
Development of a Serum Proteomic-Based Diagnostic Model for Lung Cancer Using Machine Learning Algorithms and Unveiling the Role of SLC16A4 in Tumor Progression and Immune Response


Abstract
1. Introduction
2. Materials and Methods
2.1. Data Source
2.2. Differential Protein Expression Analysis
2.3. Machine Learning Model Construction
2.4. Model Validation
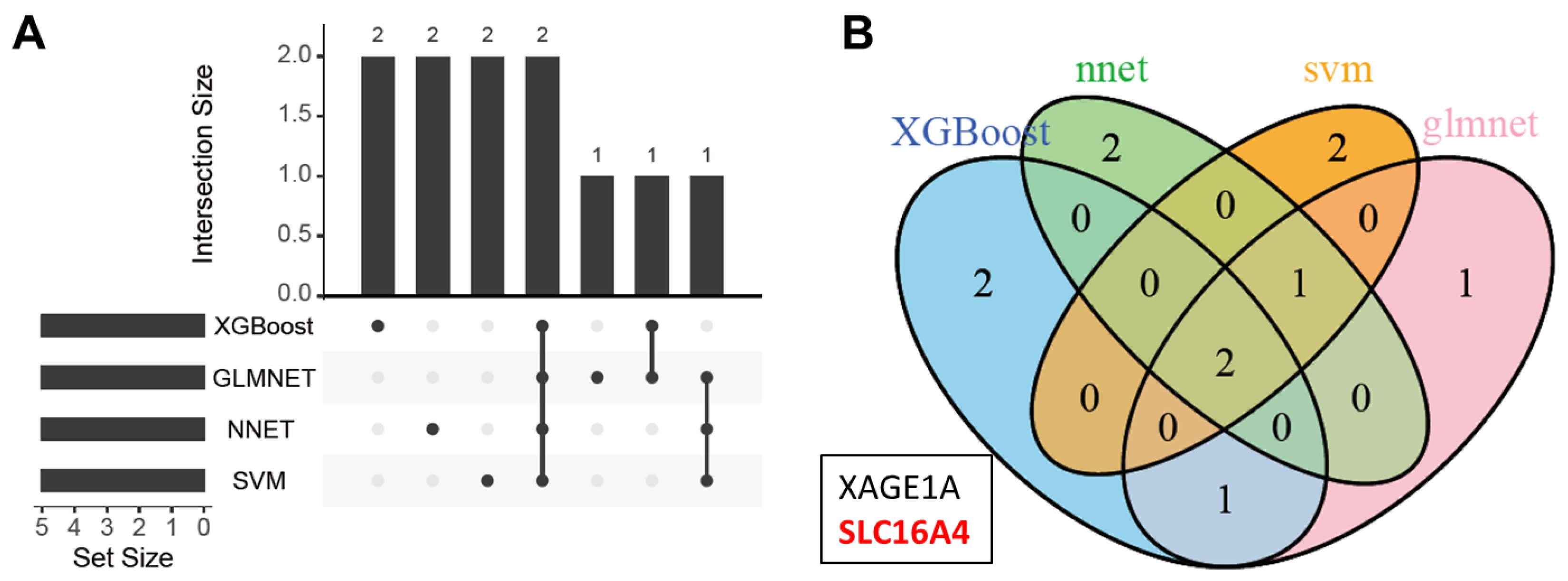
2.5. Key Protein Selection
2.6. Expression Analysis of SLC16A4
2.7. Correlation Analysis
2.8. Enrichment Analysis
2.9. Cell Culture
2.10. Overexpression Plasmid Construction
2.11. CCK8 Cell Proliferation Assay
2.12. EdU Cell Proliferation Assay
2.13. β-Galactosidase Staining for Cellular Senescence
2.14. Quantitative PCR (qPCR)
2.15. Statistical Analysis
3. Results
3.1. Dataset
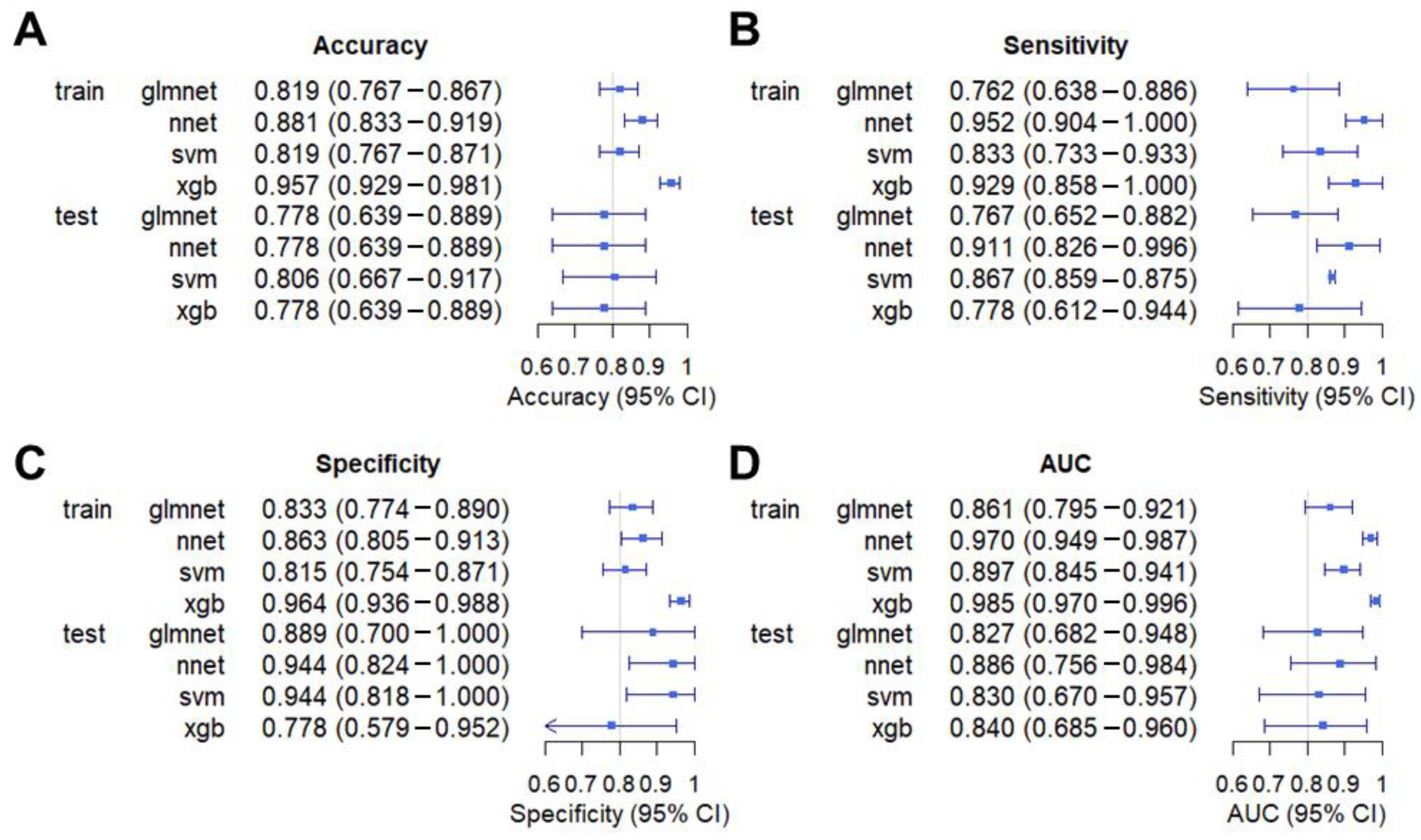
3.2. Model Construction
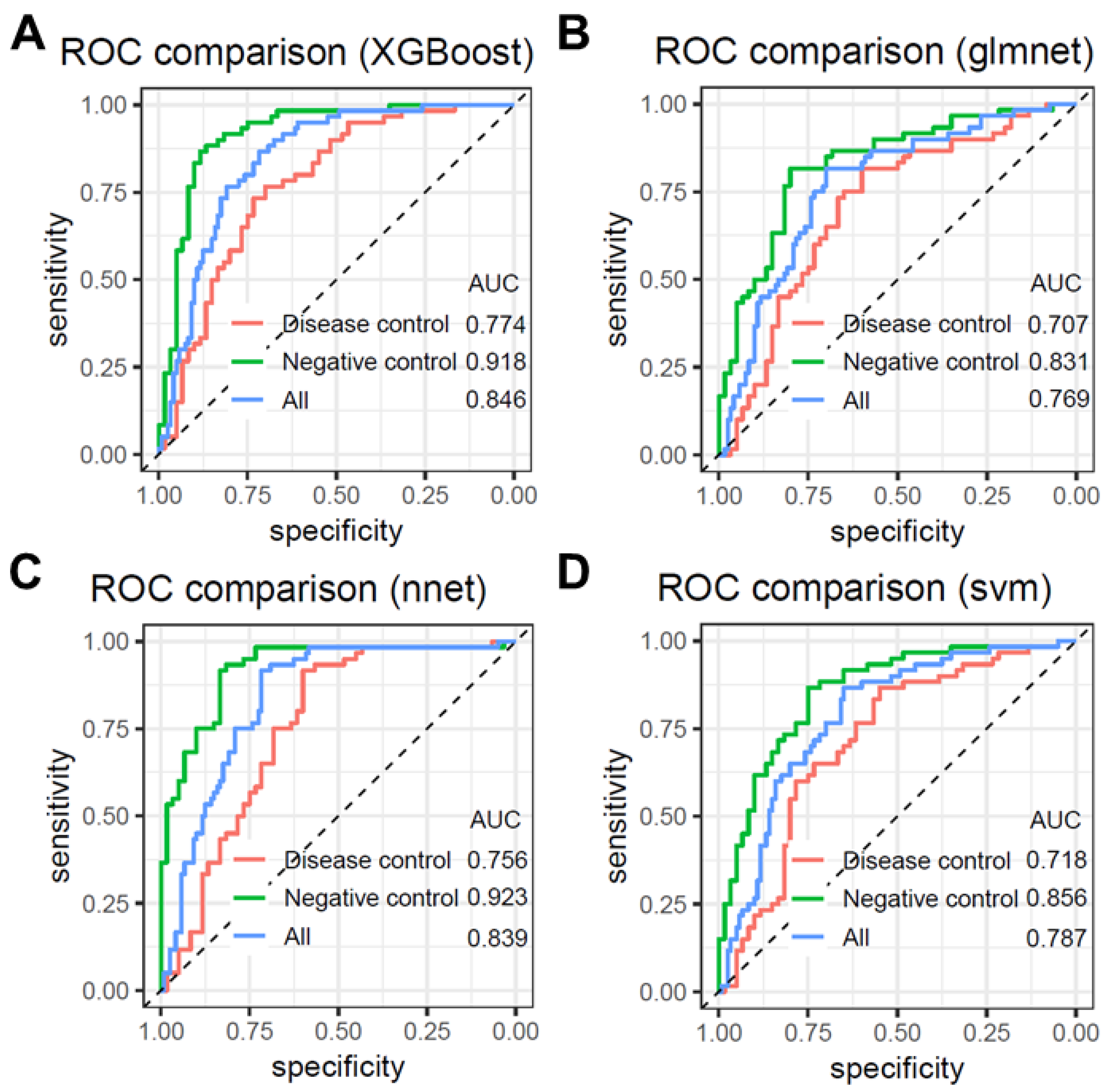
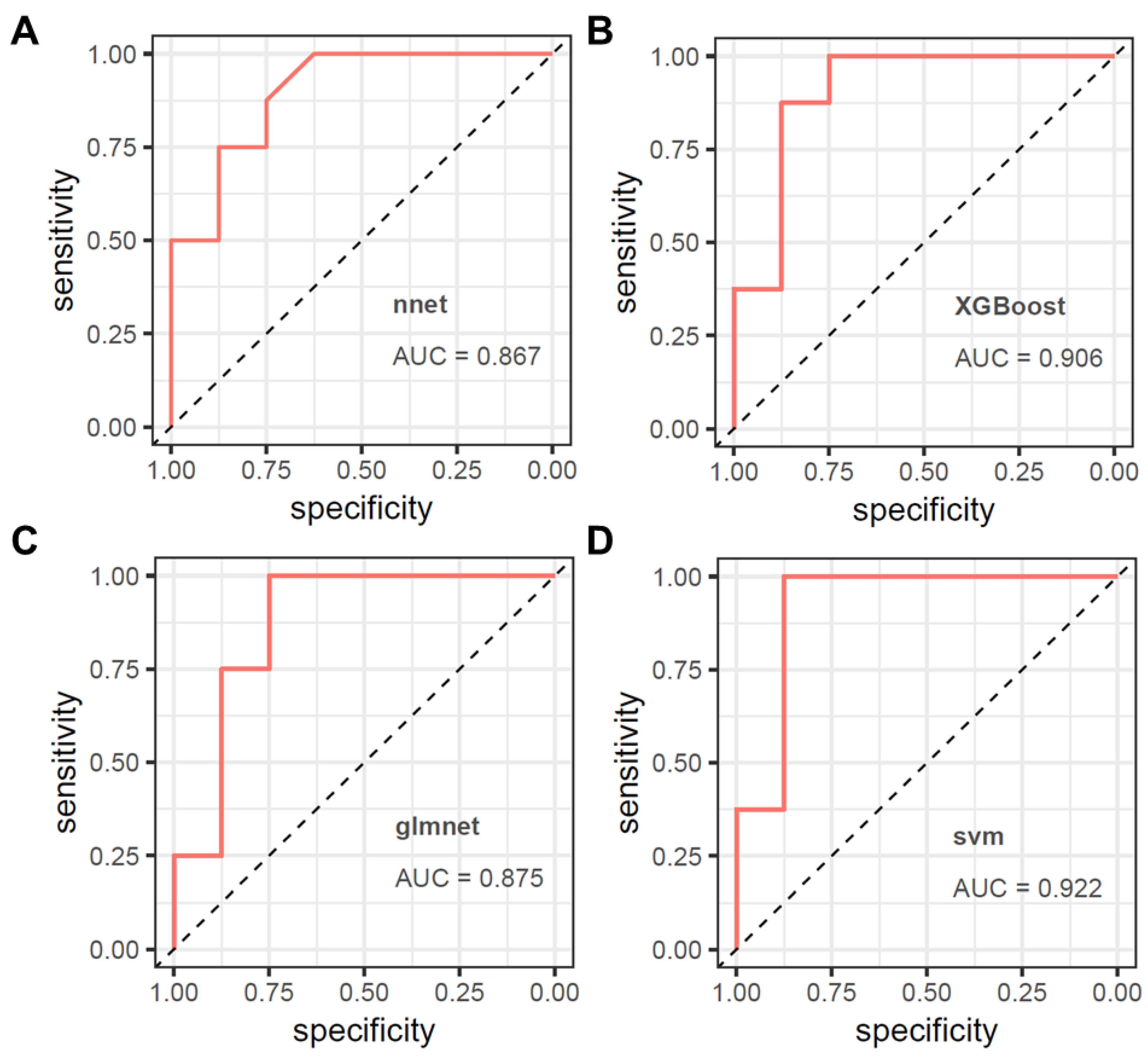
3.3. Model Validation
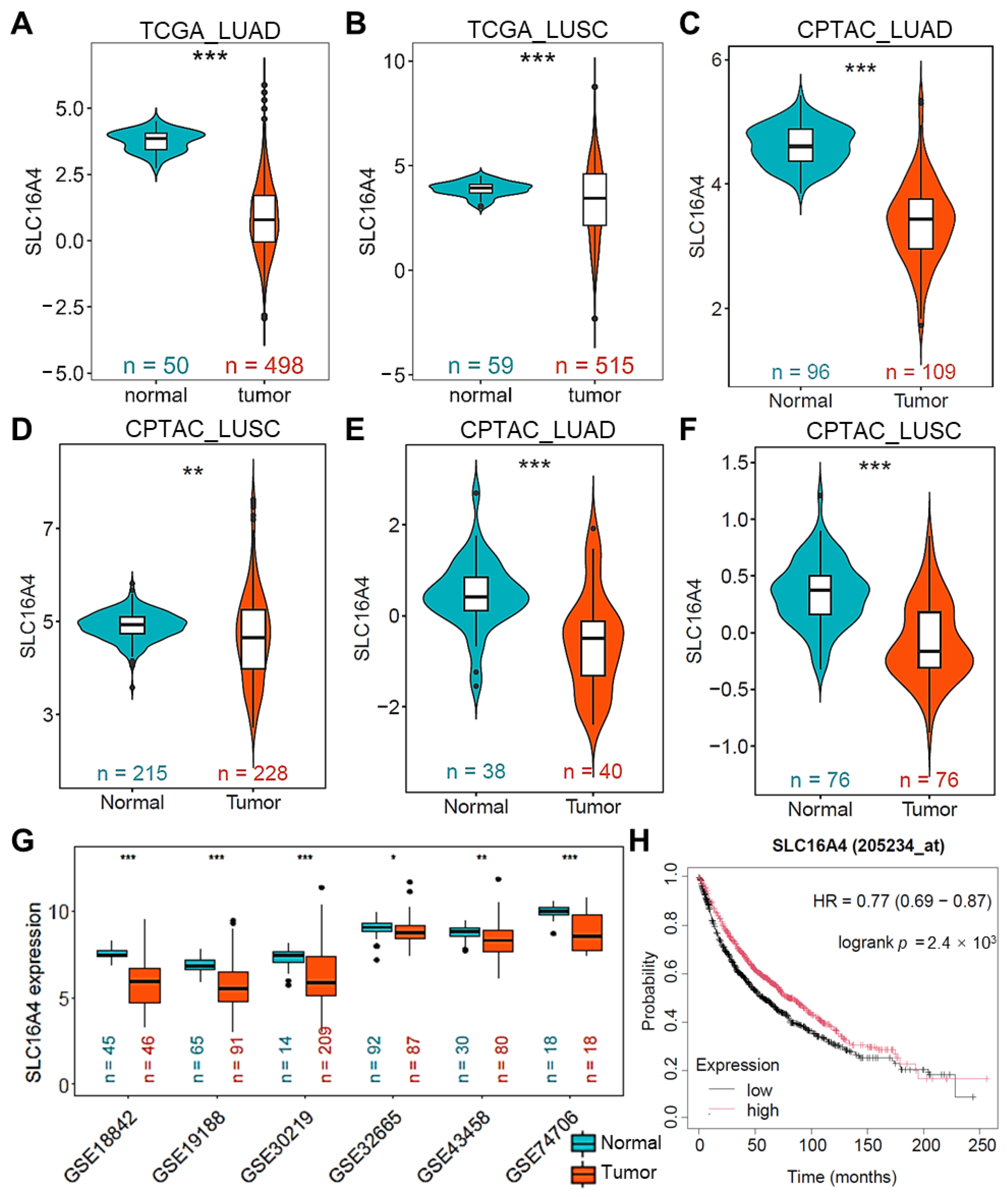
3.4. Downregulation of SLC16A4 in Lung Cancer
3.5. SLC16A4 Is Regulated by Copy Number Variation and DNA Methylation
3.6. Association of SLC16A4 with Clinical Characteristics in Lung Cancer
3.7. Association of SLC16A4 with Immune Cell Infiltration
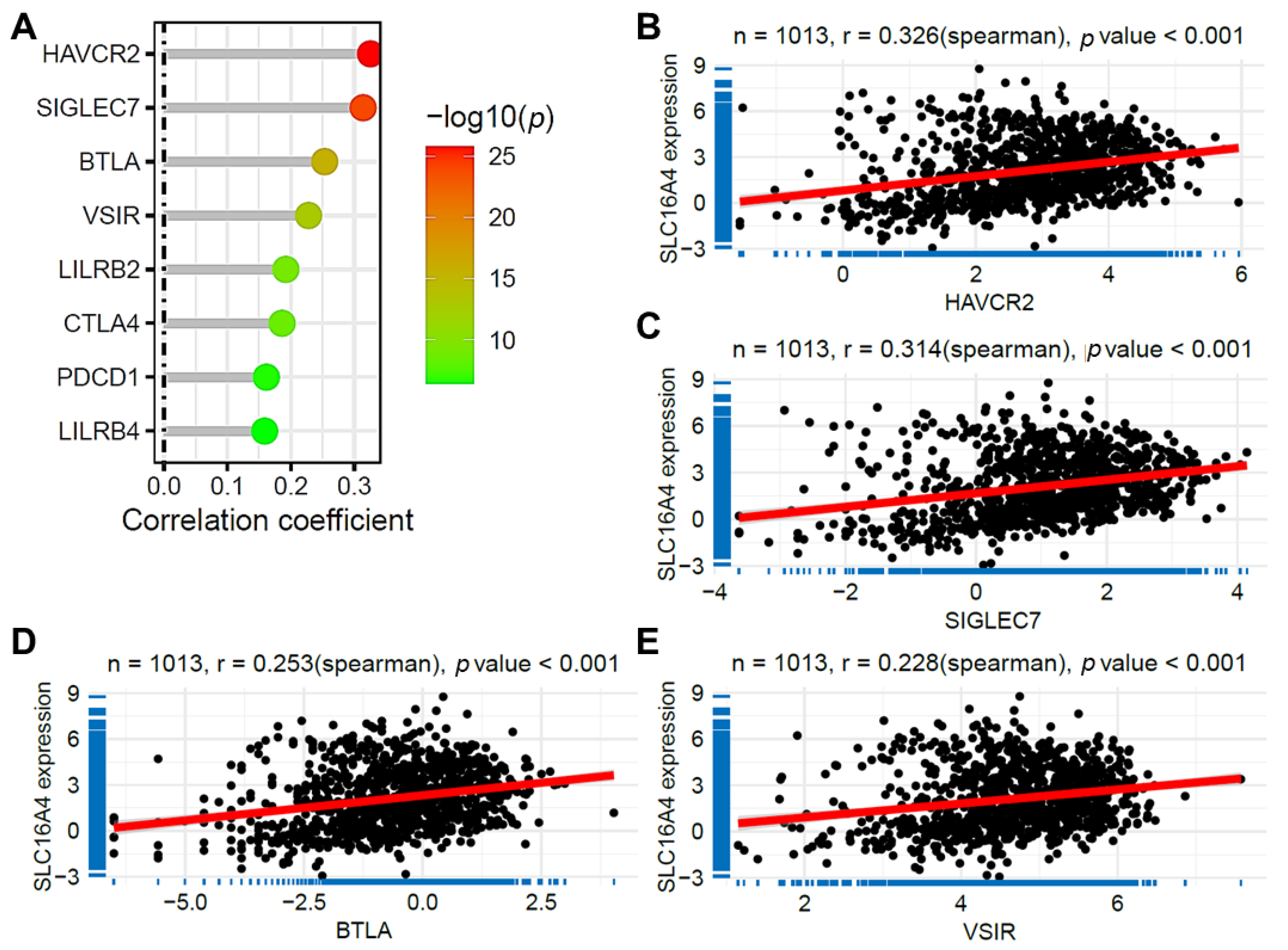
3.8. Correlation of SLC16A4 with Immune Checkpoint Gene Expression
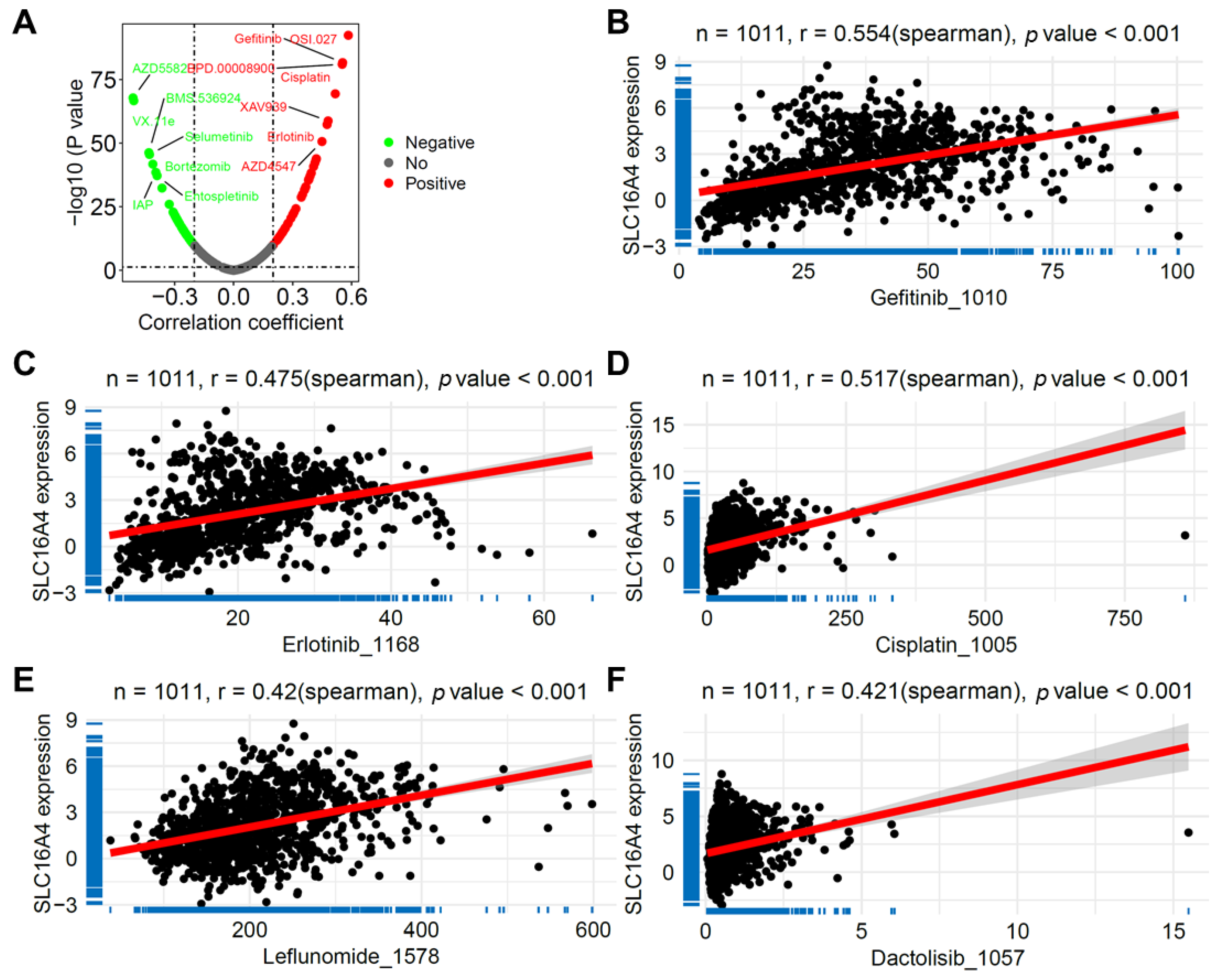
3.9. Correlation of SLC16A4 with Anticancer Drug Sensitivity
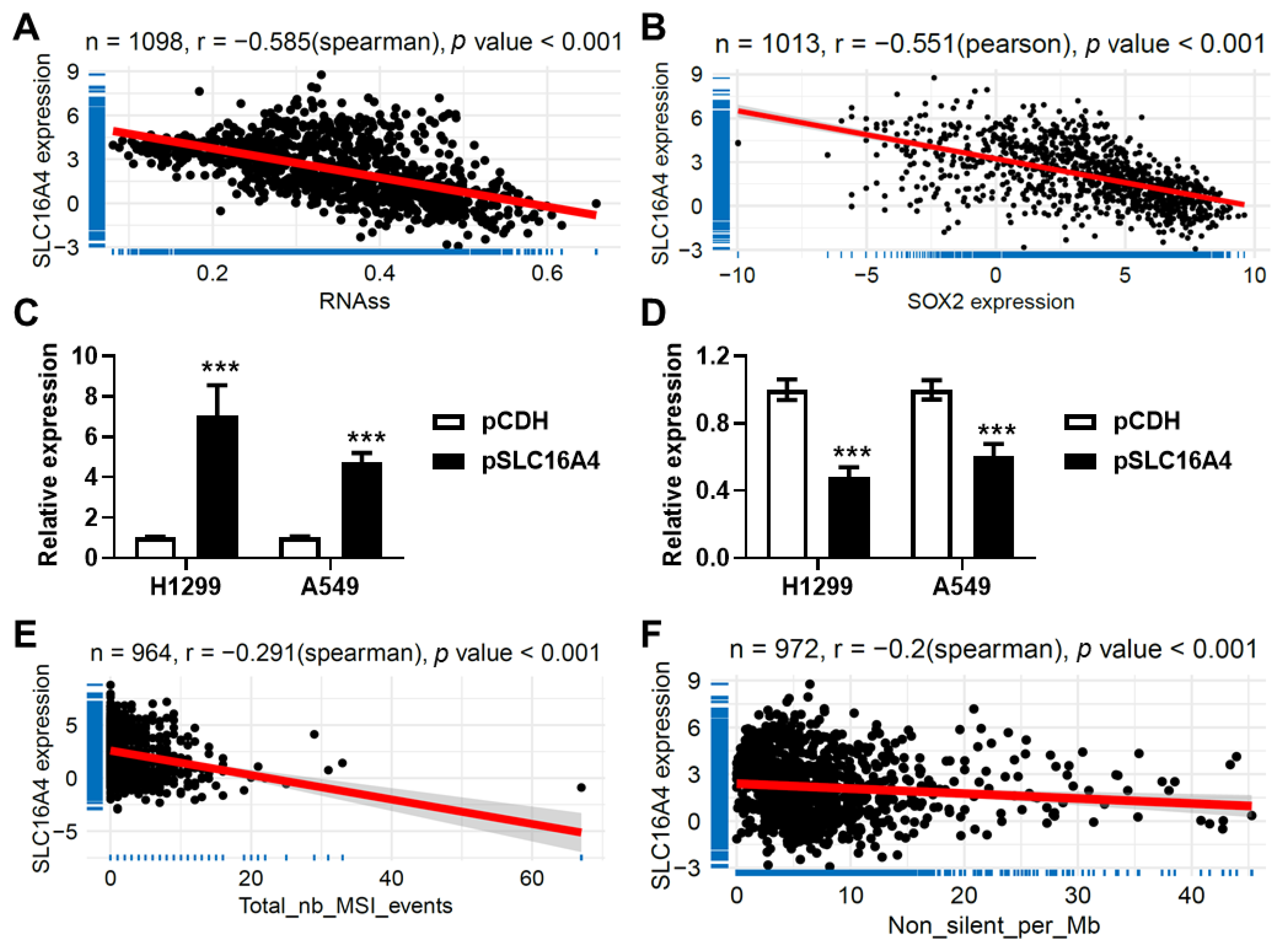
3.10. SLC16A4 Correlated with Tumor Stemness and Tumor Mutation Burden
3.11. SLC16A4 Correlated with Tumor-Related Biological Functions and Signaling Pathways
3.12. SLC16A4 Inhibits Cell Proliferation and Promotes Cell Senescence
4. Discussion
5. Conclusions
6. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ji, Y.; Zhang, Y.; Liu, S.; Li, J.; Jin, Q.; Wu, J.; Duan, H.; Liu, X.; Yang, L.; Huang, Y. The epidemiological landscape of lung cancer: Current status, temporal trend and future projections based on the latest estimates from GLOBOCAN 2022. J. Nat. Cancer Cent. 2025, 5, 278–286. [Google Scholar] [CrossRef]
- Leiter, A.; Veluswamy, R.R.; Wisnivesky, J.P. The global burden of lung cancer: Current status and future trends. Nat. Rev. Clin. Oncol. 2023, 20, 624–639. [Google Scholar] [CrossRef]
- Zhang, J.; Ijzerman, M.J.; Oberoi, J.; Karnchanachari, N.; Bergin, R.J.; Franchini, F.; Druce, P.; Wang, X.; Emery, J.D. Time to diagnosis and treatment of lung cancer: A systematic overview of risk factors, interventions and impact on patient outcomes. Lung Cancer 2022, 166, 27–39. [Google Scholar] [CrossRef]
- Prabhakar, B.; Shende, P.; Augustine, S. Current trends and emerging diagnostic techniques for lung cancer. Biomed. Pharmacother. 2018, 106, 1586–1599. [Google Scholar] [CrossRef]
- Aslam, B.; Basit, M.; Nisar, M.A.; Khurshid, M.; Rasool, M.H. Proteomics: Technologies and Their Applications. J. Chromatogr. Sci. 2017, 55, 182–196. [Google Scholar] [CrossRef]
- Chen, H.; Drapkin, B.J.; Minna, J.D. Proteomics: A new dimension to decode small cell lung cancer. Cell 2024, 187, 14–16. [Google Scholar] [CrossRef]
- Geyer, P.E.; Holdt, L.M.; Teupser, D.; Mann, M. Revisiting biomarker discovery by plasma proteomics. Mol. Syst. Biol. 2017, 13, 942. [Google Scholar] [CrossRef]
- Haug, C.J.; Drazen, J.M. Artificial Intelligence and Machine Learning in Clinical Medicine, 2023. N. Engl. J. Med. 2023, 388, 1201–1208. [Google Scholar] [CrossRef]
- Deo, R.C. Machine Learning in Medicine. Circulation 2015, 132, 1920–1930. [Google Scholar] [CrossRef]
- Obermeyer, Z.; Emanuel, E.J. Predicting the Future—Big Data, Machine Learning, and Clinical Medicine. N. Engl. J. Med. 2016, 375, 1216–1219. [Google Scholar] [CrossRef]
- Din, N.M.U.; Dar, R.A.; Rasool, M.; Assad, A. Breast cancer detection using deep learning: Datasets, methods, and challenges ahead. Comput. Biol. Med. 2022, 149, 106073. [Google Scholar] [CrossRef]
- Li, Y.; Wu, X.; Yang, P.; Jiang, G.; Luo, Y. Machine Learning for Lung Cancer Diagnosis, Treatment, and Prognosis. Genom. Proteom. Bioinform. 2022, 20, 850–866. [Google Scholar] [CrossRef]
- Jones, O.T.; Matin, R.N.; van der Schaar, M.; Prathivadi Bhayankaram, K.; Ranmuthu, C.K.I.; Islam, M.S.; Behiyat, D.; Boscott, R.; Calanzani, N.; Emery, J.; et al. Artificial intelligence and machine learning algorithms for early detection of skin cancer in community and primary care settings: A systematic review. Lancet Digit. Health 2022, 4, e466–e476. [Google Scholar] [CrossRef]
- Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Blagus, R.; Lusa, L. SMOTE for high-dimensional class-imbalanced data. BMC Bioinform. 2013, 14, 106. [Google Scholar] [CrossRef]
- Wang, J.; Song, X.; Wei, M.; Qin, L.; Zhu, Q.; Wang, S.; Liang, T.; Hu, W.; Zhu, X.; Li, J. PCAS: An Integrated Tool for Multi-Dimensional Cancer Research Utilizing Clinical Proteomic Tumor Analysis Consortium Data. Int. J. Mol. Sci. 2024, 25, 6690. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, J.; Zhao, M. ONGene: A literature-based database for human oncogenes. J. Genet. Genom. 2017, 44, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Shibru, B.; Fey, K.; Fricke, S.; Blaudszun, A.R.; Furst, F.; Weise, M.; Seiffert, S.; Weyh, M.K.; Kohl, U.; Sack, U.; et al. Detection of Immune Checkpoint Receptors—A Current Challenge in Clinical Flow Cytometry. Front. Immunol. 2021, 12, 694055. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Maeser, D.; Gruener, R.F.; Huang, R.S. oncoPredict: An R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief. Bioinform. 2021, 22, bbab260. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Spick, M.; Muazzam, A.; Pandha, H.; Michael, A.; Gethings, L.A.; Hughes, C.J.; Munjoma, N.; Plumb, R.S.; Wilson, I.D.; Whetton, A.D.; et al. Multi-omic diagnostics of prostate cancer in the presence of benign prostatic hyperplasia. Heliyon 2023, 9, e22604. [Google Scholar] [CrossRef]
- Peiffer-Smadja, N.; Rawson, T.M.; Ahmad, R.; Buchard, A.; Georgiou, P.; Lescure, F.X.; Birgand, G.; Holmes, A.H. Machine learning for clinical decision support in infectious diseases: A narrative review of current applications. Clin. Microbiol. Infect. 2020, 26, 584–595. [Google Scholar] [CrossRef]
- Guan, X.; Du, Y.; Ma, R.; Teng, N.; Ou, S.; Zhao, H.; Li, X. Construction of the XGBoost model for early lung cancer prediction based on metabolic indices. BMC Med. Inform. Decis. Mak. 2023, 23, 107. [Google Scholar] [CrossRef]
- Yu, D.; Liu, Z.; Su, C.; Han, Y.; Duan, X.; Zhang, R.; Liu, X.; Yang, Y.; Xu, S. Copy number variation in plasma as a tool for lung cancer prediction using Extreme Gradient Boosting (XGBoost) classifier. Thorac. Cancer 2020, 11, 95–102. [Google Scholar] [CrossRef]
- Wu, P.; Zhang, C.; Tang, X.; Li, D.; Zhang, G.; Zi, X.; Liu, J.; Yin, E.; Zhao, J.; Wang, P.; et al. Pan-cancer characterization of cell-free immune-related miRNA identified as a robust biomarker for cancer diagnosis. Mol. Cancer 2024, 23, 31. [Google Scholar] [CrossRef]
- Halestrap, A.P. The SLC16 gene family—Structure, role and regulation in health and disease. Mol. Asp. Med. 2013, 34, 337–349. [Google Scholar] [CrossRef]
- Felmlee, M.A.; Jones, R.S.; Rodriguez-Cruz, V.; Follman, K.E.; Morris, M.E. Monocarboxylate Transporters (SLC16): Function, Regulation, and Role in Health and Disease. Pharmacol. Rev. 2020, 72, 466–485. [Google Scholar] [CrossRef]
- Singh, M.; Afonso, J.; Sharma, D.; Gupta, R.; Kumar, V.; Rani, R.; Baltazar, F.; Kumar, V. Targeting monocarboxylate transporters (MCTs) in cancer: How close are we to the clinics? Semin. Cancer Biol. 2023, 90, 1–14. [Google Scholar] [CrossRef]
- Wang, L.; Lankhorst, L.; Bernards, R. Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer 2022, 22, 340–355. [Google Scholar] [CrossRef]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef]
- Pezzuto, A.; D’Ascanio, M.; Ricci, A.; Pagliuca, A.; Carico, E. Expression and role of p16 and GLUT1 in malignant diseases and lung cancer: A review. Thorac. Cancer 2020, 11, 3060–3070. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Training (n = 144) | Testing (n = 36) | Validating (n = 16) | All (n = 196) | p | |
|---|---|---|---|---|---|---|
| Age | >60 | 56 (42.75%) | 14 (41.18%) | 4 (25.00%) | 74 (40.88%) | 0.602 |
| ≤60 | 75 (57.25%) | 20 (58.82%) | 12 (75.00%) | 107 (59.12%) | ||
| gender | female | 49 (36.84%) | 18 (50.00%) | 10 (62.50%) | 77 (41.62%) | 0.161 |
| male | 84 (63.16%) | 18 (50.00%) | 6 (37.50%) | 108 (58.38%) | ||
| smoke | no | 24 (68.57%) | 11 (84.62%) | 8 (100.00%) | 43 (76.79%) | 0.242 |
| yes | 11 (31.43%) | 2 (15.38%) | 0 | 13 (23.21%) | ||
| surgery | no | 2 (5.71%) | 1 (7.69%) | 0 | 3 (5.36%) | 0.896 |
| yes | 33 (94.29%) | 12 (92.31%) | 8 (100.00%) | 53 (94.64%) | ||
| Disease type | Negative Control | 42 (29.17%) | 18 (50.00%) | 8 (50.00%) | 68 (34.69%) | NA |
| Disease control | 60 (41.67%) | 0 | 0 | 60 (30.61%) | ||
| Cancer | 42 (29.17%) | 18 (50.00%) | 8 (50.00%) | 68 (34.69%) | ||
| Characteristics | N | Expression | p | |
|---|---|---|---|---|
| Gender | female | 405 | 2.580 ± 1.879 | <0.001 |
| male | 608 | 1.812 ± 2.026 | ||
| Age | >60 | 720 | 2.121 ± 2.014 | 0.768 |
| ≤60 | 265 | 2.079 ± 1.986 | ||
| M | M0 | 753 | 2.049 ± 2.023 | 0.002 |
| M1 | 32 | 3.241 ± 2.021 | ||
| N | N0 | 648 | 2.103 ± 1.973 | 0.041 |
| N1 | 226 | 1.871 ± 2.013 | ||
| N2 | 114 | 2.530 ± 2.119 | ||
| N3 | 7 | 2.073 ± 1.375 | ||
| T | T1 | 282 | 2.505 ± 1.907 | 0.002 |
| T2 | 569 | 1.968 ± 1.976 | ||
| T3 | 118 | 1.911 ± 2.241 | ||
| T4 | 41 | 1.995 ± 1.992 | ||
| Stage | Stage I | 518 | 2.186 ± 1.914 | 0.001 |
| Stage II | 283 | 1.853 ± 2.053 | ||
| Stage III | 167 | 2.141 ± 2.089 | ||
| Stage IV | 33 | 3.246 ± 1.989 | ||
| Smoke.history | Current | 253 | 1.736 ± 1.910 | <0.001 |
| Never | 94 | 2.877 ± 1.819 | ||
| Reformed (>15) | 215 | 2.615 ± 2.149 | ||
| Reformed (≤15) | 416 | 1.914 ± 1.938 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, H.; Zhang, J.; Zhang, L.; Li, T.; Li, M.; Li, J.; Wang, J. Development of a Serum Proteomic-Based Diagnostic Model for Lung Cancer Using Machine Learning Algorithms and Unveiling the Role of SLC16A4 in Tumor Progression and Immune Response. Biomolecules 2025, 15, 1081. https://doi.org/10.3390/biom15081081
Hu H, Zhang J, Zhang L, Li T, Li M, Li J, Wang J. Development of a Serum Proteomic-Based Diagnostic Model for Lung Cancer Using Machine Learning Algorithms and Unveiling the Role of SLC16A4 in Tumor Progression and Immune Response. Biomolecules. 2025; 15(8):1081. https://doi.org/10.3390/biom15081081
Chicago/Turabian StyleHu, Hanqin, Jiaxin Zhang, Lisha Zhang, Tiancan Li, Miaomiao Li, Jianxiang Li, and Jin Wang. 2025. "Development of a Serum Proteomic-Based Diagnostic Model for Lung Cancer Using Machine Learning Algorithms and Unveiling the Role of SLC16A4 in Tumor Progression and Immune Response" Biomolecules 15, no. 8: 1081. https://doi.org/10.3390/biom15081081
APA StyleHu, H., Zhang, J., Zhang, L., Li, T., Li, M., Li, J., & Wang, J. (2025). Development of a Serum Proteomic-Based Diagnostic Model for Lung Cancer Using Machine Learning Algorithms and Unveiling the Role of SLC16A4 in Tumor Progression and Immune Response. Biomolecules, 15(8), 1081. https://doi.org/10.3390/biom15081081