
CD4/CD8–p56lck Induced T-Cell Receptor Signaling and Its Implications for Immunotherapy


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
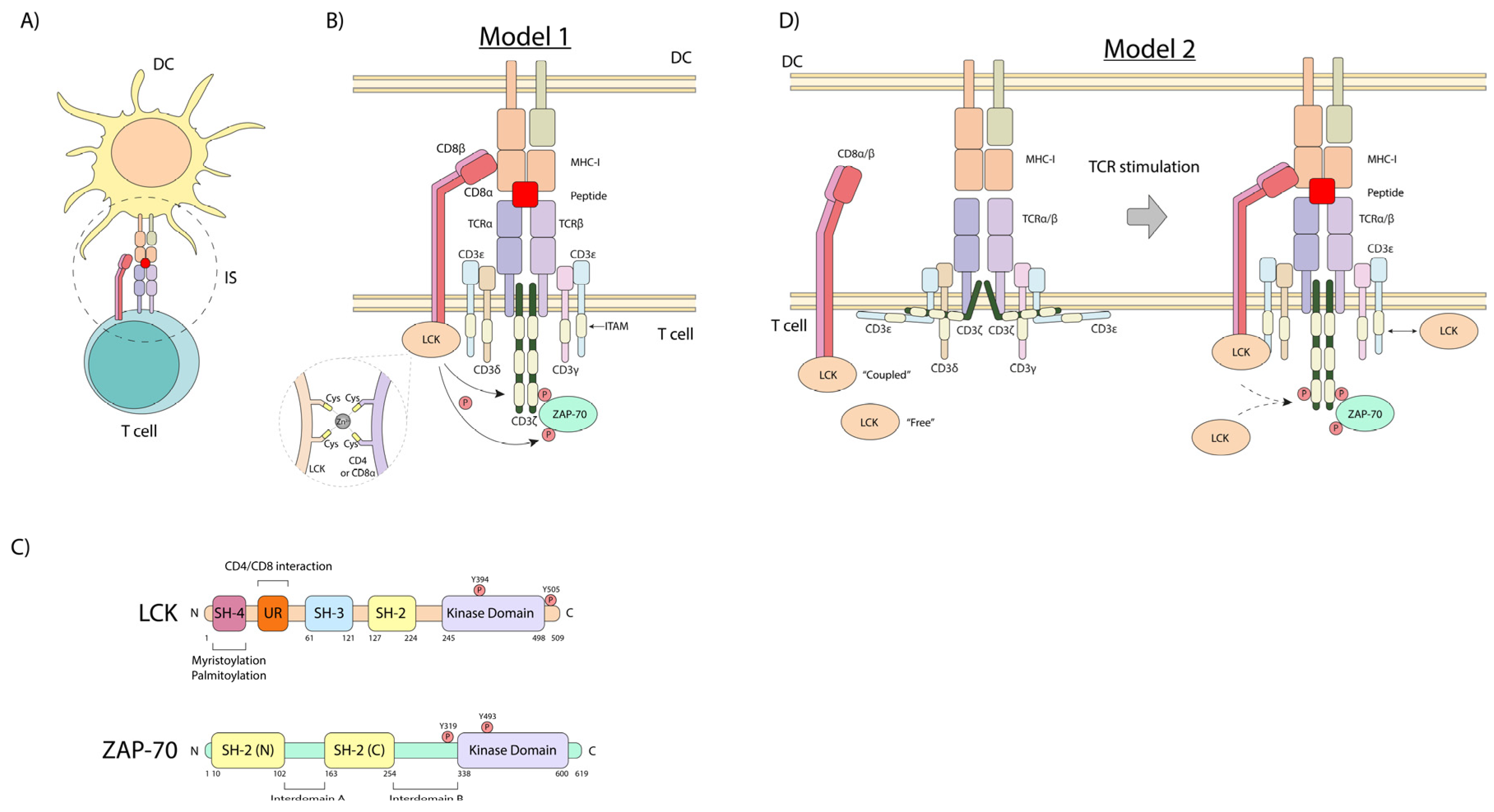
2. The First Steps in the TCR Signaling Pathway
3. Microdomains
4. T-Cell Subsets
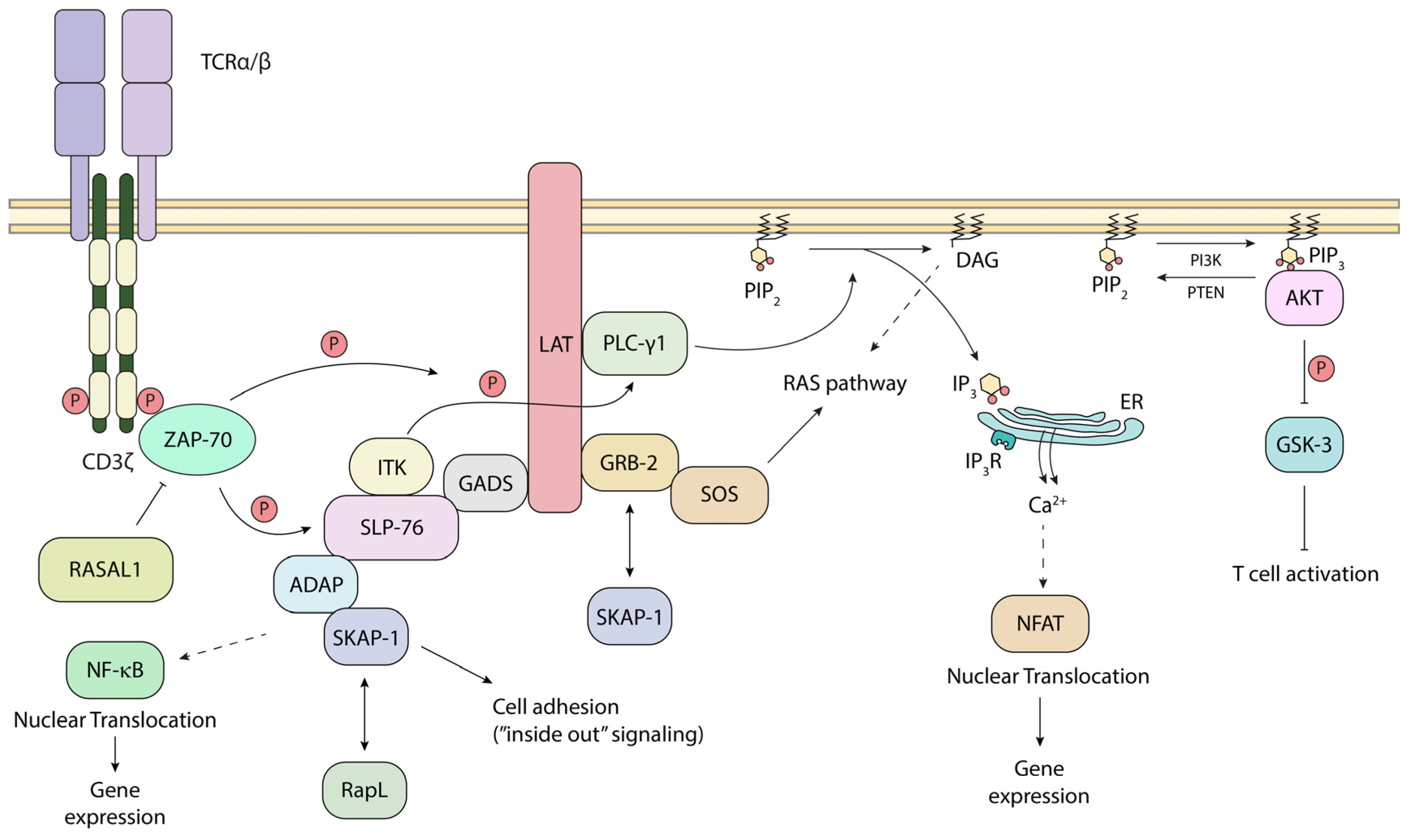
5. Steps in p56lck-Initiated TCR Downstream Signaling
6. CD4/CD8–p56lck and CAR T-Cell Immunotherapy
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, L.; Su, Y.; Jiao, A.; Wang, X.; Zhang, B. T cells in health and disease. Signal Transduct. Target. Ther. 2023, 8, 235. [Google Scholar] [CrossRef]
- Rudd, C.E. Disabled receptor signaling and new primary immunodeficiency disorders. N. Engl. J. Med. 2006, 354, 1874–1877. [Google Scholar] [CrossRef]
- Meitei, H.T.; Lal, G. T cell receptor signaling in the differentiation and plasticity of CD4(+) T cells. Cytokine Growth. Factor. Rev. 2023, 69, 14–27. [Google Scholar] [CrossRef]
- Rudd, C.E.; Trevillyan, J.M.; Dasgupta, J.D.; Wong, L.L.; Schlossman, S.F. The CD4 receptor is complexed in detergent lysates to a protein-tyrosine kinase (pp58) from human T lymphocytes. Proc. Natl. Acad. Sci. USA 1988, 85, 5190–5194, reprint in J. Immunol. 2010, 185, 2645–2649. [Google Scholar] [CrossRef] [PubMed]
- Barber, E.K.; Dasgupta, J.D.; Schlossman, S.F.; Trevillyan, J.M.; Rudd, C.E. The CD4 and CD8 antigens are coupled to a protein-tyrosine kinase (p56lck) that phosphorylates the CD3 complex. Proc. Natl. Acad. Sci. USA 1989, 86, 3277–3281. [Google Scholar] [CrossRef] [PubMed]
- Rudd, C.E. CD4, CD8 and the TCR-CD3 complex: A novel class of protein-tyrosine kinase receptor. Immunol. Today 1990, 11, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Simpson, S.C.; Bolen, J.B.; Veillette, A. CD4 and p56lck can stably associate when co-expressed in NIH3T3 cells. Oncogene 1989, 4, 1141–1143. [Google Scholar]
- Veillette, A.; Bookman, M.A.; Horak, E.M.; Bolen, J.B. The CD4 and CD8 T cell surface antigens are associated with the internal membrane tyrosine-protein kinase p56lck. Cell 1988, 55, 301–308. [Google Scholar] [CrossRef]
- Rudd, C.E.; Schneider, H. Unifying concepts in CD28, ICOS and CTLA4 co-receptor signalling. Nat. Rev. Immunol. 2003, 3, 544–556. [Google Scholar] [CrossRef]
- Schneider, H.; Schneider, H.; Cai, Y.C.; Prasad, K.V.; Schoelson, S.E.; Rudd, C.E. T-cell antigen CD28 binds to the GRB-2/SOS complex, regulators of p21ras. Eur. J. Immunol. 1995, 25, 1044–1050. [Google Scholar] [CrossRef]
- Prasad, K.V.; Cai, Y.C.; Raab, M.; Duckworth, B.; Cantley, L.; Shoelson, S.E.; Rudd, C.E. T-cell antigen CD28 interacts with the lipid kinase phosphatidylinositol 3-kinase by a cytoplasmic Tyr(P)-Met-Xaa-Met motif. Proc. Natl. Acad. Sci. USA 1994, 91, 2834–2838. [Google Scholar] [CrossRef]
- Kong, K.F.; Yokosuka, T.; Canonigo-Balancio, A.J.; Isakov, N.; Saito, T.; Altman, A. A motif in the V3 domain of the kinase PKC-theta determines its localization in the immunological synapse and functions in T cells via association with CD28. Nat. Immunol. 2011, 12, 1105–1112. [Google Scholar] [CrossRef]
- Schneider, H.; Cai, Y.-C.; Cefai, D.; Raab, M.; Rudd, C. Mechanisms of CD28 signalling. Res. Immunol. 1995, 146, 149–154. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Freeman, G.J. The B7—CD28 superfamily. Nat. Rev. Immunol. 2002, 2, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.; Downey, J.; Smith, A.; Zinselmeyer, B.H.; Rush, C.; Brewer, J.M.; Wei, B.; Hogg, N.; Garside, P.; Rudd, C.E. Reversal of the TCR stop signal by CTLA-4. Science 2006, 313, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Rudd, C.E. Upstream-downstream: CD28 cosignaling pathways and T cell function. Immunity 1996, 4, 527–534. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Bluestone, J.A.; Nadler, L.M.; Thompson, C.B. The B7 and CD28 receptor families. Immunol. Today 1994, 15, 321–331. [Google Scholar] [CrossRef]
- Rudd, C.E. How the Discovery of the CD4/CD8-p56(lck) Complexes Changed Immunology and Immunotherapy. Front. Cell. Dev. Biol. 2021, 9, 626095. [Google Scholar] [CrossRef]
- Ruella, M.; June, C.H. Chimeric Antigen Receptor T cells for B Cell Neoplasms: Choose the Right CAR for You. Curr. Hematol. Malig. Rep. 2016, 11, 368–384. [Google Scholar] [CrossRef]
- Boussiotis, V.A. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N. Engl. J. Med. 2016, 375, 1767–1778. [Google Scholar] [CrossRef]
- Liu, Y.J. Dendritic cell subsets and lineags, and their function in innate and adaptive immunity. Cell 2001, 106, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Pishesha, N.; Harmand, T.J.; Ploegh, H.L. A guide to antigen processing and presentation. Nat. Rev. Immunol. 2022, 22, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Meuer, S.C.; Schlossman, S.F.; Reinherz, E.L. Clonal analysis of human cytotoxic T lymphocytes: T4+ and T8+ effector T cells recognize products of different major histocompatibility complex regions. Proc. Natl. Acad. Sci. USA 1982, 79, 4395–4399. [Google Scholar] [CrossRef]
- Koretzky, G.A. Multiple roles of CD4 and CD8 in T cell activation. J. Immunol. 2010, 185, 2643–2644. [Google Scholar] [CrossRef]
- Teh, H.S.; Garvin, A.M.; Forbush, K.A.; Carlow, D.A.; Davis, C.B.; Littman, D.R.; Perlmutter, R.M. Participation of CD4 coreceptor molecules in T-cell repertoire selection. Nature 1991, 349, 241–243. [Google Scholar] [CrossRef]
- Kim, P.W.; Sun, Z.-Y.J.; Blacklow, S.C.; Wagner, G.; Eck, M.J. A zinc clasp structure tethers Lck to T cell coreceptors CD4 and CD8. Science 2003, 301, 1725–1728. [Google Scholar] [CrossRef]
- Rudd, C.E.; Anderson, P.; Morimoto, C.; Streuli, M.; Schlossman, S.F. Molecular interactions, T-cell subsets and a role of the CD4/CD8:p56lck complex in human T-cell activation. Immunol. Rev. 1989, 111, 225–266. [Google Scholar] [CrossRef]
- Lin, R.S.; Rodriguez, C.; Veillette, A.; Lodish, H.F. Zinc is essential for binding of p56(lck) to CD4 and CD8alpha. J. Biol. Chem. 1998, 273, 32878–32882. [Google Scholar] [CrossRef]
- Holdorf, A.D.; Lee, K.-H.; Burack, W.R.; Allen, P.M.; Shaw, A.S. Regulation of Lck activity by CD4 and CD28 in the immunological synapse. Nat. Immunol. 2002, 3, 259–264. [Google Scholar] [CrossRef]
- Rossy, J.; Williamson, D.J.; Gaus, K. How does the kinase Lck phosphorylate the T cell receptor? Spatial organization as a regulatory mechanism. Front. Immunol. 2012, 3, 167. [Google Scholar] [CrossRef]
- Samelson, L.E. Signal transduction mediated by the T cell antigen receptor: The role of adapter proteins. Annu. Rev. Immunol. 2002, 20, 371–394. [Google Scholar] [CrossRef]
- Shin, J.; Doyle, C.; Yang, Z.; Kappes, D.; Strominger, J. Structural features of the cytoplasmic region of CD4 required for internalization. EMBO J. 1990, 9, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Sleckman, B.P.; Shin, J.; E Igras, V.; Collins, T.L.; Strominger, J.L.; Burakoff, S.J. Disruption of the CD4-p56lck complex is required for rapid internalization of CD4. Proc. Natl. Acad. Sci. USA 1992, 89, 7566–7570. [Google Scholar] [CrossRef] [PubMed]
- Monks, C.R.; Freiberg, B.A.; Kupfer, H.; Sciaky, N.; Kupfer, A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature 1998, 395, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.S.; Dustin, M.L. Making the T cell receptor go the distance: A topological view of T cell activation. Immunity 1997, 6, 361–369. [Google Scholar] [CrossRef]
- Nika, K.; Tautz, L.; Arimura, Y.; Vang, T.; Williams, S.; Mustelin, T. A weak Lck tail bite is necessary for Lck function in T cell antigen receptor signaling. J. Biol. Chem. 2007, 282, 36000–36009. [Google Scholar] [CrossRef]
- Casas, J.; Brzostek, J.; Zarnitsyna, V.I.; Hong, J.-S.; Wei, Q.; Hoerter, J.A.H.; Fu, G.; Ampudia, J.; Zamoyska, R.; Zhu, C.; et al. Ligand-engaged TCR is triggered by Lck not associated with CD8 coreceptor. Nat. Commun. 2014, 5, 5624. [Google Scholar] [CrossRef]
- Wei, Q.; Brzostek, J.; Sankaran, S.; Casas, J.; Hew, L.S.-Q.; Yap, J.; Zhao, X.; Wojciech, L.; Gascoigne, N.R.J. Lck bound to coreceptor is less active than free Lck. Proc. Natl. Acad. Sci. USA 2020, 117, 15809–15817. [Google Scholar] [CrossRef]
- Irie, H.Y.; Mong, M.S.; Itano, A.; Crooks, M.E.C.; Littman, D.R.; Burakoff, S.J.; Robey, E. The cytoplasmic domain of CD8 beta regulates Lck kinase activation and CD8 T cell development. J. Immunol. 1998, 161, 183–191. [Google Scholar] [CrossRef]
- Bosselut, R.; Kubo, S.; Guinter, T.; Kopacz, J.L.; Altman, J.D.; Feigenbaum, L.; Singer, A. Role of CD8beta domains in CD8 coreceptor function: Importance for MHC I binding, signaling, and positive selection of CD8+ T cells in the thymus. Immunity 2000, 12, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Cheroutre, H.; Lambolez, F. Doubting the TCR coreceptor function of CD8alphaalpha. Immunity 2008, 28, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Kern, P.S.; Teng, M.-K.; Smolyar, A.; Liu, J.-H.; Liu, J.; Hussey, R.E.; Spoerl, R.; Chang, H.-C.; Reinherz, E.L.; Wang, J.-H. Structural basis of CD8 coreceptor function revealed by crystallographic analysis of a murine CD8alphaalpha ectodomain fragment in complex with H-2Kb. Immunity 1998, 9, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Kavathas, P.B. Comparison of the roles of CD8 alpha alpha and CD8 alpha beta in interaction with MHC class I. J. Immunol. 1997, 159, 6077–6082. [Google Scholar] [CrossRef]
- Xu, C.; Gagnon, E.; Call, M.E.; Schnell, J.R.; Schwieters, C.D.; Carman, C.V.; Chou, J.J.; Wucherpfennig, K.W. Regulation of T cell receptor activation by dynamic membrane binding of the CD3epsilon cytoplasmic tyrosine-based motif. Cell 2008, 135, 702–713. [Google Scholar] [CrossRef]
- Zhang, H.; Cordoba, S.-P.; Dushek, O.; van der Merwe, P.A. Basic residues in the T-cell receptor zeta cytoplasmic domain mediate membrane association and modulate signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 19323–19328. [Google Scholar] [CrossRef]
- Gagnon, E.; Schubert, D.A.; Gordo, S.; Chu, H.H.; Wucherpfennig, K.W. Local changes in lipid environment of TCR microclusters regulate membrane binding by the CD3epsilon cytoplasmic domain. J. Exp. Med. 2012, 209, 2423–2439. [Google Scholar] [CrossRef]
- Pizzo, P.; Viola, A. Lymphocyte lipid rafts: Structure and function. Curr. Opin. Immunol. 2003, 15, 255–260. [Google Scholar] [CrossRef]
- Wang, H.Y.; Chan, S.H.; Dey, S.; Castello-Serrano, I.; Rosen, M.K.; Ditlev, J.A.; Levental, K.R.; Levental, I. Coupling of protein condensates to ordered lipid domains determines functional membrane organization. Sci. Adv. 2023, 9, eadf6205. [Google Scholar] [CrossRef]
- Martin, M.; Schneider, H.; Azouz, A.; Rudd, C.E. Cytotoxic T lymphocyte antigen 4 and CD28 modulate cell surface raft expression in their regulation of T cell function. J. Exp. Med. 2001, 194, 1675–1681. [Google Scholar] [CrossRef]
- Chikuma, S.; Imboden, J.B.; Bluestone, J.A. Negative regulation of T cell receptor-lipid raft interaction by cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 2003, 197, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Horkova, V.; Drobek, A.; Mueller, D.; Gubser, C.; Niederlova, V.; Wyss, L.; King, C.G.; Zehn, D.; Stepanek, O. Dynamics of the Coreceptor-LCK Interactions during T Cell Development Shape the Self-Reactivity of Peripheral CD4 and CD8 T Cells. Cell Rep. 2020, 30, 1504–1514. [Google Scholar] [CrossRef] [PubMed]
- Marsh, M.; Parsons, I.J.; Reid, P.; Pelchen-Matthews, A. Endocytic regulation of the T lymphocyte co-receptor proteins CD4 and CD8. Biochem. Soc. Trans. 1993, 21, 703–706. [Google Scholar] [CrossRef] [PubMed]
- Pelchen-Matthews, A.; Parsons, I.J.; Marsh, M. Phorbol ester-induced downregulation of CD4 is a multistep process involving dissociation from p56lck, increased association with clathrin-coated pits, and altered endosomal sorting. J. Exp. Med. 1993, 178, 1209–1222. [Google Scholar] [CrossRef]
- Horkova, V.; Drobek, A.; Paprckova, D.; Niederlova, V.; Prasai, A.; Uleri, V.; Glatzova, D.; Kraller, M.; Cesnekova, M.; Janusova, S.; et al. Unique roles of co-receptor-bound LCK in helper and cytotoxic T cells. Nat. Immunol. 2023, 24, 174–185. [Google Scholar] [CrossRef]
- Weiss, A.; Littman, D.R. Signal transduction by lymphocyte antigen receptors. Cell 1994, 76, 263–274. [Google Scholar] [CrossRef]
- Weissman, A.M.; Samelson, L.E.; Klausner, R.D. A new subunit of the human T-cell antigen receptor complex. Nature 1986, 324, 480–482. [Google Scholar] [CrossRef]
- Weissman, A.M.; Ross, P.; Luong, E.T.; Garcia-Morales, P.; Jelachich, M.L.; E Biddison, W.; Klausner, R.D.; E Samelson, L. Tyrosine phosphorylation of the human T cell antigen receptor zeta-chain: Activation via CD3 but not CD2. J. Immunol. 1988, 141, 3532–3536. [Google Scholar] [CrossRef]
- Irving, B.A.; Chan, A.C.; Weiss, A. Functional characterization of a signal transducing motif present in the T cell antigen receptor zeta chain. J. Exp. Med. 1993, 177, 1093–1103. [Google Scholar] [CrossRef]
- Chan, A.C.; Dalton, M.; Johnson, R.; Kong, G.; Wang, T.; Thoma, R.; Kurosaki, T. Activation of ZAP-70 kinase activity by phosphorylation of tyrosine 493 is required for lymphocyte antigen receptor function. EMBO J. 1995, 14, 2499–2508. [Google Scholar] [CrossRef]
- Thaker, Y.R.; Raab, M.; Strebhardt, K.; Rudd, C.E. GTPase-activating protein Rasal1 associates with ZAP-70 of the TCR and negatively regulates T-cell tumor immunity. Nat. Commun. 2019, 10, 4804. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.; Kang, H.; da Silva, A.; Zhu, X.; Rudd, C.E. FYN-T-FYB-SLP-76 interactions define a T-cell receptor zeta/CD3-mediated tyrosine phosphorylation pathway that up-regulates interleukin 2 transcription in T-cells. J. Biol. Chem. 1999, 274, 21170–21179. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.; da Silva, A.J.; Findell, P.R.; Rudd, C.E. Regulation of Vav-SLP-76 binding by ZAP-70 and its relevance to TCR zeta/CD3 induction of interleukin-2. Immunity 1997, 6, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Berry, D.; Nash, P.; Pawson, T.; McGlade, C.J.; Li, S.S. Structural basis for specific binding of the Gads SH3 domain to an RxxK motif-containing SLP-76 peptide: A novel mode of peptide recognition. Mol. Cell 2003, 11, 471–481. [Google Scholar] [CrossRef]
- Berg, L.J.; Finkelstein, L.D.; Lucas, J.A.; Schwartzberg, P.L. Tec family kinases in T lymphocyte development and function. Annu. Rev. Immunol. 2005, 23, 549–600. [Google Scholar] [CrossRef]
- Oh-hora, M.; Rao, A. Calcium signaling in lymphocytes. Curr. Opin. Immunol. 2008, 20, 250–258. [Google Scholar] [CrossRef]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.-H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef]
- Liu, H.; Thaker, Y.R.; Stagg, L.; Schneider, H.; Ladbury, J.E.; Rudd, C.E. SLP-76 sterile alpha motif (SAM) and individual H5 alpha helix mediate oligomer formation for microclusters and T-cell activation. J. Biol. Chem. 2013, 288, 29539–29549. [Google Scholar] [CrossRef]
- Hu, H.; Djuretic, I.; Sundrud, M.S.; Rao, A. Transcriptional partners in regulatory T cells: Foxp3, Runx and NFAT. Trends Immunol. 2007, 28, 329–332. [Google Scholar] [CrossRef]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef]
- Wang, H.; Rudd, C.E. SKAP-55, SKAP-55-related and ADAP adaptors modulate integrin-mediated immune-cell adhesion. Trends Cell Biol. 2008, 18, 486–493. [Google Scholar] [CrossRef]
- Wang, H.; Moon, E.-Y.; Azouz, A.; Wu, X.; Smith, A.; Schneider, H.; Hogg, N.; Rudd, C.E. SKAP-55 regulates integrin adhesion and formation of T cell-APC conjugates. Nat. Immunol. 2003, 4, 366–374. [Google Scholar] [CrossRef]
- Duke-Cohan, J.S.; Kang, H.; Liu, H.; Rudd, C.E. Regulation and function of SKAP-55 non-canonical motif binding to the SH3c domain of adhesion and degranulation-promoting adaptor protein. J. Biol. Chem. 2006, 281, 13743–13750. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Freund, C.; Duke-Cohan, J.S.; Musacchio, A.; Wagner, G.; Rudd, C.E. SH3 domain recognition of a proline-independent tyrosine-based RKxxYxxY motif in immune cell adaptor SKAP55. EMBO J. 2000, 19, 2889–2899. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.; Strebhardt, K.; Rudd, C.E. Immune adaptor SKAP1 acts a scaffold for Polo-like kinase 1 (PLK1) for the optimal cell cycling of T-cells. Sci. Rep. 2019, 9, 10462. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.H.; Park, J.-E.; Yu, L.-R.; Soung, N.-K.; Yun, S.-M.; Bang, J.K.; Seong, Y.-S.; Yu, H.; Garfield, S.; Veenstra, T.D.; et al. Self-regulated Plk1 recruitment to kinetochores by the Plk1-PBIP1 interaction is critical for proper chromosome segregation. Mol. Cell 2006, 24, 409–422. [Google Scholar] [CrossRef]
- Matsumura, S.; Toyoshima, F.; Nishida, E. Polo-like kinase 1 facilitates chromosome alignment during prometaphase through BubR1. J. Biol. Chem. 2007, 282, 15217–15227. [Google Scholar] [CrossRef]
- Raab, M.; Sanhaji, M.; Matthess, Y.; Hörlin, A.; Lorenz, I.; Dötsch, C.; Habbe, N.; Waidmann, O.; Kurunci-Csacsko, E.; Firestein, R.; et al. PLK1 has tumor-suppressive potential in APC-truncated colon cancer cells. Nat. Commun. 2018, 9, 1106. [Google Scholar] [CrossRef]
- Wang, H.; Wei, B.; Bismuth, G.; Rudd, C.E. SLP-76-ADAP adaptor module regulates LFA-1 mediated costimulation and T cell motility. Proc. Natl. Acad. Sci. USA 2009, 106, 12436–12441. [Google Scholar] [CrossRef]
- Wang, H.; McCann, F.E.; Gordan, J.D.; Wu, X.; Raab, M.; Malik, T.H.; Davis, D.M.; Rudd, C.E. ADAP-SLP-76 binding differentially regulates supramolecular activation cluster (SMAC) formation relative to T cell-APC conjugation. J. Exp. Med. 2004, 200, 1063–1074. [Google Scholar] [CrossRef]
- Raab, M.; Wang, H.; Lu, Y.; Smith, X.; Wu, Z.; Strebhardt, K.; Ladbury, J.E.; Rudd, C.E. T cell receptor “inside-out” pathway via signaling module SKAP1-RapL regulates T cell motility and interactions in lymph nodes. Immunity 2010, 32, 541–556. [Google Scholar] [CrossRef]
- Wang, H.; Liu, H.; Lu, Y.; Lovatt, M.; Wei, B.; Rudd, C.E. Functional defects of SKAP-55-deficient T cells identify a regulatory role for the adaptor in LFA-1 adhesion. Mol. Cell. Biol. 2007, 27, 6863–6875. [Google Scholar] [CrossRef] [PubMed]
- Kuropka, B.; Witte, A.; Sticht, J.; Waldt, N.; Majkut, P.; Hackenberger, C.P.R.; Schraven, B.; Krause, E.; Kliche, S.; Freund, C. Analysis of Phosphorylation-dependent Protein Interactions of Adhesion and Degranulation Promoting Adaptor Protein (ADAP) Reveals Novel Interaction Partners Required for Chemokine-directed T cell Migration. Mol. Cell. Proteomics 2015, 14, 2961–2972. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; June, C.H. Going viral: Chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol. Rev. 2015, 263, 68–89. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Brentjens, R.; Riviere, I. The promise and potential pitfalls of chimeric antigen receptors. Curr. Opin. Immunol. 2009, 21, 215–223. [Google Scholar] [CrossRef]
- Cai, Y.C.; Cefai, D.; Schneider, H.; Raab, M.; Nabavi, N.; Rudd, C.E. Selective CD28pYMNM mutations implicate phosphatidylinositol 3-kinase in CD86-CD28-mediated costimulation. Immunity 1995, 3, 417–426. [Google Scholar] [CrossRef]
- Okkenhaug, K.; Wu, L.; Garza, K.M.; La Rose, J.; Khoo, W.; Odermatt, B.; Mak, T.W.; Ohashi, P.S.; Rottapel, R. A point mutation in CD28 distinguishes proliferative signals from survival signals. Nat. Immunol. 2001, 2, 325–332. [Google Scholar] [CrossRef]
- Schneider, H.; Valk, E.; Leung, R.; Rudd, C.E.; Wölfl, S. CTLA-4 activation of phosphatidylinositol 3-kinase (PI 3-K) and protein kinase B (PKB/AKT) sustains T-cell anergy without cell death. PLoS ONE 2008, 3, e3842. [Google Scholar] [CrossRef]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef]
- Guo, X.; Kazanova, A.; Thurmond, S.; Saragovi, H.U.; Rudd, C.E. Effective chimeric antigen receptor T cells against SARS-CoV-2. iScience 2021, 24, 103295. [Google Scholar] [CrossRef]
- Hudecek, M.; Sommermeyer, D.; Kosasih, P.L.; Silva-Benedict, A.; Liu, L.; Rader, C.; Jensen, M.C.; Riddell, S.R. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol. Res. 2015, 3, 125–135. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Tousley, A.M.; Rotiroti, M.C.; Labanieh, L.; Rysavy, L.W.; Kim, W.-J.; Lareau, C.; Sotillo, E.; Weber, E.W.; Rietberg, S.P.; Dalton, G.N.; et al. Co-opting signalling molecules enables logic-gated control of CAR T cells. Nature 2023, 615, 507–516. [Google Scholar] [CrossRef]
- Sun, C.; Shou, P.; Du, H.; Hirabayashi, K.; Chen, Y.; Herring, L.E.; Ahn, S.; Xu, Y.; Suzuki, K.; Li, G.; et al. THEMIS-SHP1 Recruitment by 4-1BB Tunes LCK-Mediated Priming of Chimeric Antigen Receptor-Redirected T Cells. Cancer Cell 2020, 37, 216–225.e6. [Google Scholar] [CrossRef]
- Raab, M.; Cai, Y.C.; Bunnell, S.C.; Heyeck, S.D.; Berg, L.J.; E Rudd, C. p56Lck and p59Fyn regulate CD28 binding to phosphatidylinositol 3-kinase, growth factor receptor-bound protein GRB-2, and T cell-specific protein-tyrosine kinase ITK: Implications for T-cell costimulation. Proc. Natl. Acad. Sci. USA 1995, 92, 8891–8895. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.A.; Beck-Garcìa, E.; Woessner, N.M.; Flachsmann, L.J.; Cárdenas, R.M.-H.V.; Brandl, S.M.; Taromi, S.; Fiala, G.J.; Morath, A.; Mishra, P.; et al. Noncanonical binding of Lck to CD3epsilon promotes TCR signaling and CAR function. Nat. Immunol. 2020, 21, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Filby, A.; Seddon, B.; Kleczkowska, J.; Salmond, R.; Tomlinson, P.; Smida, M.; Lindquist, J.A.; Schraven, B.; Zamoyska, R. Fyn regulates the duration of TCR engagement needed for commitment to effector function. J. Immunol. 2007, 179, 4635–4644. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Brzostek, J.; Sakthi Vale, P.D.; Wei, Q.; Koh, C.K.T.; Ong, J.X.H.; Wu, L.Z.; Tan, J.C.; Chua, Y.L.; Yap, J.; et al. CD28-CAR-T cell activation through FYN kinase signaling rather than LCK enhances therapeutic performance. Cell Rep. Med. 2023, 4, 100917. [Google Scholar] [CrossRef]
- Ritmeester-Loy, S.A.; Draper, I.H.; Bueter, E.C.; Lautz, J.D.; Zhang-Wong, Y.; Gustafson, J.A.; Wilson, A.L.; Lin, C.; Gafken, P.R.; Jensen, M.C.; et al. Differential protein-protein interactions underlie signaling mediated by the TCR and a 4-1BB domain-containing CAR. Sci. Signal. 2024, 17, eadd4671. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oroya, A.; Rudd, C.E. CD4/CD8–p56lck Induced T-Cell Receptor Signaling and Its Implications for Immunotherapy. Biomolecules 2025, 15, 1096. https://doi.org/10.3390/biom15081096
Oroya A, Rudd CE. CD4/CD8–p56lck Induced T-Cell Receptor Signaling and Its Implications for Immunotherapy. Biomolecules. 2025; 15(8):1096. https://doi.org/10.3390/biom15081096
Chicago/Turabian StyleOroya, Andres, and Christopher E. Rudd. 2025. "CD4/CD8–p56lck Induced T-Cell Receptor Signaling and Its Implications for Immunotherapy" Biomolecules 15, no. 8: 1096. https://doi.org/10.3390/biom15081096
APA StyleOroya, A., & Rudd, C. E. (2025). CD4/CD8–p56lck Induced T-Cell Receptor Signaling and Its Implications for Immunotherapy. Biomolecules, 15(8), 1096. https://doi.org/10.3390/biom15081096