

The Role of Alarmins in the Pathogenesis of Asthma

Abstract
1. Introduction
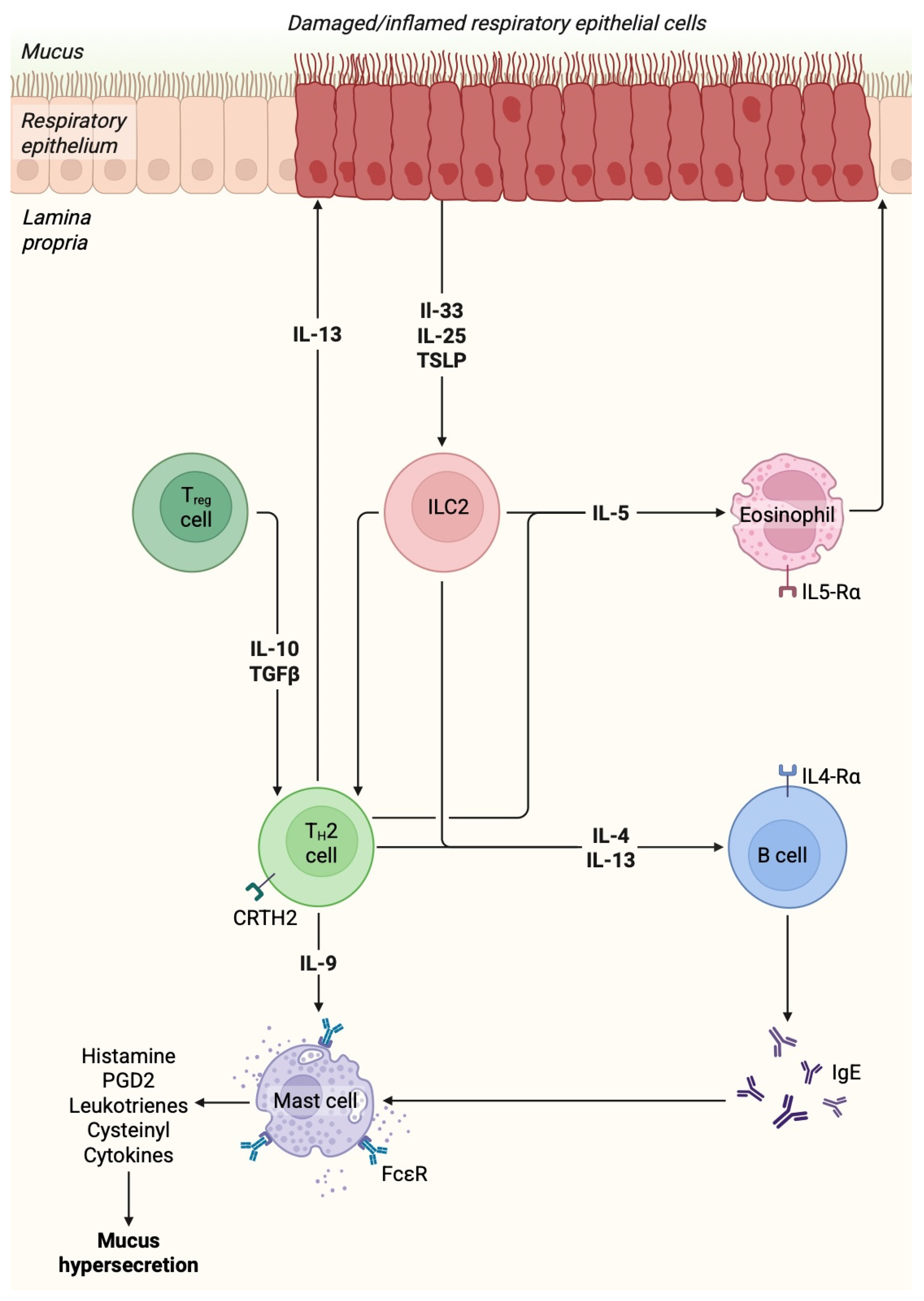
2. Pathogenesis and Pathophysiology of Asthma
3. Alarmins and Asthma
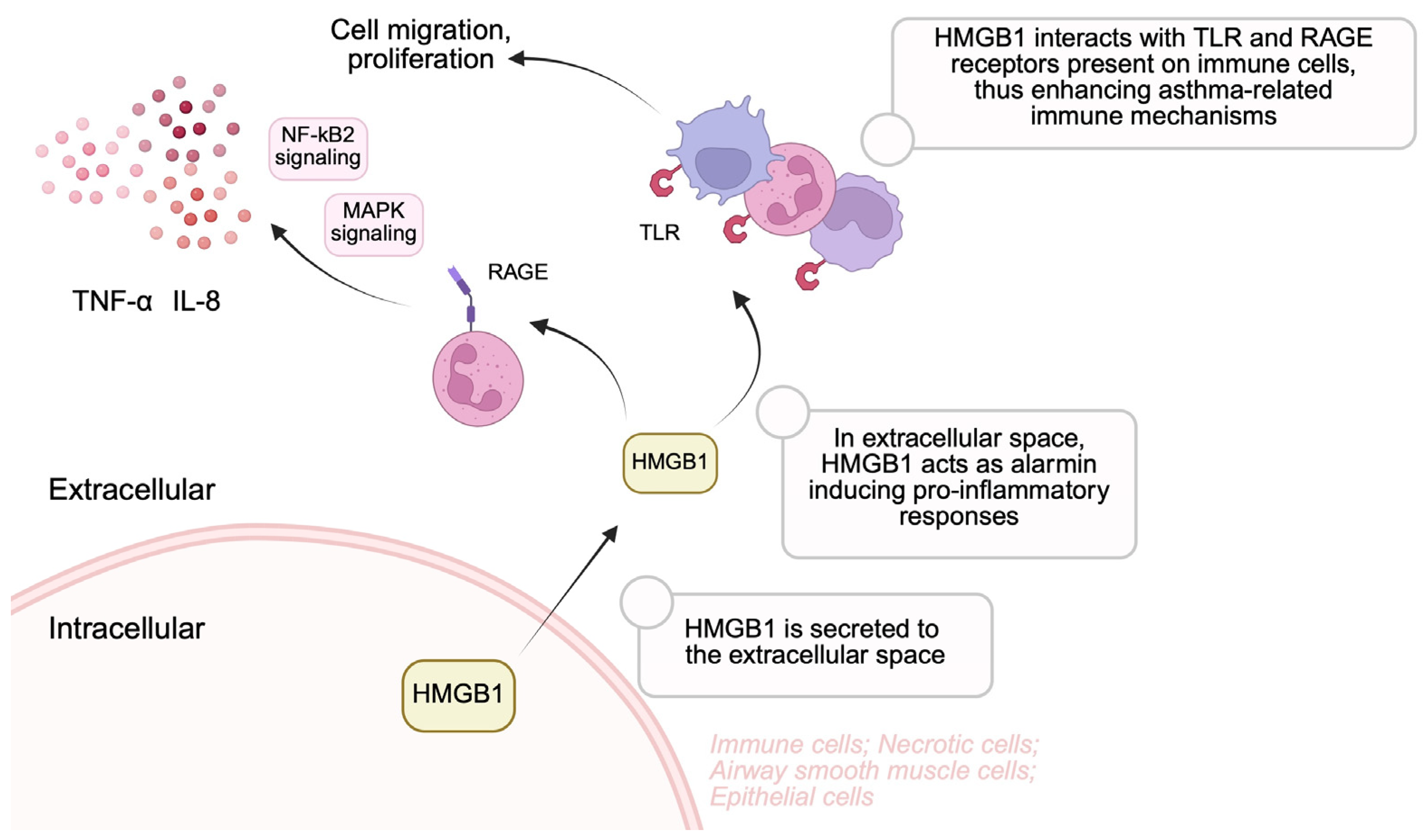
3.1. High-Mobility Group Box-1
3.2. S100
3.3. Interleukin-33
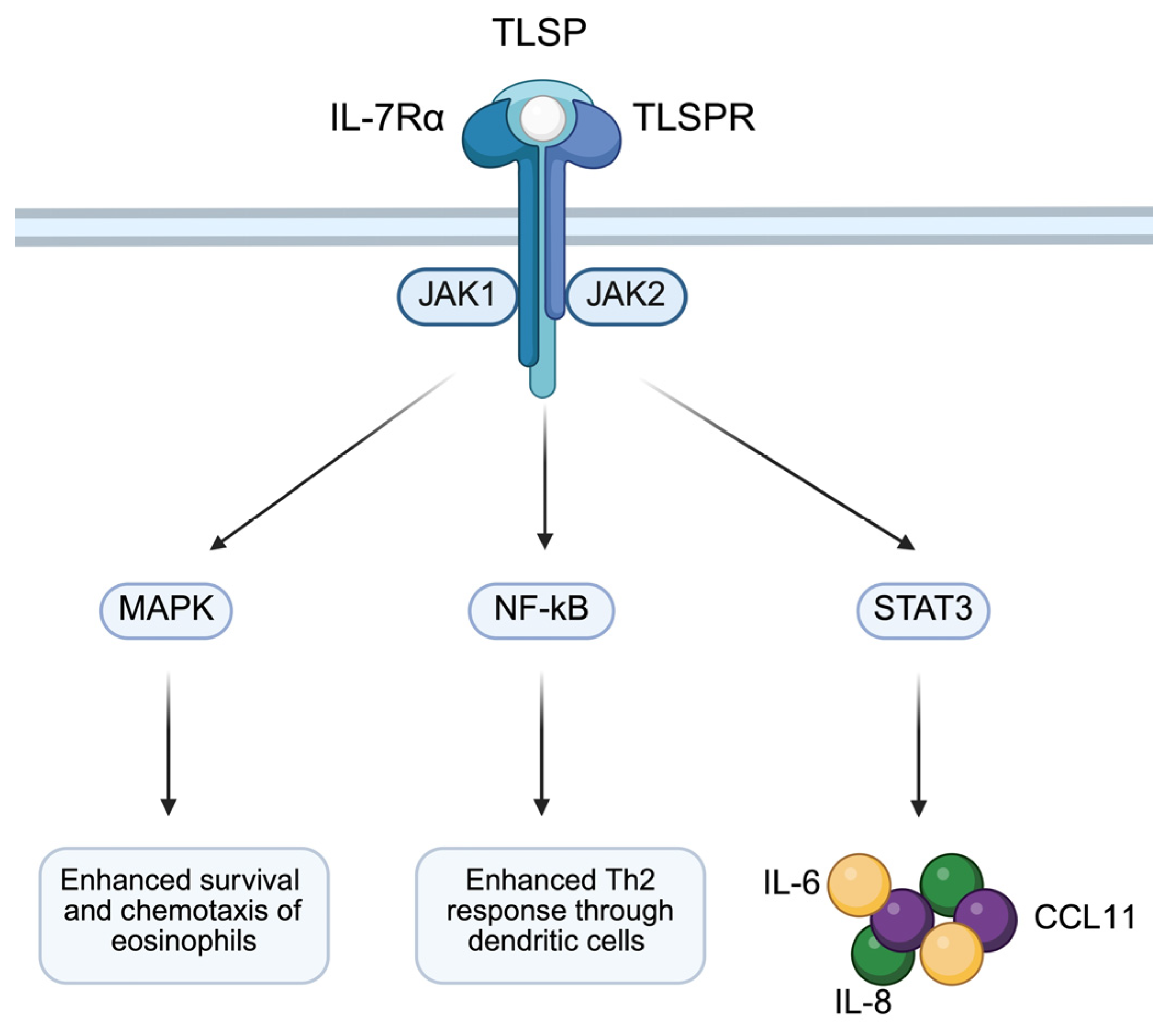
3.4. TSLP
3.5. Interleukin-25
3.6. Cathelicidin and Defensins
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| HMGB1 | High-mobility group box-1 |
| HMGN1 | High-mobility group nucleosome-binding domain 1 |
| HSP | Heat shock protein |
| TLR | Toll-like receptor |
| NF-κB | Nuclear factor kappa B |
| ILC2 | Group 2 innate lymphoid cells |
| IL | Interleukin |
| EEP | Eosinophil extracellular traps |
| RAGE | Receptor for advanced glycation end products |
References
- Porsbjerg, C.; Melen, E.; Lehtimaki, L.; Shaw, D. Asthma. Lancet 2023, 401, 858–873. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, M.; Rogliani, P.; Ora, J.; Calzetta, L.; Matera, M.G. Asthma and comorbidities: Recent advances. Pol. Arch. Intern. Med. 2022, 132, 16250. [Google Scholar] [CrossRef] [PubMed]
- Kuruvilla, M.E.; Lee, F.E.; Lee, G.B. Understanding Asthma Phenotypes, Endotypes, and Mechanisms of Disease. Clin. Rev. Allergy Immunol. 2019, 56, 219–233. [Google Scholar] [CrossRef]
- Taunk, S.T.; Cardet, J.C.; Ledford, D.K. Clinical implications of asthma endotypes and phenotypes. Allergy Asthma Proc. 2022, 43, 375–382. [Google Scholar] [CrossRef]
- Popović-Grle, S.; Štajduhar, A.; Lampalo, M.; Rnjak, D. Biomarkers in Different Asthma Phenotypes. Genes 2021, 12, 801. [Google Scholar] [CrossRef] [PubMed]
- Ricciardolo, F.L.M.; Sprio, A.E.; Baroso, A.; Gallo, F.; Riccardi, E.; Bertolini, F.; Carriero, V.; Arrigo, E.; Ciprandi, G. Characterization of T2-Low and T2-High Asthma Phenotypes in Real-Life. Biomedicines 2021, 9, 1684. [Google Scholar] [CrossRef]
- Nguyen, H.; Nasir, M. Management of Chronic Asthma in Adults. Prim. Care 2023, 50, 179–190. [Google Scholar] [CrossRef]
- Agache, I.; Akdis, C.A.; Akdis, M.; Canonica, G.W.; Casale, T.; Chivato, T.; Corren, J.; Chu, D.K.; Del Giacco, S.; Eiwegger, T.; et al. EAACI Biologicals Guidelines-Recommendations for severe asthma. Allergy 2021, 76, 14–44. [Google Scholar] [CrossRef]
- Yang, D.; Han, Z.; Oppenheim, J.J. Alarmins and immunity. Immunol. Rev. 2017, 280, 41–56. [Google Scholar] [CrossRef]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef]
- Kielbowski, K.; Skorka, P.; Plewa, P.; Bakinowska, E.; Pawlik, A. The Role of Alarmins in the Pathogenesis of Atherosclerosis and Myocardial Infarction. Curr. Issues Mol. Biol. 2024, 46, 8995–9015. [Google Scholar] [CrossRef] [PubMed]
- Kielbowski, K.; Stanska, W.; Bakinowska, E.; Rusinski, M.; Pawlik, A. The Role of Alarmins in the Pathogenesis of Rheumatoid Arthritis, Osteoarthritis, and Psoriasis. Curr. Issues Mol. Biol. 2024, 46, 3640–3675. [Google Scholar] [CrossRef] [PubMed]
- Rao, Z.; Liu, S.; Li, Z.; Wang, Q.; Gao, F.; Peng, H.; Ren, D.; Zang, Y.; Li, H.; Li, Y.; et al. Alarmin-loaded extracellular lipid droplets induce airway neutrophil infiltration during type 2 inflammation. Immunity 2024, 57, 2514–2529.e2517. [Google Scholar] [CrossRef]
- Werder, R.B.; Ullah, M.A.; Rahman, M.M.; Simpson, J.; Lynch, J.P.; Collinson, N.; Rittchen, S.; Rashid, R.B.; Sikder, M.A.A.; Handoko, H.Y.; et al. Targeting the P2Y(13) Receptor Suppresses IL-33 and HMGB1 Release and Ameliorates Experimental Asthma. Am. J. Respir. Crit. Care Med. 2022, 205, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Fahy, J.V. Type 2 inflammation in asthma--present in most, absent in many. Nat. Rev. Immunol. 2015, 15, 57–65. [Google Scholar] [CrossRef]
- Wechsler, M.E.; Scelo, G.; Larenas-Linnemann, D.E.S.; Torres-Duque, C.A.; Maspero, J.; Tran, T.N.; Murray, R.B.; Martin, N.; Menzies-Gow, A.N.; Hew, M.; et al. Association Between T2-related Comorbidities and Effectiveness of Biologics in Severe Asthma. Am. J. Respir. Crit. Care Med. 2024, 209, 262–272. [Google Scholar] [CrossRef]
- Lu, Y.; Huang, Y.; Li, J.; Huang, J.; Zhang, L.; Feng, J.; Li, J.; Xia, Q.; Zhao, Q.; Huang, L.; et al. Eosinophil extracellular traps drive asthma progression through neuro-immune signals. Nat. Cell Biol. 2021, 23, 1060–1072. [Google Scholar] [CrossRef]
- Du, X.; Li, F.; Zhang, C.; Li, N.; Huang, H.; Shao, Z.; Zhang, M.; Zhan, X.; He, Y.; Ju, Z.; et al. Eosinophil-derived chemokine (hCCL15/23, mCCL6) interacts with CCR1 to promote eosinophilic airway inflammation. Signal Transduct. Target. Ther. 2021, 6, 91. [Google Scholar] [CrossRef]
- Choi, Y.; Kim, Y.M.; Lee, H.R.; Mun, J.; Sim, S.; Lee, D.H.; Pham, D.L.; Kim, S.H.; Shin, Y.S.; Lee, S.W.; et al. Eosinophil extracellular traps activate type 2 innate lymphoid cells through stimulating airway epithelium in severe asthma. Allergy 2020, 75, 95–103. [Google Scholar] [CrossRef]
- Scott, G.; Asrat, S.; Allinne, J.; Keat Lim, W.; Nagashima, K.; Birchard, D.; Srivatsan, S.; Ajithdoss, D.K.; Oyejide, A.; Ben, L.H.; et al. IL-4 and IL-13, not eosinophils, drive type 2 airway inflammation, remodeling and lung function decline. Cytokine 2023, 162, 156091. [Google Scholar] [CrossRef]
- Olsthoorn, S.E.M.; van Krimpen, A.; Hendriks, R.W.; Stadhouders, R. Chronic Inflammation in Asthma: Looking Beyond the Th2 Cell. Immunol. Rev. 2025, 330, e70010. [Google Scholar] [CrossRef] [PubMed]
- Cheema, N.A.; Castagna, A.; Ambrosani, F.; Argentino, G.; Friso, S.; Zurlo, M.; Beri, R.; Maule, M.; Vaia, R.; Senna, G.; et al. Extracellular Vesicles in Asthma: Intercellular Cross-Talk in TH2 Inflammation. Cells 2025, 14, 542. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, T.; Iwata, A.; Furuya, H.; Kato, K.; Okabe, A.; Toda, Y.; Kanai, M.; Fujimura, L.; Sakamoto, A.; Kageyama, T.; et al. A distal enhancer of GATA3 regulates Th2 differentiation and allergic inflammation. Proc. Natl. Acad. Sci. USA 2024, 121, e2320727121. [Google Scholar] [CrossRef]
- Baba, R.; Kabata, H.; Shirasaki, Y.; Kamatani, T.; Yamagishi, M.; Irie, M.; Watanabe, R.; Matsusaka, M.; Masaki, K.; Miyata, J.; et al. Upregulation of IL-4 receptor signaling pathway in circulating ILC2s from asthma patients. J. Allergy Clin. Immunol. Glob. 2022, 1, 299–304. [Google Scholar] [CrossRef]
- Matera, M.G.; Ora, J.; Calzetta, L.; Rogliani, P.; Cazzola, M. Investigational anti IL-13 asthma treatments: A 2023 update. Expert. Opin. Investig. Drugs 2023, 32, 373–386. [Google Scholar] [CrossRef]
- Duffus, E.K.; Holguin, F.; Rastogi, D. Non-T2 asthma. Curr. Opin. Pulm. Med. 2025, 31, 287–293. [Google Scholar] [CrossRef]
- Goodwin, G.H.; Johns, E.W. Isolation and characterisation of two calf-thymus chromatin non-histone proteins with high contents of acidic and basic amino acids. Eur. J. Biochem. 1973, 40, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Kang, R.; Tang, D. The mechanism of HMGB1 secretion and release. Exp. Mol. Med. 2022, 54, 91–102. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, R. HMGB1 is a promising therapeutic target for asthma. Cytokine 2023, 165, 156171. [Google Scholar] [CrossRef]
- Furci, F.; Murdaca, G.; Pelaia, C.; Imbalzano, E.; Pelaia, G.; Caminati, M.; Allegra, A.; Senna, G.; Gangemi, S. TSLP and HMGB1: Inflammatory Targets and Potential Biomarkers for Precision Medicine in Asthma and COPD. Biomedicines 2023, 11, 437. [Google Scholar] [CrossRef]
- Yuan, S.; Liu, Z.; Xu, Z.; Liu, J.; Zhang, J. High mobility group box 1 (HMGB1): A pivotal regulator of hematopoietic malignancies. J. Hematol. Oncol. 2020, 13, 91. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Gauthier, A.; Daley, L.; Dial, K.; Wu, J.; Woo, J.; Lin, M.; Ashby, C.; Mantell, L.L. The Role of HMGB1, a Nuclear Damage-Associated Molecular Pattern Molecule, in the Pathogenesis of Lung Diseases. Antioxid. Redox Signal 2019, 31, 954–993. [Google Scholar] [CrossRef] [PubMed]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Balasubramaniam, V.R.M.T.; Othman, I.; Shaikh, M.F. Enlightening the role of high mobility group box 1 (HMGB1) in inflammation: Updates on receptor signalling. Eur. J. Pharmacol. 2019, 858, 172487. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Erlandsson-Harris, H.; Yang, H.; Tracey, K.J. HMGB1 as a DNA-binding cytokine. J. Leukoc. Biol. 2002, 72, 1084–1091. [Google Scholar] [CrossRef]
- Han, Z.; Junxu; Zhong, N. Expression of matrix metalloproteinases MMP-9 within the airways in asthma. Respir. Med. 2003, 97, 563–567. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.W.; Farkas, A.; Loughran, P.; Hoppe, G.; Bianchi, M.E.; Tracey, K.J.; Zeh, H.J.; et al. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 2010, 190, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Keyel, P.A. How is inflammation initiated? Individual influences of IL-1, IL-18 and HMGB1. Cytokine 2014, 69, 136–145. [Google Scholar] [CrossRef]
- Yang, Q.; Li, M.; Hou, Y.; He, H.; Sun, S. High-mobility group box 1 emerges as a therapeutic target for asthma. Immun. Inflamm. Dis. 2023, 11, e1124. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xu, Y.; Xiong, P.; Tan, Z.; Gong, F.; Hou, X.; Zheng, F. Effects of mimicked acetylated HMGB1 on macrophages and dendritic cells. Mol. Med. Rep. 2018, 18, 5527–5535. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, Z.A.; Armour, C.L.; Phipps, S.; Sukkar, M.B. RAGE and TLRs: Relatives, friends or neighbours? Mol. Immunol. 2013, 56, 739–744. [Google Scholar] [CrossRef]
- Perkins, T.N.; Donnell, M.L.; Oury, T.D. The axis of the receptor for advanced glycation endproducts in asthma and allergic airway disease. Allergy 2021, 76, 1350–1366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, J.; Pan, H.; Yang, K.; Hu, C. Astragaloside IV promotes the pyroptosis of airway smooth muscle cells in childhood asthma by suppressing HMGB1/RAGE axis to inactivate NF-κb pathway. Autoimmunity 2024, 57, 2387100. [Google Scholar] [CrossRef]
- Watanabe, T.; Asai, K.; Fujimoto, H.; Tanaka, H.; Kanazawa, H.; Hirata, K. Increased levels of HMGB-1 and endogenous secretory RAGE in induced sputum from asthmatic patients. Respir. Med. 2011, 105, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Chen, Q.; Yang, H.; Tracey, K.J.; Bustin, M.; Oppenheim, J.J. High mobility group box-1 protein induces the migration and activation of human dendritic cells and acts as an alarmin. J. Leukoc. Biol. 2007, 81, 59–66. [Google Scholar] [CrossRef]
- Xu, S.; Liu, W.; Zhang, L.; He, Q.; Ma, C.; Jiang, J.; Ye, S.; Ge, L.; Chen, Z.; Zhou, L. High mobility group box 1 levels as potential predictors of asthma severity. Chin. Med. J. 2023, 136, 1606–1608. [Google Scholar] [CrossRef] [PubMed]
- Manti, S.; Leonardi, S.; Parisi, G.F.; De Vivo, D.; Salpietro, A.; Spinuzza, A.; Arrigo, T.; Salpietro, C.; Cuppari, C. High mobility group box 1: Biomarker of inhaled corticosteroid treatment response in children with moderate-severe asthma. Allergy Asthma Proc. 2017, 38, 197–203. [Google Scholar] [CrossRef]
- Liu, W.; Li, L.; Piao, Y.; Wang, Z.; Dai, L.; Li, Y.; Piao, H.; Song, Y.; Cui, Q.; Wang, C.; et al. Mechanism of action of miR-15b-5p in alleviating asthma airway remodeling through the HMGB1/TLR4/IL-33 signaling axis. Int. Immunopharmacol. 2025, 145, 113753. [Google Scholar] [CrossRef]
- Shim, E.J.; Chun, E.; Lee, H.S.; Bang, B.R.; Kim, T.W.; Cho, S.H.; Min, K.U.; Park, H.W. The role of high-mobility group box-1 (HMGB1) in the pathogenesis of asthma. Clin. Exp. Allergy 2012, 42, 958–965. [Google Scholar] [CrossRef]
- Stenfeldt, A.L.; Wennerås, C. Danger signals derived from stressed and necrotic epithelial cells activate human eosinophils. Immunology 2004, 112, 605–614. [Google Scholar] [CrossRef]
- Brandt, E.B.; Lewkowich, I.P. RAGE-induced asthma: A role for the receptor for advanced glycation end-products in promoting allergic airway disease. J. Allergy Clin. Immunol. 2019, 144, 651–653. [Google Scholar] [CrossRef]
- Sun, J.; Jiang, Y.; Li, L.; Li, R.; Ling, F.; Du, X.; Han, Q.; Chu, S.; Liang, Y.; Mai, L.; et al. HMGB1/RAGE Signaling Regulates Th17/IL-17 and Its Role in Bronchial Epithelial-Mesenchymal Transformation. Curr. Mol. Med. 2024, 24, 1401–1412. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Hou, C.; Kong, J.; Wen, H.; Zheng, X.; Wu, L.; Huang, H.; Chen, Y. HMGB1 binding to receptor for advanced glycation end products enhances inflammatory responses of human bronchial epithelial cells by activating p38 MAPK and ERK1/2. Mol. Cell Biochem. 2015, 405, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Popa, C.; Netea, M.G.; van Riel, P.L.; van der Meer, J.W.; Stalenhoef, A.F. The role of TNF-alpha in chronic inflammatory conditions, intermediary metabolism, and cardiovascular risk. J. Lipid Res. 2007, 48, 751–762. [Google Scholar] [CrossRef]
- Asai, K.; Kanazawa, H.; Kamoi, H.; Shiraishi, S.; Hirata, K.; Yoshikawa, J. Increased levels of vascular endothelial growth factor in induced sputum in asthmatic patients. Clin. Exp. Allergy 2003, 33, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; O’Connor, B.; Ratoff, J.; Meng, Q.; Fang, C.; Cousins, D.; Zhang, G.; Gu, S.; Gao, Z.; Shamji, B.; et al. Expression and cellular provenance of thymic stromal lymphopoietin and chemokines in patients with severe asthma and chronic obstructive pulmonary disease. J. Immunol. 2008, 181, 2790–2798. [Google Scholar] [CrossRef]
- Zhou, Y.; Jiang, Y.Q.; Wang, W.X.; Zhou, Z.X.; Wang, Y.G.; Yang, L.; Ji, Y.L. HMGB1 and RAGE levels in induced sputum correlate with asthma severity and neutrophil percentage. Hum. Immunol. 2012, 73, 1171–1174. [Google Scholar] [CrossRef]
- Davies, D.E. The bronchial epithelium in chronic and severe asthma. Curr. Allergy Asthma Rep. 2001, 1, 127–133. [Google Scholar] [CrossRef]
- Yamasaki, A.; Okazaki, R.; Harada, T. Neutrophils and Asthma. Diagnostics 2022, 12, 1175. [Google Scholar] [CrossRef]
- Chang, J.; Xia, Y.; Wasserloos, K.; Deng, M.; Blose, K.J.; Vorp, D.A.; Turnquist, H.R.; Billiar, T.R.; Pitt, B.A.; Zhang, M.Z.; et al. Cyclic stretch induced IL-33 production through HMGB1/TLR-4 signaling pathway in murine respiratory epithelial cells. PLoS ONE 2017, 12, e0184770. [Google Scholar] [CrossRef]
- Fu, J.; Lin, S.H.; Wang, C.J.; Li, S.Y.; Feng, X.Y.; Liu, Q.; Xu, F. HMGB1 regulates IL-33 expression in acute respiratory distress syndrome. Int. Immunopharmacol. 2016, 38, 267–274. [Google Scholar] [CrossRef]
- Lee, C.C.; Lai, Y.T.; Chang, H.T.; Liao, J.W.; Shyu, W.C.; Li, C.Y.; Wang, C.N. Inhibition of high-mobility group box 1 in lung reduced airway inflammation and remodeling in a mouse model of chronic asthma. Biochem. Pharmacol. 2013, 86, 940–949. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.H.; Lee, Y.; Paik, M.J.; Yee, S.T. Inhibitions of HMGB1 and TLR4 alleviate DINP-induced asthma in mice. Toxicol. Res. 2019, 8, 621–629. [Google Scholar] [CrossRef]
- Hou, C.; Kong, J.; Liang, Y.; Huang, H.; Wen, H.; Zheng, X.; Wu, L.; Chen, Y. HMGB1 contributes to allergen-induced airway remodeling in a murine model of chronic asthma by modulating airway inflammation and activating lung fibroblasts. Cell Mol. Immunol. 2015, 12, 409–423. [Google Scholar] [CrossRef]
- Tang, H.; Zhao, H.; Song, J.; Dong, H.; Yao, L.; Liang, Z.; LV, Y.; Zou, F.; Cai, S. Ethyl pyruvate decreases airway neutrophil infiltration partly through a high mobility group box 1-dependent mechanism in a chemical-induced murine asthma model. Int. Immunopharmacol. 2014, 21, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, N.; Wang, T.; Dai, B.; Shang, Y. Vitamin D reduces inflammatory response in asthmatic mice through HMGB1/TLR4/NF-κB signaling pathway. Mol. Med. Rep. 2018, 17, 2915–2920. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.W. A soluble protein characteristic of the nervous system. Biochem. Biophys. Res. Commun. 1965, 19, 739–744. [Google Scholar] [CrossRef]
- Sattar, Z.; Lora, A.; Jundi, B.; Railwah, C.; Geraghty, P. The S100 Protein Family as Players and Therapeutic Targets in Pulmonary Diseases. Pulm. Med. 2021, 2021, 5488591. [Google Scholar] [CrossRef]
- Kozlyuk, N.; Monteith, A.J.; Garcia, V.; Damo, S.M.; Skaar, E.P.; Chazin, W.J. S100 Proteins in the Innate Immune Response to Pathogens. Methods Mol. Biol. 2019, 1929, 275–290. [Google Scholar] [CrossRef]
- Gonzalez, L.L.; Garrie, K.; Turner, M.D. Role of S100 proteins in health and disease. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118677. [Google Scholar] [CrossRef]
- Marenholz, I.; Heizmann, C.W.; Fritz, G. S100 proteins in mouse and man: From evolution to function and pathology (including an update of the nomenclature). Biochem. Biophys. Res. Commun. 2004, 322, 1111–1122. [Google Scholar] [CrossRef]
- Singh, P.; Ali, S.A. Multifunctional Role of S100 Protein Family in the Immune System: An Update. Cells 2022, 11, 2274. [Google Scholar] [CrossRef] [PubMed]
- Cerón, J.J.; Ortín-Bustillo, A.; López-Martínez, M.J.; Martínez-Subiela, S.; Eckersall, P.D.; Tecles, F.; Tvarijonaviciute, A.; Muñoz-Prieto, A. S-100 Proteins: Basics and Applications as Biomarkers in Animals with Special Focus on Calgranulins (S100A8, A9, and A12). Biology 2023, 12, 881. [Google Scholar] [CrossRef] [PubMed]
- Gilston, B.A.; Skaar, E.P.; Chazin, W.J. Binding of transition metals to S100 proteins. Sci. China Life Sci. 2016, 59, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, L.C.; Donor, M.T.; Prell, J.S.; Harms, M.J. Multiple Evolutionary Origins of Ubiquitous Cu2+ and Zn2+ Binding in the S100 Protein Family. PLoS ONE 2016, 11, e0164740. [Google Scholar] [CrossRef]
- Sedaghat, F.; Notopoulos, A. S100 protein family and its application in clinical practice. Hippokratia 2008, 12, 198–204. [Google Scholar]
- Zimmer, D.B.; Eubanks, J.O.; Ramakrishnan, D.; Criscitiello, M.F. Evolution of the S100 family of calcium sensor proteins. Cell Calcium 2013, 53, 170–179. [Google Scholar] [CrossRef]
- Schenten, V.; Plançon, S.; Jung, N.; Hann, J.; Bueb, J.L.; Bréchard, S.; Tschirhart, E.J.; Tolle, F. Secretion of the Phosphorylated Form of S100A9 from Neutrophils Is Essential for the Proinflammatory Functions of Extracellular S100A8/A9. Front. Immunol. 2018, 9, 447. [Google Scholar] [CrossRef]
- Sprenkeler, E.G.G.; Zandstra, J.; van Kleef, N.D.; Goetschalckx, I.; Verstegen, B.; Aarts, C.E.M.; Janssen, H.; Tool, A.T.J.; van Mierlo, G.; van Bruggen, R.; et al. S100A8/A9 Is a Marker for the Release of Neutrophil Extracellular Traps and Induces Neutrophil Activation. Cells 2022, 11, 236. [Google Scholar] [CrossRef]
- Quoc, Q.L.; Choi, Y.; Thi Bich, T.C.; Yang, E.M.; Shin, Y.S.; Park, H.S. S100A9 in adult asthmatic patients: A biomarker for neutrophilic asthma. Exp. Mol. Med. 2021, 53, 1170–1179. [Google Scholar] [CrossRef]
- Huang, C.; Zheng, D.; Fu, C.; Cai, Z.; Zhang, H.; Xie, Z.; Luo, L.; Li, H.; Huang, Y.; Chen, J. Secreted S100A4 causes asthmatic airway epithelial barrier dysfunction induced by house dust mite extracts via activating VEGFA/VEGFR2 pathway. Environ. Toxicol. 2023, 38, 1431–1444. [Google Scholar] [CrossRef]
- Shihui, M.; Shirong, Y.; Jing, L.; Jingjing, H.; Tongqian, W.; Tian, T.; Chenyu, W.; Fang, Y. S100A4 reprofiles lipid metabolism in mast cells via RAGE and PPAR-γ signaling pathway. Int. Immunopharmacol. 2024, 128, 111555. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, W.; Gunst, S.J. S100A4 is secreted by airway smooth muscle tissues and activates inflammatory signaling pathways via receptors for advanced glycation end products. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 319, L185–L195. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Liu, S.; Li, F.; Li, C.; Chen, S.; Gao, X.; Wang, X. The miR-124-3p regulates the allergic airway inflammation and remodeling in an ovalbumin-asthmatic mouse model by inhibiting S100A4. Immun. Inflamm. Dis. 2023, 11, e730. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Ma, L.; Jin, X.; He, J.; Chen, K.; Zhang, D.; Yuan, R.; Yang, J.; Zhong, Q.; Zhou, H.; et al. S100A4 Is Critical for a Mouse Model of Allergic Asthma by Impacting Mast Cell Activation. Front. Immunol. 2021, 12, 692733. [Google Scholar] [CrossRef]
- Cheng, M.; Shi, Y.L.; Shang, P.P.; Chen, Y.J.; Xu, Y.D. Inhibitory Effect of S100A11 on Airway Smooth Muscle Contraction and Airway Hyperresponsiveness. Curr. Med. Sci. 2022, 42, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Gray, R.D.; MacGregor, G.; Noble, D.; Imrie, M.; Dewar, M.; Boyd, A.C.; Innes, J.A.; Porteous, D.J.; Greening, A.P. Sputum proteomics in inflammatory and suppurative respiratory diseases. Am. J. Respir. Crit. Care Med. 2008, 178, 444–452. [Google Scholar] [CrossRef]
- Lee, T.H.; Chang, H.S.; Bae, D.J.; Song, H.J.; Kim, M.S.; Park, J.S.; Jun, J.A.; Lee, S.Y.; Uh, S.T.; Kim, S.H.; et al. Role of S100A9 in the development of neutrophilic inflammation in asthmatics and in a murine model. Clin. Immunol. 2017, 183, 158–166. [Google Scholar] [CrossRef]
- Eguíluz-Gracia, I.; Malmstrom, K.; Dheyauldeen, S.A.; Lohi, J.; Sajantila, A.; Aaløkken, R.; Sundaram, A.Y.M.; Gilfillan, G.D.; Makela, M.; Baekkevold, E.S.; et al. Monocytes accumulate in the airways of children with fatal asthma. Clin. Exp. Allergy 2018, 48, 1631–1639. [Google Scholar] [CrossRef]
- Aoki, T.; Matsumoto, Y.; Hirata, K.; Ochiai, K.; Okada, M.; Ichikawa, K.; Shibasaki, M.; Arinami, T.; Sumazaki, R.; Noguchi, E. Expression profiling of genes related to asthma exacerbations. Clin. Exp. Allergy 2009, 39, 213–221. [Google Scholar] [CrossRef]
- Wang, C.H.; Punde, T.H.; Huang, C.D.; Chou, P.C.; Huang, T.T.; Wu, W.H.; Liu, C.H.; Chung, K.F.; Kuo, H.P. Fibrocyte trafficking in patients with chronic obstructive asthma and during an acute asthma exacerbation. J. Allergy Clin. Immunol. 2015, 135, 1154–1162.e1151–1155. [Google Scholar] [CrossRef]
- Yang, Z.; Yan, W.X.; Cai, H.; Tedla, N.; Armishaw, C.; Di Girolamo, N.; Wang, H.W.; Hampartzoumian, T.; Simpson, J.L.; Gibson, P.G.; et al. S100A12 provokes mast cell activation: A potential amplification pathway in asthma and innate immunity. J. Allergy Clin. Immunol. 2007, 119, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Tsubokawa, D.; Satoh, M. Strongyloides venezuelensis-derived venestatin ameliorates asthma pathogenesis by suppressing receptor for advanced glycation end-products-mediated signaling. Pulm. Pharmacol. Ther. 2022, 75, 102148. [Google Scholar] [CrossRef] [PubMed]
- Tabaa, M.M.E.; Fattah, A.M.K.; Shaalan, M.; Rashad, E.; El Mahdy, N.A. Dapagliflozin mitigates ovalbumin-prompted airway inflammatory-oxidative successions and associated bronchospasm in a rat model of allergic asthma. Expert. Opin. Ther. Targets 2022, 26, 487–506. [Google Scholar] [CrossRef]
- Lee, J.U.; Park, J.S.; Jun, J.A.; Kim, M.K.; Chang, H.S.; Baek, D.G.; Song, H.J.; Kim, M.S.; Park, C.S. Inhibitory Effect of Paquinimod on a Murine Model of Neutrophilic Asthma Induced by Ovalbumin with Complete Freund’s Adjuvant. Can. Respir. J. 2021, 2021, 8896108. [Google Scholar] [CrossRef]
- Huang, X.; Qu, D.; Liang, Y.; Huang, Q.; Li, M.; Hou, C. Elevated S100A4 in asthmatics and an allergen-induced mouse asthma model. J. Cell Biochem. 2019, 120, 9667–9676. [Google Scholar] [CrossRef] [PubMed]
- Pinto, S.M.; Subbannayya, Y.; Rex, D.A.B.; Raju, R.; Chatterjee, O.; Advani, J.; Radhakrishnan, A.; Keshava Prasad, T.S.; Wani, M.R.; Pandey, A. A network map of IL-33 signaling pathway. J. Cell Commun. Signal. 2018, 12, 615–624. [Google Scholar] [CrossRef]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef]
- Liang, Y.; Wang, X.; Wang, H.; Yang, W.; Yi, P.; Soong, L.; Cong, Y.; Cai, J.; Fan, X.; Sun, J. IL-33 activates mTORC1 and modulates glycolytic metabolism in CD8(+) T cells. Immunology 2022, 165, 61–73. [Google Scholar] [CrossRef]
- Marx, A.F.; Kallert, S.M.; Brunner, T.M.; Villegas, J.A.; Geier, F.; Fixemer, J.; Abreu-Mota, T.; Reuther, P.; Bonilla, W.V.; Fadejeva, J.; et al. The alarmin interleukin-33 promotes the expansion and preserves the stemness of Tcf-1(+) CD8(+) T cells in chronic viral infection. Immunity 2023, 56, 813–828.e810. [Google Scholar] [CrossRef]
- Zou, L.; Dang, W.; Tao, Y.; Zhao, H.; Yang, B.; Xu, X.; Li, Y. The Il-33/St2 Axis Promotes Acute Respiratory Distress Syndrome by Natural Killer T Cells. Shock. 2023, 59, 902–911. [Google Scholar] [CrossRef]
- Mok, M.Y.; Luo, C.Y.; Huang, F.P.; Kong, W.Y.; Chan, G.C.F. IL-33 Orchestrated the Interaction and Immunoregulatory Functions of Alternatively Activated Macrophages and Regulatory T Cells In Vitro. J. Immunol. 2023, 211, 1134–1143. [Google Scholar] [CrossRef] [PubMed]
- Ketelaar, M.E.; Portelli, M.A.; Dijk, F.N.; Shrine, N.; Faiz, A.; Vermeulen, C.J.; Xu, C.J.; Hankinson, J.; Bhaker, S.; Henry, A.P.; et al. Phenotypic and functional translation of IL33 genetics in asthma. J. Allergy Clin. Immunol. 2021, 147, 144–157. [Google Scholar] [CrossRef]
- Ji, Y.; Wang, E.; Mohammed, M.T.; Hameed, N.; Christodoulou, M.I.; Liu, X.; Zhou, W.; Fang, Z.; Jia, N.; Yu, H.; et al. Selective production of IL-33-neutralizing autoantibody ameliorates asthma responses and severity. Clin. Immunol. 2024, 264, 110234. [Google Scholar] [CrossRef]
- Poulsen, N.N.; Bjerregaard, A.; Khoo, S.K.; Laing, I.A.; Le Souëf, P.; Backer, V.; Rapley, L.; Cohen, S.E.; Barrett, L.; Thompson, P.; et al. Airway Interleukin-33 and type 2 cytokines in adult patients with acute asthma. Respir. Med. 2018, 140, 50–56. [Google Scholar] [CrossRef]
- Du, J.; Liu, Y.; Lan, G.; Zhou, Y.; Ni, Y.; Liao, K.; Zheng, F.; Cheng, Q.; Shi, G.; Su, X. PTRF-IL33-ZBP1 signaling mediating macrophage necroptosis contributes to HDM-induced airway inflammation. Cell Death Dis. 2023, 14, 432. [Google Scholar] [CrossRef] [PubMed]
- Schuijs, M.J.; Brenis Gomez, C.M.; Bick, F.; Van Moorleghem, J.; Vanheerswynghels, M.; van Loo, G.; Beyaert, R.; Voehringer, D.; Locksley, R.M.; Hammad, H.; et al. Interleukin-33-activated basophils promote asthma by regulating Th2 cell entry into lung tissue. J. Exp. Med. 2024, 221, e20240103. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Zheng, X.; Huang, J.; Hu, X. Association of. Front. Cell Dev. Biol. 2021, 9, 759542. [Google Scholar] [CrossRef]
- Badi, Y.E.; Salcman, B.; Taylor, A.; Rana, B.; Kermani, N.Z.; Riley, J.H.; Worsley, S.; Mumby, S.; Dahlen, S.E.; Cousins, D.; et al. IL1RAP expression and the enrichment of IL-33 activation signatures in severe neutrophilic asthma. Allergy 2023, 78, 156–167. [Google Scholar] [CrossRef]
- van der Ploeg, E.K.; Krabbendam, L.; Vroman, H.; van Nimwegen, M.; de Bruijn, M.J.W.; de Boer, G.M.; Bergen, I.M.; Kool, M.; Tramper-Standers, G.A.; Braunstahl, G.J.; et al. Type-2 CD8(+) T-cell formation relies on interleukin-33 and is linked to asthma exacerbations. Nat. Commun. 2023, 14, 5137. [Google Scholar] [CrossRef]
- Wechsler, M.E.; Ruddy, M.K.; Pavord, I.D.; Israel, E.; Rabe, K.F.; Ford, L.B.; Maspero, J.F.; Abdulai, R.M.; Hu, C.C.; Martincova, R.; et al. Efficacy and Safety of Itepekimab in Patients with Moderate-to-Severe Asthma. N. Engl. J. Med. 2021, 385, 1656–1668. [Google Scholar] [CrossRef]
- Kelsen, S.G.; Agache, I.O.; Soong, W.; Israel, E.; Chupp, G.L.; Cheung, D.S.; Theess, W.; Yang, X.; Staton, T.L.; Choy, D.F.; et al. Astegolimab (anti-ST2) efficacy and safety in adults with severe asthma: A randomized clinical trial. J. Allergy Clin. Immunol. 2021, 148, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Jing, X.; Yu, L.; Jiang, Z.; Lu, Y.; Peng, J.; Xu, X.; Liu, H.; Li, R.; Tang, H. Metformin alleviates inflammatory responses in acute allergic asthma by inhibiting ILC2s function. Int. Immunopharmacol. 2025, 159, 114897. [Google Scholar] [CrossRef] [PubMed]
- Friend, S.L.; Hosier, S.; Nelson, A.; Foxworthe, D.; Williams, D.E.; Farr, A. A thymic stromal cell line supports in vitro development of surface IgM+ B cells and produces a novel growth factor affecting B and T lineage cells. Exp. Hematol. 1994, 22, 321–328. [Google Scholar] [PubMed]
- Zhong, J.; Sharma, J.; Raju, R.; Palapetta, S.M.; Prasad, T.S.; Huang, T.C.; Yoda, A.; Tyner, J.W.; van Bodegom, D.; Weinstock, D.M.; et al. TSLP signaling pathway map: A platform for analysis of TSLP-mediated signaling. Database 2014, 2014, bau007. [Google Scholar] [CrossRef]
- Sebastian, K.; Borowski, A.; Kuepper, M.; Friedrich, K. Signal transduction around thymic stromal lymphopoietin (TSLP) in atopic asthma. Cell Commun. Signal. 2008, 6, 5. [Google Scholar] [CrossRef]
- Smolinska, S.; Antolín-Amérigo, D.; Popescu, F.D.; Jutel, M. Thymic Stromal Lymphopoietin (TSLP), Its Isoforms and the Interplay with the Epithelium in Allergy and Asthma. Int. J. Mol. Sci. 2023, 24, 12725. [Google Scholar] [CrossRef]
- Verstraete, K.; Peelman, F.; Braun, H.; Lopez, J.; Van Rompaey, D.; Dansercoer, A.; Vandenberghe, I.; Pauwels, K.; Tavernier, J.; Lambrecht, B.N.; et al. Structure and antagonism of the receptor complex mediated by human TSLP in allergy and asthma. Nat. Commun. 2017, 8, 14937. [Google Scholar] [CrossRef]
- Tsilingiri, K.; Fornasa, G.; Rescigno, M. Thymic Stromal Lymphopoietin: To Cut a Long Story Short. Cell Mol. Gastroenterol. Hepatol. 2017, 3, 174–182. [Google Scholar] [CrossRef]
- Park, J.H.; Jeong, D.Y.; Peyrin-Biroulet, L.; Eisenhut, M.; Shin, J.I. Insight into the role of TSLP in inflammatory bowel diseases. Autoimmun. Rev. 2017, 16, 55–63. [Google Scholar] [CrossRef]
- Xie, Y.; Takai, T.; Chen, X.; Okumura, K.; Ogawa, H. Long TSLP transcript expression and release of TSLP induced by TLR ligands and cytokines in human keratinocytes. J. Dermatol. Sci. 2012, 66, 233–237. [Google Scholar] [CrossRef]
- Borowski, A.; Vetter, T.; Kuepper, M.; Wohlmann, A.; Krause, S.; Lorenzen, T.; Virchow, J.C.; Luttmann, W.; Friedrich, K. Expression analysis and specific blockade of the receptor for human thymic stromal lymphopoietin (TSLP) by novel antibodies to the human TSLPRα receptor chain. Cytokine 2013, 61, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Matera, M.G.; Rogliani, P.; Calzetta, L.; Cazzola, M. TSLP Inhibitors for Asthma: Current Status and Future Prospects. Drugs 2020, 80, 449–458. [Google Scholar] [CrossRef]
- Scheeren, F.A.; van Lent, A.U.; Nagasawa, M.; Weijer, K.; Spits, H.; Legrand, N.; Blom, B. Thymic stromal lymphopoietin induces early human B-cell proliferation and differentiation. Eur. J. Immunol. 2010, 40, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Allakhverdi, Z.; Comeau, M.R.; Jessup, H.K.; Yoon, B.R.; Brewer, A.; Chartier, S.; Paquette, N.; Ziegler, S.F.; Sarfati, M.; Delespesse, G. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J. Exp. Med. 2007, 204, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Siracusa, M.C.; Saenz, S.A.; Hill, D.A.; Kim, B.S.; Headley, M.B.; Doering, T.A.; Wherry, E.J.; Jessup, H.K.; Siegel, L.A.; Kambayashi, T.; et al. TSLP promotes interleukin-3-independent basophil haematopoiesis and type 2 inflammation. Nature 2011, 477, 229–233. [Google Scholar] [CrossRef]
- Hui, C.C.; Rusta-Sallehy, S.; Asher, I.; Heroux, D.; Denburg, J.A. The effects of thymic stromal lymphopoietin and IL-3 on human eosinophil-basophil lineage commitment: Relevance to atopic sensitization. Immun. Inflamm. Dis. 2014, 2, 44–55. [Google Scholar] [CrossRef]
- Leyva-Castillo, J.M.; Hener, P.; Michea, P.; Karasuyama, H.; Chan, S.; Soumelis, V.; Li, M. Skin thymic stromal lymphopoietin initiates Th2 responses through an orchestrated immune cascade. Nat. Commun. 2013, 4, 2847. [Google Scholar] [CrossRef]
- Varricchi, G.; Pecoraro, A.; Marone, G.; Criscuolo, G.; Spadaro, G.; Genovese, A. Thymic Stromal Lymphopoietin Isoforms, Inflammatory Disorders, and Cancer. Front. Immunol. 2018, 9, 1595. [Google Scholar] [CrossRef]
- Rochman, Y.; Kashyap, M.; Robinson, G.W.; Sakamoto, K.; Gomez-Rodriguez, J.; Wagner, K.U.; Leonard, W.J. Thymic stromal lymphopoietin-mediated STAT5 phosphorylation via kinases JAK1 and JAK2 reveals a key difference from IL-7-induced signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 19455–19460. [Google Scholar] [CrossRef]
- Yu, X.; Li, H.; Ren, X. Signaling cascades initiated by TSLP-mediated signals in different cell types. Cell Immunol. 2012, 279, 174–179. [Google Scholar] [CrossRef]
- Parnes, J.R.; Molfino, N.A.; Colice, G.; Martin, U.; Corren, J.; Menzies-Gow, A. Targeting TSLP in Asthma. J. Asthma Allergy 2022, 15, 749–765. [Google Scholar] [CrossRef]
- Chorvinsky, E.; Nino, G.; Salka, K.; Gaviria, S.; Gutierrez, M.J.; Pillai, D.K. TSLP bronchoalveolar lavage levels at baseline are linked to clinical disease severity and reduced lung function in children with asthma. Front. Pediatr. 2022, 10, 971073. [Google Scholar] [CrossRef] [PubMed]
- Canè, L.; Poto, R.; Palestra, F.; Pirozzi, M.; Parashuraman, S.; Iacobucci, I.; Ferrara, A.L.; La Rocca, A.; Mercadante, E.; Pucci, P.; et al. TSLP is localized in and released from human lung macrophages activated by T2-high and T2-low stimuli: Relevance in asthma and COPD. Eur. J. Intern. Med. 2024, 124, 89–98. [Google Scholar] [CrossRef]
- Murrison, L.B.; Ren, X.; Preusse, K.; He, H.; Kroner, J.; Chen, X.; Jenkins, S.; Johansson, E.; Biagini, J.M.; Weirauch, M.T.; et al. TSLP disease-associated genetic variants combined with airway TSLP expression influence asthma risk. J. Allergy Clin. Immunol. 2022, 149, 79–88. [Google Scholar] [CrossRef]
- Ying, S.; O’Connor, B.; Ratoff, J.; Meng, Q.; Mallett, K.; Cousins, D.; Robinson, D.; Zhang, G.; Zhao, J.; Lee, T.H.; et al. Thymic stromal lymphopoietin expression is increased in asthmatic airways and correlates with expression of Th2-attracting chemokines and disease severity. J. Immunol. 2005, 174, 8183–8190. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, Y.; Lv, Z.; Chen, Y.; Huang, K.; Corrigan, C.J.; Ying, S. Bronchial Allergen Challenge of Patients with Atopic Asthma Triggers an Alarmin (IL-33, TSLP, and IL-25) Response in the Airways Epithelium and Submucosa. J. Immunol. 2018, 201, 2221–2231. [Google Scholar] [CrossRef] [PubMed]
- Paplińska-Goryca, M.; Nejman-Gryz, P.; Proboszcz, M.; Kwiecień, I.; Hermanowicz-Salamon, J.; Grabczak, E.M.; Krenke, R. Expression of TSLP and IL-33 receptors on sputum macrophages of asthma patients and healthy subjects. J. Asthma 2020, 57, 1–10. [Google Scholar] [CrossRef]
- Toki, S.; Goleniewska, K.; Zhang, J.; Zhou, W.; Newcomb, D.C.; Zhou, B.; Kita, H.; Boyd, K.L.; Peebles, R.S. TSLP and IL-33 reciprocally promote each other’s lung protein expression and ILC2 receptor expression to enhance innate type-2 airway inflammation. Allergy 2020, 75, 1606–1617. [Google Scholar] [CrossRef]
- Pelaia, C.; Pelaia, G.; Crimi, C.; Maglio, A.; Gallelli, L.; Terracciano, R.; Vatrella, A. Tezepelumab: A Potential New Biological Therapy for Severe Refractory Asthma. Int. J. Mol. Sci. 2021, 22, 4369. [Google Scholar] [CrossRef]
- Sverrild, A.; Cerps, S.; Nieto-Fontarigo, J.J.; Ramu, S.; Hvidtfeldt, M.; Menzel, M.; Kearley, J.; Griffiths, J.M.; Parnes, J.R.; Porsbjerg, C.; et al. Tezepelumab decreases airway epithelial IL-33 and T2-inflammation in response to viral stimulation in patients with asthma. Allergy 2024, 79, 656–666. [Google Scholar] [CrossRef]
- Corren, J.; Pham, T.H.; Garcia Gil, E.; Sałapa, K.; Ren, P.; Parnes, J.R.; Colice, G.; Griffiths, J.M. Baseline type 2 biomarker levels and response to tezepelumab in severe asthma. Allergy 2022, 77, 1786–1796. [Google Scholar] [CrossRef] [PubMed]
- Diver, S.; Khalfaoui, L.; Emson, C.; Wenzel, S.E.; Menzies-Gow, A.; Wechsler, M.E.; Johnston, J.; Molfino, N.; Parnes, J.R.; Megally, A.; et al. Effect of tezepelumab on airway inflammatory cells, remodelling, and hyperresponsiveness in patients with moderate-to-severe uncontrolled asthma (CASCADE): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir. Med. 2021, 9, 1299–1312. [Google Scholar] [CrossRef]
- Greig, R.; Chan, R.; Fardon, T.C.; Lipworth, B.J. Real-world effects of tezepelumab on small airway dysfunction in severe refractory asthma. Ann. Allergy Asthma Immunol. 2025, 134, P484–P485. [Google Scholar] [CrossRef] [PubMed]
- Carpagnano, G.E.; Dragonieri, S.; Resta, E.; Lulaj, E.; Montagnolo, F.; Portacci, A.; Magaletti, P.; Soccio, P.; Lacedonia, D.; Scioscia, G. Short-term Tezepelumab effectiveness in patients with severe asthma: A multicenter study. J. Asthma 2025, 62, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Wang, G.; Han, D. Long-term safety of tezepelumab in patients with asthma: A systematic review and meta-analysis of randomized controlled trials. J. Asthma 2025, 62, 4–13. [Google Scholar] [CrossRef]
- Portacci, A.; Scioscia, G.; Dragonieri, S.; Aliani, M.; Lulaj, E.; Montagnolo, F.; Magaletti, P.; Soccio, P.; Salerno, L.; Lacedonia, D.; et al. The impact of tezepelumab therapy on perceived asthma triggers: A multicenter real-life study. J. Asthma 2025. ahead of print. [Google Scholar] [CrossRef]
- Gauvreau, G.M.; Hohlfeld, J.M.; FitzGerald, J.M.; Boulet, L.P.; Cockcroft, D.W.; Davis, B.E.; Korn, S.; Kornmann, O.; Leigh, R.; Mayers, I.; et al. Inhaled anti-TSLP antibody fragment, ecleralimab, blocks responses to allergen in mild asthma. Eur. Respir. J. 2023, 61, 2201193. [Google Scholar] [CrossRef]
- Deiteren, A.; Bontinck, L.; Conickx, G.; Vigan, M.; Dervaux, N.; Gassiot, M.; Bas, S.; Suratt, B.; Staudinger, H.; Krupka, E. A first-in-human, single and multiple dose study of lunsekimig, a novel anti-TSLP/anti-IL-13 NANOBODY® compound, in healthy volunteers. Clin. Transl. Sci. 2024, 17, e13864. [Google Scholar] [CrossRef]
- Bao, C.; Liu, C.; Liu, Q.; Hua, L.; Hu, J.; Li, Z.; Xu, S. Liproxstatin-1 alleviates LPS/IL-13-induced bronchial epithelial cell injury and neutrophilic asthma in mice by inhibiting ferroptosis. Int. Immunopharmacol. 2022, 109, 108770. [Google Scholar] [CrossRef]
- Cheng, D.T.; Ma, C.; Niewoehner, J.; Dahl, M.; Tsai, A.; Zhang, J.; Gonsiorek, W.; Apparsundaram, S.; Pashine, A.; Ravindran, P.; et al. Thymic stromal lymphopoietin receptor blockade reduces allergic inflammation in a cynomolgus monkey model of asthma. J. Allergy Clin. Immunol. 2013, 132, 455–462. [Google Scholar] [CrossRef]
- Murphy, M.B.; Vitale, L.; O’Neill, T.; Maurer, D.M.; Malenchek, L.; Crocker, A.; Patterson, C.; Mills-Chen, L.; Saley, V.; Antczak, N.M.; et al. Dual Inhibition of Mast Cells and Thymic Stromal Lymphopoietin Using a Novel Bispecific Antibody, CDX-622. Allergy 2025. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Deiteren, A.; Krupka, E.; Bontinck, L.; Imberdis, K.; Conickx, G.; Bas, S.; Patel, N.; Staudinger, H.W.; Suratt, B.T. A proof-of-mechanism trial in asthma with lunsekimig, a bispecific NANOBODY molecule. Eur. Respir. J. 2025, 65, 2401461. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Ruan, Y.; Ying, H.; Wang, J.; Wang, H.; Chen, S. Baicalin Attenuates Type 2 Immune Responses in a Mouse Allergic Asthma Model through Inhibiting the Production of Thymic Stromal Lymphopoietin. Int. Arch. Allergy Immunol. 2025, 186, 203–211. [Google Scholar] [CrossRef]
- Xu, M.; Dong, C. IL-25 in allergic inflammation. Immunol. Rev. 2017, 278, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Chen, Q.; Wang, X.; Liu, X.; Zhang, L. IL-25 induces airway remodeling in asthma by orchestrating the phenotypic changes of epithelial cell and fibrocyte. Respir. Res. 2023, 24, 212. [Google Scholar] [CrossRef]
- Peng, B.; Sun, L.; Zhang, M.; Yan, H.; Shi, G.; Xia, Z.; Dai, R.; Tang, W. Role of IL-25 on Eosinophils in the Initiation of Th2 Responses in Allergic Asthma. Front. Immunol. 2022, 13, 842500. [Google Scholar] [CrossRef]
- Zhang, K.; Feng, Y.; Liang, Y.; Wu, W.; Chang, C.; Chen, D.; Chen, S.; Gao, J.; Chen, G.; Yi, L.; et al. Epithelial miR-206 targets CD39/extracellular ATP to upregulate airway IL-25 and TSLP in type 2-high asthma. JCI Insight 2021, 6, e148103. [Google Scholar] [CrossRef]
- Ballantyne, S.J.; Barlow, J.L.; Jolin, H.E.; Nath, P.; Williams, A.S.; Chung, K.F.; Sturton, G.; Wong, S.H.; McKenzie, A.N. Blocking IL-25 prevents airway hyperresponsiveness in allergic asthma. J. Allergy Clin. Immunol. 2007, 120, 1324–1331. [Google Scholar] [CrossRef]
- Williams, T.C.; Loo, S.L.; Nichol, K.S.; Reid, A.T.; Veerati, P.C.; Esneau, C.; Wark, P.A.B.; Grainge, C.L.; Knight, D.A.; Vincent, T.; et al. IL-25 blockade augments antiviral immunity during respiratory virus infection. Commun. Biol. 2022, 5, 415. [Google Scholar] [CrossRef]
- An, G.; Wang, W.; Zhang, X.; Huang, Q.; Li, Q.; Chen, S.; Du, X.; Corrigan, C.J.; Huang, K.; Wang, W.; et al. Combined blockade of IL-25, IL-33 and TSLP mediates amplified inhibition of airway inflammation and remodelling in a murine model of asthma. Respirology 2020, 25, 603–612. [Google Scholar] [CrossRef]
- Xu, X.; Luo, S.; Li, B.; Dai, H.; Zhang, J. IL-25 contributes to lung fibrosis by directly acting on alveolar epithelial cells and fibroblasts. Exp. Biol. Med. 2019, 244, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.J.; Huang, K.W.; Li, Y.; Zhang, Q.; Wang, J.J.; Wang, W.; Liu, J.; Lv, Z.; An, Y.Q.; Ding, Y.Z.; et al. Direct comparison of the dynamics of IL-25- and ‘allergen’-induced airways inflammation, remodelling and hypersensitivity in a murine asthma model. Clin. Exp. Allergy 2014, 44, 765–777. [Google Scholar] [CrossRef]
- Chang, C.; Chen, G.; Wu, W.; Chen, D.; Chen, S.; Gao, J.; Feng, Y.; Zhen, G. Exogenous IL-25 ameliorates airway neutrophilia via suppressing macrophage M1 polarization and the expression of IL-12 and IL-23 in asthma. Respir. Res. 2023, 24, 260. [Google Scholar] [CrossRef] [PubMed]
- Kalinauskaite-Zukauske, V.; Januskevicius, A.; Janulaityte, I.; Miliauskas, S.; Malakauskas, K. Serum Levels of Epithelial-Derived Cytokines as Interleukin-25 and Thymic Stromal Lymphopoietin after a Single Dose of Mepolizumab in Patients with Severe Non-Allergic Eosinophilic Asthma: A Short Report. Can. Respir. J. 2019, 2019, 8607657. [Google Scholar] [CrossRef]
- Palacionyte, J.; Januskevicius, A.; Vasyle, E.; Rimkunas, A.; Miliauskas, S.; Malakauskas, K. Clinical Remission Criteria and Serum Levels of Type 2 Inflammation Mediators during 24 Weeks of Treatment with the Anti-IL-5 Drug Mepolizumab in Patients with T2-High Severe Asthma. Diagnostics 2024, 14, 1345. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.L.; Tsai, M.K.; Tsai, Y.G.; Lin, Y.C.; Hsu, Y.L.; Chen, Y.T.; Lin, Y.C.; Hung, C.H. Montelukast Increased IL-25, IL-33, and TSLP via Epigenetic Regulation in Airway Epithelial Cells. Int. J. Mol. Sci. 2023, 24, 1227. [Google Scholar] [CrossRef]
- Arora, S.; Dev, K.; Agarwal, B.; Das, P.; Syed, M.A. Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology 2018, 223, 383–396. [Google Scholar] [CrossRef]
- Tsai, M.L.; Tsai, Y.G.; Lin, Y.C.; Hsu, Y.L.; Chen, Y.T.; Tsai, M.K.; Liao, W.T.; Lin, Y.C.; Hung, C.H. IL-25 Induced ROS-Mediated M2 Macrophage Polarization via AMPK-Associated Mitophagy. Int. J. Mol. Sci. 2021, 23, 3. [Google Scholar] [CrossRef]
- Altieri, A.; Marshall, C.L.; Ramotar, P.; Lloyd, D.; Hemshekhar, M.; Spicer, V.; van der Does, A.M.; Mookherjee, N. Human Host Defense Peptide LL-37 Suppresses TNFalpha-Mediated Matrix Metalloproteinases MMP9 and MMP13 in Human Bronchial Epithelial Cells. J. Innate Immun. 2024, 16, 203–215. [Google Scholar] [CrossRef]
- Pinkerton, J.W.; Kim, R.Y.; Koeninger, L.; Armbruster, N.S.; Hansbro, N.G.; Brown, A.C.; Jayaraman, R.; Shen, S.; Malek, N.; Cooper, M.A.; et al. Human beta-defensin-2 suppresses key features of asthma in murine models of allergic airways disease. Clin. Exp. Allergy 2021, 51, 120–131. [Google Scholar] [CrossRef]
- Borchers, N.S.; Santos-Valente, E.; Toncheva, A.A.; Wehkamp, J.; Franke, A.; Gaertner, V.D.; Nordkild, P.; Genuneit, J.; Jensen, B.A.H.; Kabesch, M. Human beta-Defensin 2 Mutations Are Associated with Asthma and Atopy in Children and Its Application Prevents Atopic Asthma in a Mouse Model. Front. Immunol. 2021, 12, 636061. [Google Scholar] [CrossRef]
- Chen, G.; Zheng, Y.; Wu, N.; Yang, X.; Qu, S. Human beta defensin 3 knockdown inhibits the proliferation and migration of airway smooth muscle cells through regulating the PI3K/AKT signaling pathway. Mol. Immunol. 2024, 168, 38–46. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Alarmin | Involvement in the Pathophysiology of Asthma | Therapeutic Implications | References |
|---|---|---|---|
| HMGB1 | ↑ Eosinophils, IL-5, IL-13 in sputum; ↑ Th17, IL-4, IgG, IgE; ↑ TGF-β1, TNFα, TSLP, MM-9, VEGF, IL-6, IL-8, IL-17; Th17 and neutrophils—interaction with RAGE signaling; ↑ IL-1 β, IL-33—interaction with TLR4 signaling; ↑ TNFα, IL-1, IL-17, IFNγ—interaction with JAK/STAT signaling. | Inhaled glucocorticoid therapy—↓ HMGB1 in the squatum; HMGB1 neutralizing antibody—↓ HMGB and Th1, Th2, Th17’ vitamin D—↓ HMGB1 and suppressed inflammation within the respiratory tract in animal models; Astragaloside IV—↓ HMGB1 and RAGE signaling. | [38,41,42,46,47,48,51,58,59,60,61,65] |
| S100 proteins | ↑ Neutrophils in the sputum; interaction with RAGE, MAPK signaling; ↑ Th2. | Dapagliflozin—↓ S100, leading to mitigated bronchospasm; Paquinimod—↓ inflammatory cells. | [67,79,80,93,94] |
| IL-33 | ↑ Eosinophils | Itepekimab—↑ lung function; Astegolimab—↓ exacerbation rate in patients suffering from low blood eosinophil asthma. | [102,110,111] |
| IL-25 | ↑ Fibrosis | Mepolizumab—↓ IL-25; Montelukast combined with corticosteroid—↓ IL-25 | [155,158,159,160,165,166] |
| TSLP | ↑ DC; ↑ Th2, IL-4, IL-5, IL-9, IL-13; ↑ Th17, neutrophils. | Tezepelumab—↓ IL-5, IL-13, IL-33, T2, eosinophils. Ecleralimab—↓ airway inflammation and bronchospasm; Lunsekimig—↓ TSLP, IL-13 Lipoxstatin-1 (Lip-1) —↓ TSLP. | [139,140,141,147,148,149] |
| LL-37 | LL-37 reduces the expression of MMPs induced by TNF in bronchial epithelial cells. | - | [169] |
| Defensins | The expression of hBD2 is reduced in asthmatic children, while that of hBD1 and hBD3 increased. | The use of hBD2 as a treatment agent was shown to induce beneficial effects in animal studies. Silencing hBD3 could represent a promising treatment strategy in asthma. | [170,171,172] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plewa, P.; Pokwicka, J.; Bakinowska, E.; Kiełbowski, K.; Pawlik, A. The Role of Alarmins in the Pathogenesis of Asthma. Biomolecules 2025, 15, 996. https://doi.org/10.3390/biom15070996
Plewa P, Pokwicka J, Bakinowska E, Kiełbowski K, Pawlik A. The Role of Alarmins in the Pathogenesis of Asthma. Biomolecules. 2025; 15(7):996. https://doi.org/10.3390/biom15070996
Chicago/Turabian StylePlewa, Paulina, Julia Pokwicka, Estera Bakinowska, Kajetan Kiełbowski, and Andrzej Pawlik. 2025. "The Role of Alarmins in the Pathogenesis of Asthma" Biomolecules 15, no. 7: 996. https://doi.org/10.3390/biom15070996
APA StylePlewa, P., Pokwicka, J., Bakinowska, E., Kiełbowski, K., & Pawlik, A. (2025). The Role of Alarmins in the Pathogenesis of Asthma. Biomolecules, 15(7), 996. https://doi.org/10.3390/biom15070996