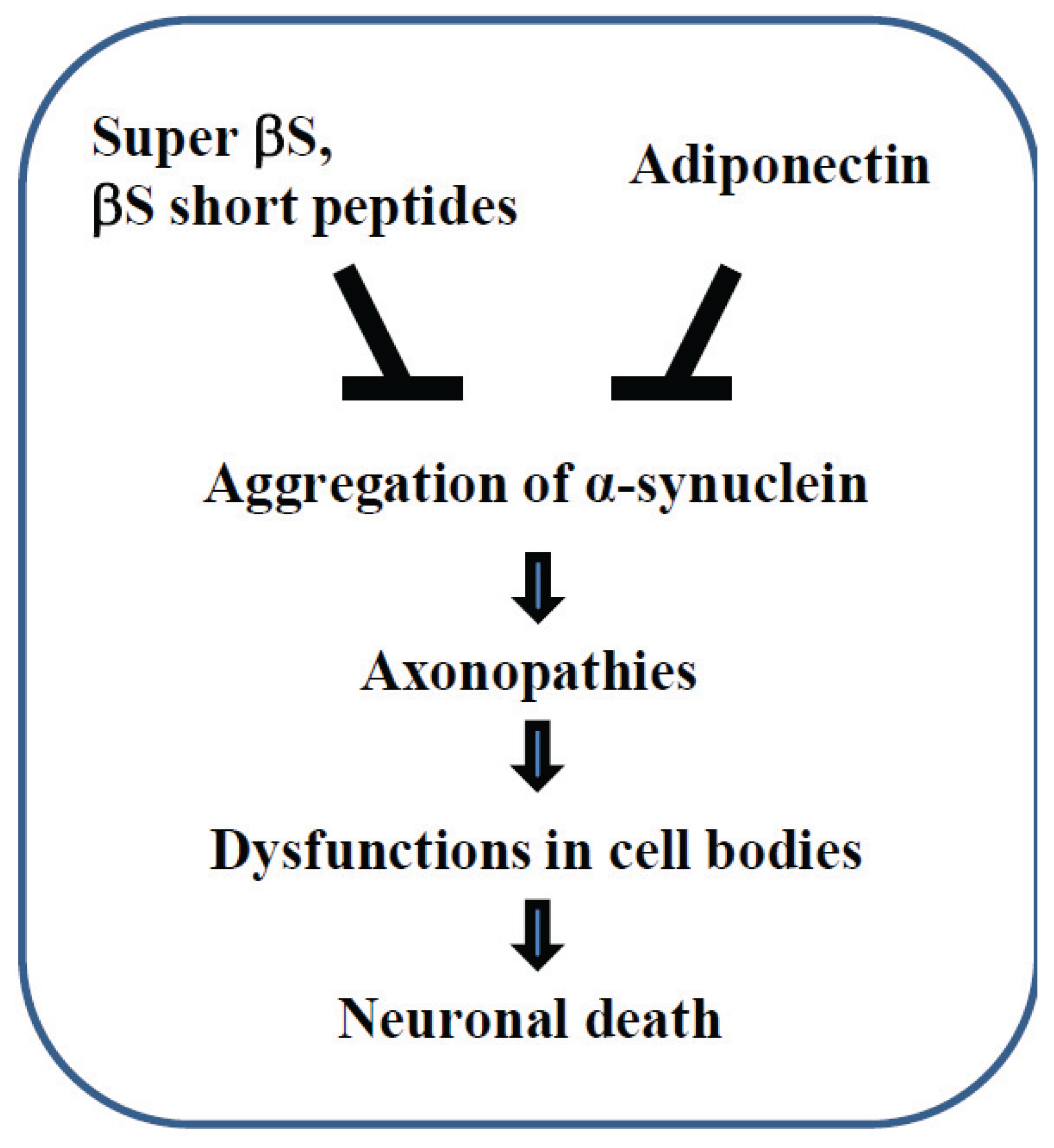
4. Axonal Protection Strategy Based on βS Actions
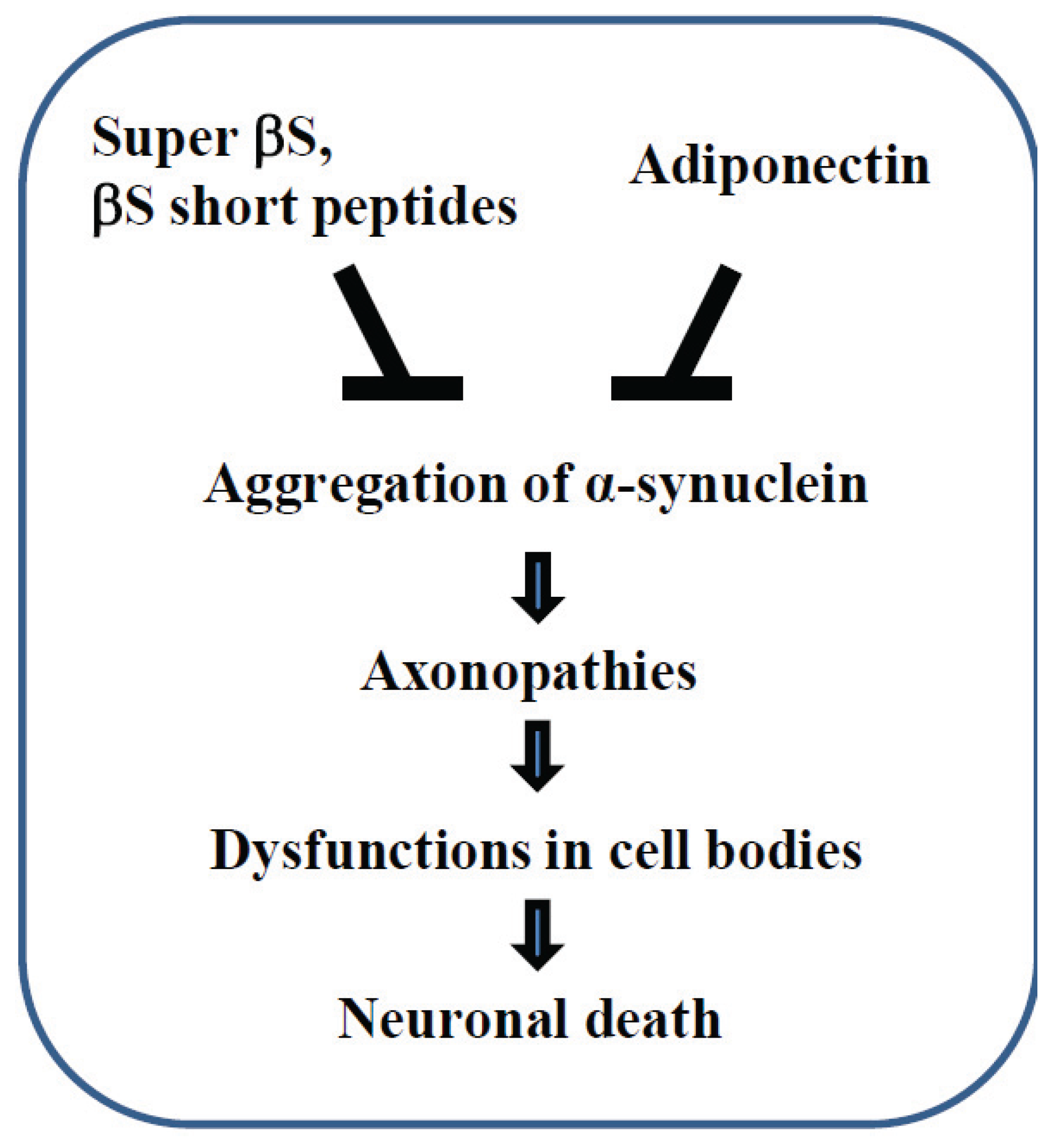
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Hashimoto, M.; Masliah, E. Alpha-synuclein in Lewy body disease and alzheimer’s disease. Brain Pathol. 1999, 9, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Trojanowski, J.Q.; Goedert, M.; Iwatsubo, T.; Lee, V.M. Fatal attractions: Abnormal protein aggregation and neuron death in Parkinson’s disease and Lewy body dementia. Cell Death Differ. 1998, 5, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Norris, E.H.; Giasson, B.I.; Lee, V.M. Alpha-synuclein: Normal function and role in neurodegenerative diseases. Curr. Top. Dev. Biol. 2004, 60, 17–54. [Google Scholar] [PubMed]
- Jenner, P.; Morris, H.R.; Robbins, T.W.; Goedert, M.; Hardy, J.; Ben-Shlomo, Y.; Bolam, P.; Burn, D.; Hindle, J.V.; Brooks, D. Parkinson’s disease—The debate on the clinical phenomenology, aetiology, pathology and pathogenesis. J. Parkinson’s Dis. 2013, 3, 1–11. [Google Scholar]
- Sekiyama, K.; Takamatsu, Y.; Waragai, M.; Hashimoto, M. Role of genomics in translational research for Parkinson’s disease. Biochem. Biophys. Res. Commun. 2014, 452, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Formation and development of Lewy pathology: A critical update. J. Neurol. 2009, 256, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Tanji, K.; Odagiri, S.; Miki, Y.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol. Neurobiol. 2013, 47, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Langbaum, J.B.; Fleisher, A.S.; Caselli, R.J.; Chen, K.; Ayutyanont, N.; Quiroz, Y.T.; Kosik, K.S.; Lopera, F.; Tariot, P.N. Alzheimer’s prevention initiative: A plan to accelerate the evaluation of presymptomatic treatments. J. Alzheimer’s Dis. 2011, 26, 321–329. [Google Scholar]
- Stokin, G.B.; Lillo, C.; Falzone, T.L.; Brusch, R.G.; Rockenstein, E.; Mount, S.L.; Raman, R.; Davies, P.; Masliah, E.; Williams, D.S.; et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 2005, 307, 1282–1288. [Google Scholar] [CrossRef]
- Llorens, J. Toxic neurofilamentous axonopathies—Accumulation of neurofilaments and axonal degeneration. J. Intern. Med. 2013, 273, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Hubner, C.A.; Kurth, I. Membrane-shaping disorders: A common pathway in axon degeneration. Brain 2014, 137, 3109–3121. [Google Scholar] [CrossRef] [PubMed]
- LoPachin, R.M.; Gavin, T. Toxic neuropathies: Mechanistic insights based on a chemical perspective. Neurosci. Lett. 2015, 596, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Sekigawa, A.; Takamatsu, Y.; Sekiyama, K.; Takenouchi, T.; Sugama, S.; Waragai, M.; Fujita, M.; Hashimoto, M. Diversity of mitochondrial pathology in a mouse model of axonal degeneration in synucleinopathies. Oxidative Med. Cell. Longev. 2013. [Google Scholar] [CrossRef]
- Adalbert, R.; Coleman, M.P. Axon pathology in age-related neurodegenerative disorders. Neuropathol. Appl. Neurobiol. 2012. [Google Scholar] [CrossRef]
- Braak, H.; de Vos, R.A.; Bohl, J.; del Tredici, K. Gastric α-synuclein immunoreactive inclusions in meissner’s and auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 2006, 396, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Ghebremedhin, E.; Rub, U.; Bratzke, H.; del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Orimo, S.; Uchihara, T.; Nakamura, A.; Mori, F.; Kakita, A.; Wakabayashi, K.; Takahashi, H. Axonal α-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008, 131, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Galvin, J.E.; Giasson, B.; Hurtig, H.I.; Lee, V.M.; Trojanowski, J.Q. Neurodegeneration with brain iron accumulation, type 1 is characterized by α-, β-, and γ-synuclein neuropathology. Am. J. Pathol. 2000, 157, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Galvin, J.E.; Uryu, K.; Lee, V.M.; Trojanowski, J.Q. Axon pathology in Parkinson’s disease and Lewy body dementia hippocampus contains alpha-, beta-, and gamma-synuclein. Proc. Natl. Acad. Sci. USA 1999, 96, 13450–13455. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Stirling, W.; Xu, Y.; Xu, X.; Qui, D.; Mandir, A.S.; Dawson, T.M.; Copeland, N.G.; Jenkins, N.A.; Price, D.L. Human alpha-synuclein-harboring familial Parkinson’s disease-linked Ala-53 → Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 8968–8973. [Google Scholar] [CrossRef] [PubMed]
- Ninkina, N.; Peters, O.; Millership, S.; Salem, H.; van der Putten, H.; Buchman, V.L. γ-Synucleinopathy: Neurodegeneration associated with overexpression of the mouse protein. Hum. Mol. Genet. 2009, 18, 1779–1794. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Sugama, S.; Sekiyama, K.; Sekigawa, A.; Tsukui, T.; Nakai, M.; Waragai, M.; Takenouchi, T.; Takamatsu, Y.; Wei, J.; et al. A beta-synuclein mutation linked to dementia produces neurodegeneration when expressed in mouse brain. Nat. Commun. 2010. [Google Scholar] [CrossRef]
- Sekigawa, A.; Fujita, M.; Sekiyama, K.; Takamatsu, Y.; Rockenstein, E.; la Spada, A.R.; Masliah, E.; Hashimoto, M. Distinct mechanisms of axonal globule formation in mice expressing human wild type alpha-synuclein or dementia with Lewy bodies-linked p123h β-synuclein. Mol. Brain 2012. [Google Scholar] [CrossRef]
- Mori, F.; Tanji, K.; Zhang, H.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Alpha-synuclein pathology in the neostriatum in Parkinson’s disease. Acta Neuropathol. 2008, 115, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Hamano, K.; Hayashi, M.; Shioda, K.; Fukatsu, R.; Mizutani, S. Mechanisms of neurodegeneration in mucopolysaccharidoses II and IIIB: Analysis of human brain tissue. Acta Neuropathol. 2008, 115, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.L. Maturation of autophagic vacuoles in mammalian cells. Autophagy 2005, 1, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Stekhoven, J.H.; van Haelst, U.J. Ultrastructural study of so-called curvilinear bodies and fingerprint structures in lymphocytes in late-infantile amaurotic idiocy. Acta Neuropathol. 1976, 35, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Sandhoff, K.; Harzer, K. Gangliosides and gangliosidoses: Principles of molecular and metabolic pathogenesis. J. Neurosci. 2013, 33, 10195–10208. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Fujita, M.; Nakai, M.; Waragai, M.; Watabe, K.; Akatsu, H.; Rockenstein, E.; Masliah, E.; Hashimoto, M. Enhanced lysosomal pathology caused by β-synuclein mutants linked to dementia with Lewy bodies. J. Biol. Chem. 2007, 282, 28904–28914. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Fujita, M.; Nakai, M.; Waragai, M.; Sekigawa, A.; Sugama, S.; Takenouchi, T.; Masliah, E.; Hashimoto, M. Protective role of endogenous gangliosides for lysosomal pathology in a cellular model of synucleinopathies. Am. J. Pathol. 2009, 174, 1891–1909. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. Autophagy, amyloidogenesis and alzheimer disease. J. Cell Sci. 2007, 120, 4081–4091. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.J.; Ding, Y.; Kohtz, D.S.; Mizushima, N.; Cristea, I.M.; Rout, M.P.; Chait, B.T.; Zhong, Y.; Heintz, N.; Yue, Z. Induction of autophagy in axonal dystrophy and degeneration. J. Neurosci. 2006, 26, 8057–8068. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Sato, Y.; Nixon, R.A. Primary lysosomal dysfunction causes cargo-specific deficits of axonal transport leading to Alzheimer-like neuritic dystrophy. Autophagy 2011, 7, 1562–1563. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Bosco, D.A.; Fowler, D.M.; Zhang, Q.; Nieva, J.; Powers, E.T.; Wentworth, P., Jr.; Lerner, R.A.; Kelly, J.W. Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate alpha-synuclein fibrilization. Nat. Chem. Biol. 2006, 2, 249–253. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, D.; Dowman, J.; Hammond, R.; Leete, T.; Inoue, K.; Abeliovich, A. The familial parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 2006, 52, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Aliaga, L.; Cai, H. Alpha-synuclein, LRRK2 and their interplay in Parkinson’s disease. Futur. Neurol. 2012, 7, 145–153. [Google Scholar] [CrossRef]
- Lin, X.; Parisiadou, L.; Gu, X.L.; Wang, L.; Shim, H.; Sun, L.; Xie, C.; Long, C.X.; Yang, W.J.; Ding, J.; et al. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron.
- Narendra, D.; Walker, J.E.; Youle, R. Mitochondrial quality control mediated by PINK1 and Parkin: Links to parkinsonism. Cold Spring Harbor Perspect. Biol. 2012. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Souillac, P.; Millett, I.S.; Doniach, S.; Jakes, R.; Goedert, M.; Fink, A.L. Biophysical properties of the synucleins and their propensities to fibrillate: Inhibition of alpha-synuclein assembly by β- and γ-synucleins. J. Biol. Chem. 2002, 277, 11970–11978. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Rockenstein, E.; Mante, M.; Mallory, M.; Masliah, E. Beta-synuclein inhibits α-synuclein aggregation: A possible role as an anti-parkinsonian factor. Neuron 2001, 32, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Limprasert, P.; Murray, I.V.; Smith, A.C.; Lee, V.M.; Trojanowski, J.Q.; Sopher, B.L.; la Spada, A.R. Beta-synuclein modulates alpha-synuclein neurotoxicity by reducing alpha-synuclein protein expression. Hum. Mol. Genet. 2006, 15, 3002–3011. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; la Spada, A. Beta-synuclein in the pathogenesis of Parkinson’s disease and related alpha-synucleinopathies: Emerging roles and new directions. Futur. Neurol. 2012, 7, 155–163. [Google Scholar] [CrossRef]
- Hashimoto, M.; Rockenstein, E.; Mante, M.; Crews, L.; Bar-On, P.; Gage, F.H.; Marr, R.; Masliah, E. An antiaggregation gene therapy strategy for Lewy body disease utilizing beta-synuclein lentivirus in a transgenic model. Gene Ther. 2004, 11, 1713–1723. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, H.; Limprasert, P.; Fan, Y.; Onodera, O.; Kakita, A.; Takahashi, H.; Bonner, L.T.; Tsuang, D.W.; Murray, I.V.; Lee, V.M.; et al. β-Synuclein gene alterations in dementia with Lewy bodies. Neurology 2004, 63, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Taschenberger, G.; Toloe, J.; Tereshchenko, J.; Akerboom, J.; Wales, P.; Benz, R.; Becker, S.; Outeiro, T.; Looger, L.; Bahr, M.; et al. β-Synuclein aggregates and induces neurodegeneration in dopaminergic neurons. Ann. Neurol. 2013, 74, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Yamin, G.; Munishkina, L.A.; Karymov, M.A.; Lyubchenko, Y.L.; Uversky, V.N.; Fink, A.L. Forcing nonamyloidogenic beta-synuclein to fibrillate. Biochemistry 2005, 44, 9096–9107. [Google Scholar] [CrossRef] [PubMed]
- Windisch, M.; Hutter-Paier, B.; Rockenstein, E.; Hashimoto, M.; Mallory, M.; Masliah, E. Development of a new treatment for alzheimer’s disease and Parkinson’s disease using anti-aggregatory beta-synuclein-derived peptides. J. Mol. Neurosci. 2002, 19, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Shaltiel-Karyo, R.; Frenkel-Pinter, M.; Egoz-Matia, N.; Frydman-Marom, A.; Shalev, D.E.; Segal, D.; Gazit, E. Inhibiting α-synuclein oligomerization by stable cell-penetrating β-synuclein fragments recovers phenotype of Parkinson’s disease model flies. PLoS ONE 2010, 5, e13863. [Google Scholar] [CrossRef] [PubMed]
- Sekiyama, K.; Waragai, M.; Akatsu, H.; Sugama, S.; Takenouchi, T.; Takamatsu, Y.; Fujita, M.; Sekigawa, A.; Rockenstein, E.; Inoue, S.; et al. Disease-modifying effect of adiponectin in model of alpha-synucleinopathies. Ann. Clin. Transl. Neurol. 2014, 1, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G.; Davies, S.W. Filamentous nerve cell inclusions in neurodegenerative diseases. Curr. Opin. Neurobiol. 1998, 8, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Gold, B.G. The pathophysiology of proximal neurofilamentous giant axonal swellings: Implications for the pathogenesis of amyotrophic lateral sclerosis. Toxicology 1987, 46, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Leonhardt, H. “Axonal spheroids” in the spinal cord of normal rabbits. Cell Tissue Res. 1976, 174, 99–108. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sekigawa, A.; Takamatsu, Y.; Sekiyama, K.; Hashimoto, M. Role of α- and β-Synucleins in the Axonal Pathology of Parkinson’s Disease and Related Synucleinopathies. Biomolecules 2015, 5, 1000-1011. https://doi.org/10.3390/biom5021000
Sekigawa A, Takamatsu Y, Sekiyama K, Hashimoto M. Role of α- and β-Synucleins in the Axonal Pathology of Parkinson’s Disease and Related Synucleinopathies. Biomolecules. 2015; 5(2):1000-1011. https://doi.org/10.3390/biom5021000
Chicago/Turabian StyleSekigawa, Akio, Yoshiki Takamatsu, Kazunari Sekiyama, and Makoto Hashimoto. 2015. "Role of α- and β-Synucleins in the Axonal Pathology of Parkinson’s Disease and Related Synucleinopathies" Biomolecules 5, no. 2: 1000-1011. https://doi.org/10.3390/biom5021000
APA StyleSekigawa, A., Takamatsu, Y., Sekiyama, K., & Hashimoto, M. (2015). Role of α- and β-Synucleins in the Axonal Pathology of Parkinson’s Disease and Related Synucleinopathies. Biomolecules, 5(2), 1000-1011. https://doi.org/10.3390/biom5021000