
Molecular Fingerprint of Cold Adaptation in Antarctic Icefish PepT1 (Chionodraco hamatus): A Comparative Molecular Dynamics Study


Abstract
1. Introduction
2. Materials and Methods
2.1. Lipid Interaction Analysis and Order Parameters
2.2. Dynamic Cross-Correlation Analyses
3. Results
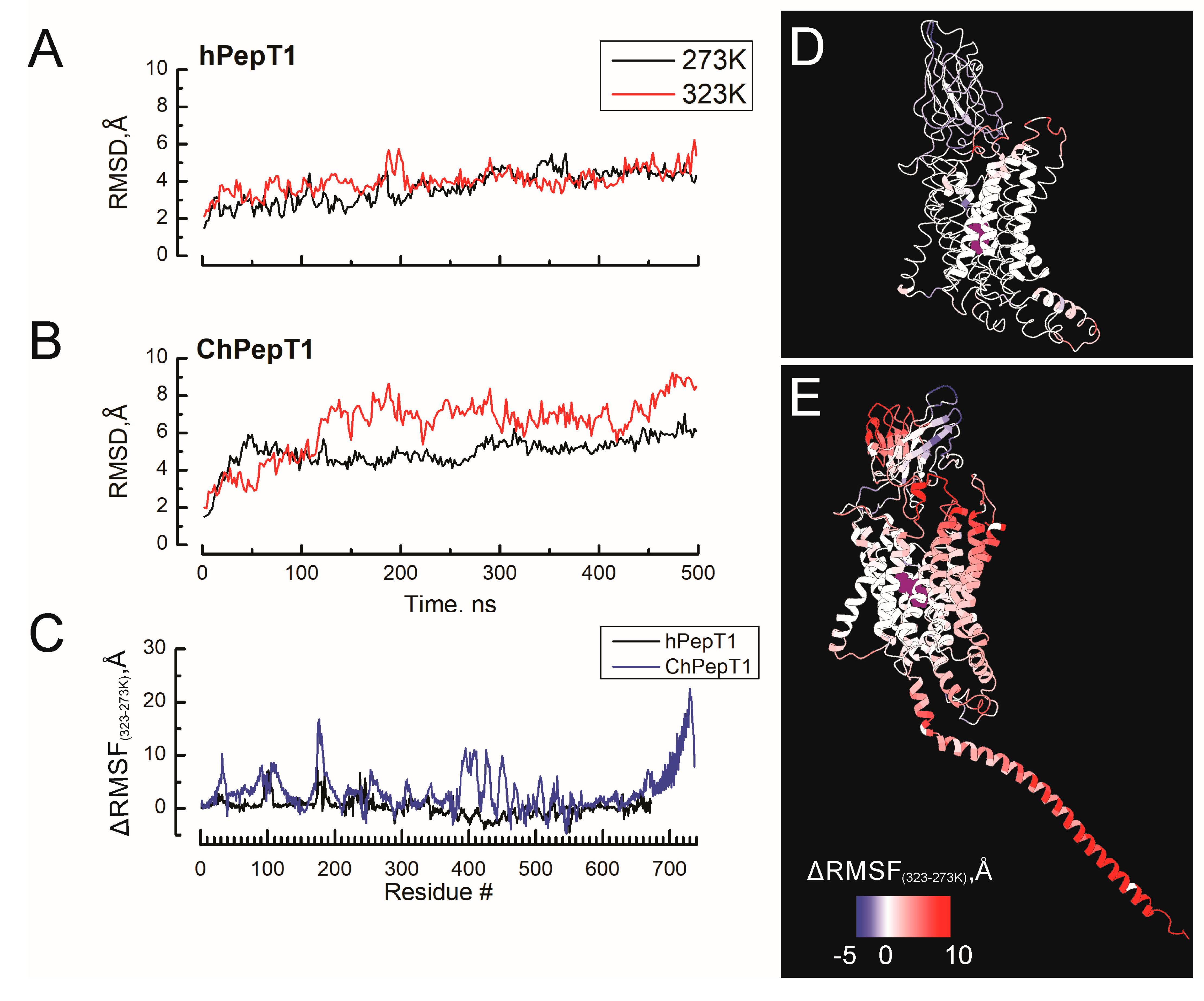
3.1. Enhanced Global Flexibility and Temperature-Responsive Dynamics in the Cold-Adapted ChPepT1
3.2. Thermodynamic Buffering of Substrate Binding Affinity in ChPepT1 Across Temperatures
3.3. Cold-Dependent Proteolipid Coupling and Membrane Flexibility in ChPepT1
3.4. Temperature-Induced Structural Remodeling and Membrane Decoupling of the C-Terminal Domain in ChPepT1
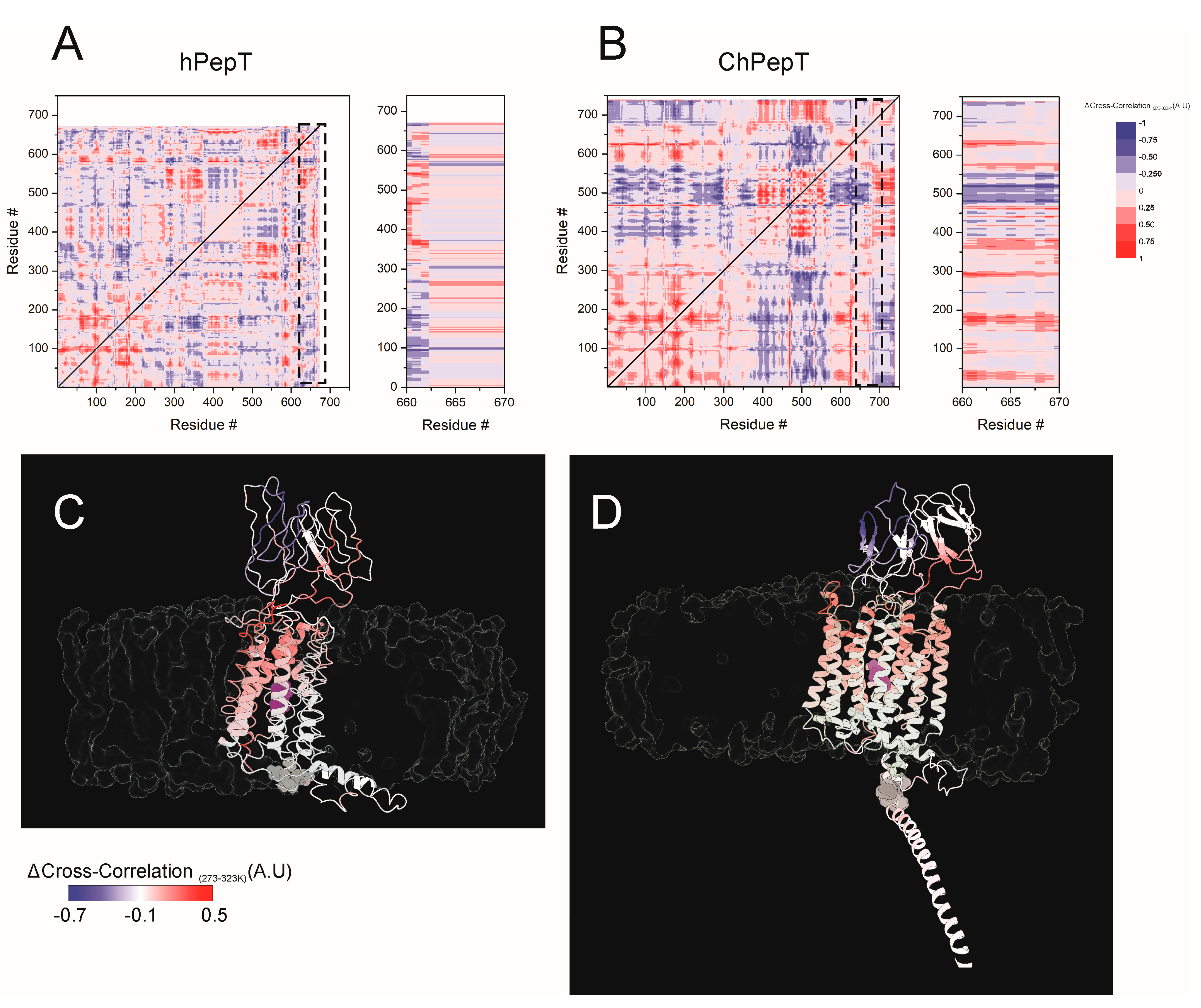
3.5. Temperature-Modulated Interdomain Coupling Reveals Dynamic Network Reorganization in ChPepT1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ChPepT1 | Chionodraco hamatus peptide transporter 1 |
| hPepT1 | Human peptide transporter 1 |
| CTD | C-terminal domain |
| ECD | Extracellular domain |
| TRS | Temperature-responsive segment |
| TM | Transmembrane (e.g., TM1, TM5) |
| MM/GBSA | Molecular Mechanics/Generalized Born Surface Area |
| RMSD | Root Mean Square Deviation |
| RMSF | Root Mean Square Fluctuation |
| MD | Molecular Dynamics |
| DCC | Dynamic Cross-Correlation |
| DCCM | Dynamic Cross-Correlation Matrix |
| SCD | Lipid order parameter (deuterium order parameter) |
| PME | Particle Mesh Ewald |
| LCPO | Linear Combination of Pairwise Overlaps |
| POPC | Palmitoyl-oleoyl-phosphatidylcholine |
| TIP3P | Transferable Intermolecular Potential with 3 Points |
| CHARMM-GUI | Chemistry at HARvard Macromolecular Mechanics—Graphical User Interface |
| VMD | Visual Molecular Dynamics |
| PDB | Protein Data Bank |
| NPT | Constant Number, Pressure, Temperature ensemble |
| NVT | Constant Number, Volume, Temperature ensemble |
| hASA | Hydrophobic Accessible Surface Area |
| Ea | Activation energy |
| ΔG | Gibbs free energy |
| ΔG‡ | Activation free energy barrier |
| ΔCC | Change in cross-correlation |
| ICD | Intracellular domain |
| pLLDT | Predicted Local Distance Difference Test (confidence score from AlphaFold) |
References
- Nguyen, V.; Wilson, C.; Hoemberger, M.; Stiller, J.B.; Agafonov, R.V.; Kutter, S.; English, J.; Theobald, D.L.; Kern, D. Evolutionary drivers of thermoadaptation in enzyme catalysis. Science 2017, 355, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Arcus, V.L.; Mulholland, A.J. Temperature, dynamics, and enzyme-catalyzed reaction rates. Annu. Rev. Biophys. 2020, 49, 163–180. [Google Scholar] [CrossRef] [PubMed]
- Fields, P.A.; Somero, G.N. Hot spots in cold adaptation: Localized increases in conformational flexibility in lactate dehydrogenase A4 orthologs of Antarctic notothenioid fishes. Proc. Natl. Acad. Sci. USA 1998, 95, 11476–11481. [Google Scholar] [CrossRef] [PubMed]
- Olufsen, M.; Smalås, A.O.; Moe, E.; Brandsdal, B.O. Increased flexibility as a strategy for cold adaptation: A comparative molecular dynamics study of cold- and warm-active uracil DNA glycosylase. J. Biol. Chem. 2005, 280, 18042–18048. [Google Scholar] [CrossRef] [PubMed]
- Fields, P.A. Review: Protein function at thermal extremes: Balancing stability and flexibility. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2001, 129, 417–431. [Google Scholar] [CrossRef] [PubMed]
- Tokuriki, N.; Tawfik, D.S. Protein dynamism and evolvability. Science 2009, 324, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Somero, G.N. Adaptation of enzymes to temperature: Searching for basic “strategies”. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2004, 139, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Cossins, A.R.; Behan, M.; Jones, G.; Bowler, K. Lipid-protein interactions in the adaptive regulation of membrane function. Biochem. Soc. Trans. 1987, 15, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Cossins, A.R.; Bowler, K.; Prosser, C.L. Homeoviscous adaptation and its effect upon membrane-bound proteins. J. Therm. Biol. 1981, 6, 183–187. [Google Scholar] [CrossRef]
- Else, P.L.; Wu, B.J. What role for membranes in determining the higher sodium pump molecular activity of mammals compared to ectotherms? J. Comp. Physiol. B Biochem. Syst. Environ. Physiol. 1999, 169, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.; Elias, C.; Xue, X.H.; Le, H.D.; Omelchenko, A.; Hryshko, L.V.; Tibbits, G.F. Determinants of cardiac Na+/Ca2+ exchanger temperature dependence: NH2-terminal transmembrane segments. Am. J. Physiol. Cell Physiol. 2002, 283, C512–C520. [Google Scholar] [CrossRef] [PubMed]
- Dode, L.; Van Baelen, K.; Wuytack, F.; Dean, W.L. Low Temperature Molecular Adaptation of the Skeletal Muscle Sarco(endo)plasmic Reticulum Ca2+-ATPase 1 (SERCA 1) in the Wood Frog (Rana sylvatica). J. Biol. Chem. 2001, 276, 3911–3919. [Google Scholar] [CrossRef] [PubMed]
- Galarza-Muñoz, G.; Soto-Morales, S.I.; Holmgren, M.; Rosenthal, J.J. Physiological adaptation of an Antarctic Na+/K+-ATPase to the cold. J. Exp. Biol. 2011, 214, 2164–2174. [Google Scholar] [CrossRef] [PubMed]
- Galarza-Muñoz, G.; Soto-Morales, S.I.; Jiao, S.; Holmgren, M.; Rosenthal, J.J.C. Molecular determinants for cold adaptation in an Antarctic Na+/K+-ATPase. Proc. Natl. Acad. Sci. USA 2023, 120, e2301207120. [Google Scholar] [CrossRef] [PubMed]
- Fei, Y.J.; Kanai, Y.; Nussberger, S.; Ganapathy, V.; Leibach, F.H.; Romero, M.F.; Singh, S.K.; Boron, W.F.; Hediger, M.A. Expression cloning of a mammalian proton-coupled oligopeptide transporter. Nature 1994, 368, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Rizzello, A.; Romano, A.; Kottra, G.; Acierno, R.; Storelli, C.; Verri, T.; Daniel, H.; Maffia, M. Protein cold adaptation strategy via a unique seven-amino acid domain in the icefish (Chionodraco hamatus) PEPT1 transporter. Proc. Natl. Acad. Sci. USA 2013, 110, 7068–7073. [Google Scholar] [CrossRef] [PubMed]
- Killer, M.; Wald, J.; Pieprzyk, J.; Marlovits, T.C.; Löw, C. Structural snapshots of human PepT1 and PepT2 reveal mechanistic insights into substrate and drug transport across epithelial membranes. Sci. Adv. 2021, 7, eabk3259. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell Jr, A.D. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [PubMed]
- Purisima, E.O.; Nilar, S.H. A simple yet accurate boundary element method for continuum dielectric calculations. J. Comput. Chem. 1995, 16, 681–689. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. Publ. Protein Soc. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Bai, Q.; Tan, S.; Xu, T.; Liu, H.; Huang, J.; Yao, X. MolAICal: A soft tool for 3D drug design of protein targets by artificial intelligence and classical algorithm. Brief. Bioinform. 2021, 22, bbaa161. [Google Scholar] [CrossRef] [PubMed]
- Douliez, J.P.; Léonard, A.; Dufourc, E.J. Restatement of order parameters in biomembranes: Calculation of C-C bond order parameters from C-D quadrupolar splittings. Biophys. J. 1995, 68, 1727–1739. [Google Scholar] [CrossRef] [PubMed]
- Ichiye, T.; Karplus, M. Collective motions in proteins: A covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins Struct. Funct. Bioinform. 1991, 11, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Arnold, G.E.; Ornstein, R.L. Molecular dynamics study of time-correlated protein domain motions and molecular flexibility: Cytochrome P450BM-3. Biophys. J. 1997, 73, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.J.; Rodrigues, A.P.C.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S.D. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, K.S.; Cavicchioli, R. Cold-adapted enzymes. Annu. Rev. Biochem. 2006, 75, 403–433. [Google Scholar] [CrossRef] [PubMed]
- Marx, J.C.; Collins, T.; D’Amico, S.; Feller, G.; Gerday, C. Cold-adapted enzymes from marine Antarctic microorganisms. Mar. Biotechnol. 2007, 9, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Zamora, R.A.; Ramirez-Sarmiento, C.A.; Castro-Fernández, V.; Villalobos, P.; Maturana, P.; Herrera-Morande, A.; Komives, E.A.; Guixé, V. Tuning of Conformational Dynamics Through Evolution-Based Design Modulates the Catalytic Adaptability of an Extremophilic Kinase. ACS Catal. 2020, 10, 10847–10857. [Google Scholar] [CrossRef]
- DasSarma, S.; Capes, M.D.; Karan, R.; DasSarma, P. Amino acid substitutions in cold-adapted proteins from Halorubrum lacusprofundi, an extremely halophilic microbe from antarctica. PLoS ONE 2013, 8, e58587. [Google Scholar] [CrossRef] [PubMed]
- Gourlay, L.J.; Mangiagalli, M.; Moroni, E.; Lotti, M.; Nardini, M. Structural determinants of cold activity and glucose tolerance of a family 1 glycoside hydrolase (GH1) from Antarctic Marinomonas sp. ef1. FEBS J. 2024, 291, 2897–2917. [Google Scholar] [CrossRef] [PubMed]
- Bossi, E.; Cherubino, F.; Margheritis, E.; Oyadeyi, A.; Vollero, A.; Peres, A. Temperature effects on the kinetic properties of the rabbit intestinal oligopeptide cotransporter PepT1. Pflügers Arch. Eur. J. Physiol. 2012, 464, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Renard, K.; Byrne, B. Insights into the Role of Membrane Lipids in the Structure, Function and Regulation of Integral Membrane Proteins. Int. J. Mol. Sci. 2021, 22, 9026. [Google Scholar] [CrossRef] [PubMed]
- Martens, C.; Stein, R.A.; Masureel, M.; Roth, A.; Mishra, S.; Dawaliby, R.; Konijnenberg, A.; Sobott, F.; Govaerts, C.; McHaourab, H.S. Lipids modulate the conformational dynamics of a secondary multidrug transporter. Nat. Struct. Mol. Biol. 2016, 23, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R.; Ursell, T.; Wiggins, P.; Sens, P. Emerging roles for lipids in shaping membrane-protein function. Nature 2009, 459, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Khameneh, A.J.; Kordzadeh, A.; Rastgoo, A.; Hadi, A. Investigation of the temperature effect on the properties of biological nanomembranes with different concentrations of cholesterol using molecular dynamics simulation. J. Mol. Graph. Model. 2025, 139, 109074. [Google Scholar] [CrossRef] [PubMed]
- Rath, S.L.; Tripathy, M.; Mandal, N. How Does Temperature Affect the Dynamics of SARS-CoV-2 M Proteins? Insights from Molecular Dynamics Simulations. J. Membr. Biol. 2022, 255, 341–356. [Google Scholar] [CrossRef] [PubMed]
- Sinensky, M. Homeoviscous adaptation—A homeostatic process that regulates the viscosity of membrane lipids in Escherichia coli. Proc. Natl. Acad. Sci. USA 1974, 71, 522–525. [Google Scholar] [CrossRef] [PubMed]
- D’Annessa, I.; Raniolo, S.; Limongelli, V.; Di Marino, D.; Colombo, G. Ligand Binding, Unbinding, and Allosteric Effects: Deciphering Small-Molecule Modulation of HSP90. J. Chem. Theory Comput. 2019, 15, 6368–6381. [Google Scholar] [CrossRef] [PubMed]
- Galdadas, I.; Qu, S.; Oliveira, A.S.F.; Olehnovics, E.; Mack, A.R.; Mojica, M.F.; Agarwal, P.K.; Tooke, C.L.; Gervasio, F.L.; Spencer, J.; et al. Allosteric communication in class A β-lactamases occurs via cooperative coupling of loop dynamics. eLife 2021, 10, e66567. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, B.; Li, J.; Yang, M.; Bai, Y.; Chen, K.; Chen, Z.; Mao, N. Mechanistic insights into the role of calcium in the allosteric regulation of the calmodulin-regulated death-associated protein kinase. Front. Mol. Biosci. 2022, 9, 1104942. [Google Scholar] [CrossRef] [PubMed]
- Feller, G.; Gerday, C. Psychrophilic enzymes: Molecular basis of cold adaptation. Cell. Mol. Life Sci. CMLS 1997, 53, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Hu, M.; Fan, X.; Ren, Z.; Portioli, C.; Yan, X.; Rong, M.; Zhou, M. Extracellular domain of PepT1 interacts with TM1 to facilitate substrate transport. Structure 2022, 30, 1035–1041.e1033. [Google Scholar] [CrossRef] [PubMed]
- Hochachka, P.W.; Somero, G.N. Biochemical Adaptation: Mechanism and Process in Physiological Evolution; Oxford University Press: Oxford, UK, 2002. [Google Scholar]
- Saavedra, H.G.; Wrabl, J.O.; Anderson, J.A.; Li, J.; Hilser, V.J. Dynamic allostery can drive cold adaptation in enzymes. Nature 2018, 558, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Apell, H.J.; Roudna, M. Partial Reactions of the Na,K-ATPase: Determination of Activation Energies and an Approach to Mechanism. J. Membr. Biol. 2020, 253, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.P.; De Giorgis, D.; Basilio, D.; Gadsby, D.C.; Rosenthal, J.J.; Latorre, R.; Holmgren, M.; Bezanilla, F. Energy landscape of the reactions governing the Na+ deeply occluded state of the Na+/K+-ATPase in the giant axon of the Humboldt squid. Proc. Natl. Acad. Sci. USA 2011, 108, 20556–20561. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| hPepT1 (kcal/mol) | chPepT1 (kcal/mol) | |||
|---|---|---|---|---|
| Energy Component | 273 K | 323 K | 273 K | 323 K |
| ∆E (Internal) | 0 | 0 | 0 | 0 |
| ∆E (Electrostatic) + ∆G(Solvation) | −9.3 | 0.23 | −15.9 | −16.05 |
| ∆E (VDW) | −15.4 | −16.06 | −16.17 | −11.9 |
| G (Binding) | −24.7 ± 0.14 | −15.8 ± 0.16 | −32 ± 0.52 | −27.9 ± 0.46 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrasco-Faus, G.; Márquez-Miranda, V.; Diaz-Franulic, I. Molecular Fingerprint of Cold Adaptation in Antarctic Icefish PepT1 (Chionodraco hamatus): A Comparative Molecular Dynamics Study. Biomolecules 2025, 15, 1058. https://doi.org/10.3390/biom15081058
Carrasco-Faus G, Márquez-Miranda V, Diaz-Franulic I. Molecular Fingerprint of Cold Adaptation in Antarctic Icefish PepT1 (Chionodraco hamatus): A Comparative Molecular Dynamics Study. Biomolecules. 2025; 15(8):1058. https://doi.org/10.3390/biom15081058
Chicago/Turabian StyleCarrasco-Faus, Guillermo, Valeria Márquez-Miranda, and Ignacio Diaz-Franulic. 2025. "Molecular Fingerprint of Cold Adaptation in Antarctic Icefish PepT1 (Chionodraco hamatus): A Comparative Molecular Dynamics Study" Biomolecules 15, no. 8: 1058. https://doi.org/10.3390/biom15081058
APA StyleCarrasco-Faus, G., Márquez-Miranda, V., & Diaz-Franulic, I. (2025). Molecular Fingerprint of Cold Adaptation in Antarctic Icefish PepT1 (Chionodraco hamatus): A Comparative Molecular Dynamics Study. Biomolecules, 15(8), 1058. https://doi.org/10.3390/biom15081058