
Evaluating Host Defense Peptides: A Comparative Analysis of Synthetic Peptides and Recombinant Concatemers

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains
2.2. Synthesis of Host Defense Peptides
2.3. Synthetic Peptides
2.4. Recombinant Production of Proteins
2.4.1. Protein Design and Cloning
2.4.2. Protein Expression and Purification
2.5. Antimicrobial Activity
2.5.1. Antimicrobial Killing Assay
2.5.2. Determination of the Minimal Inhibitory Concentrations
2.5.3. Antibiofilm Assay
2.6. Lipopolysaccharide Binding Assay
2.7. Dynamic Light Scattering (DLS)
2.8. Secondary Structure of Peptides and Proteins
2.8.1. Circular Dichroism Spectroscopy (CD)
2.8.2. ATR-FTIR Spectroscopy
3. Results
3.1. Protein Design and Physicochemical Characteristics
3.2. Bioactivity Analysis
3.2.1. Antimicrobial and Antibiofilm Activity
3.2.2. Antiendotoxin Activity
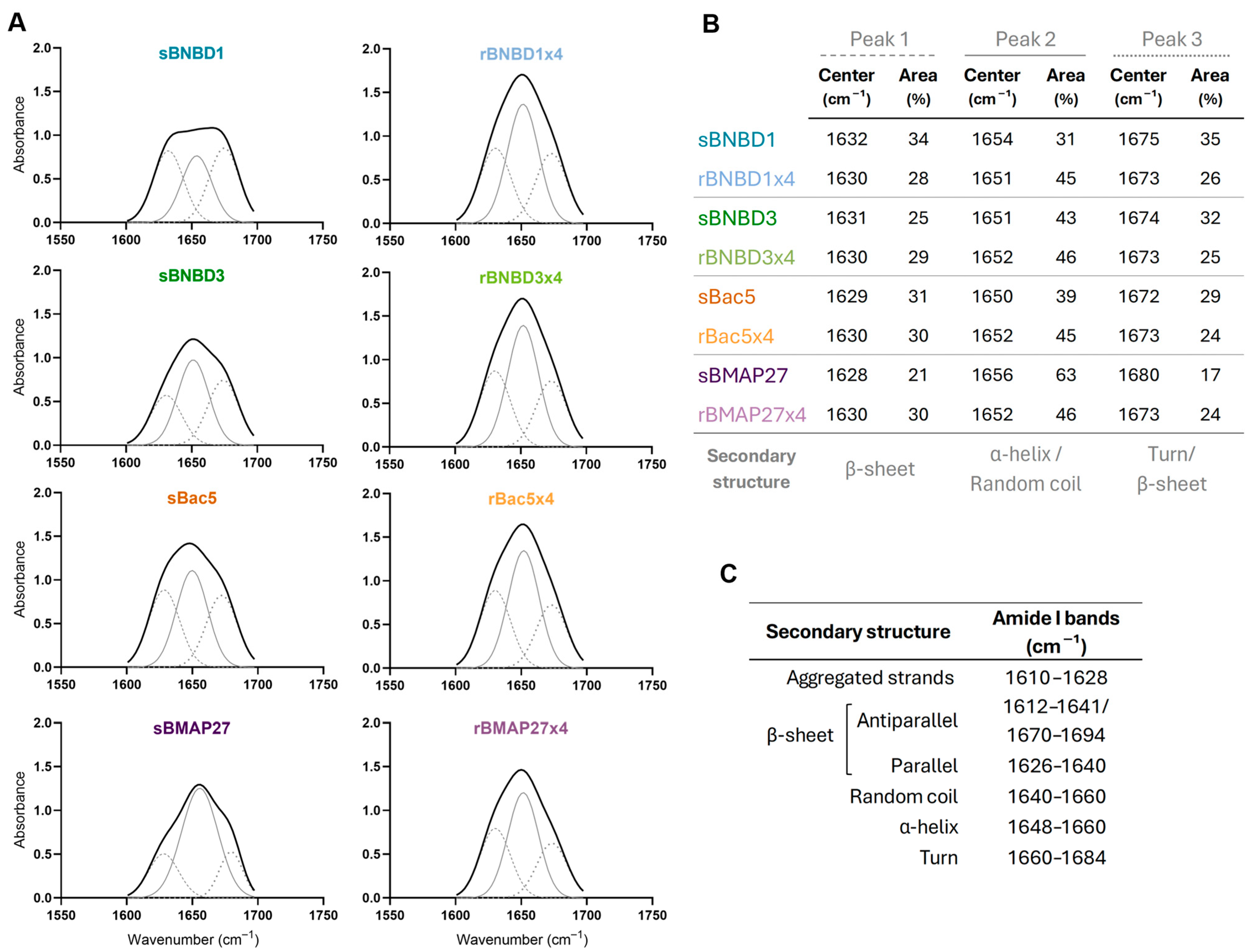
3.3. Structural Characterization and Conformational Analysis
3.3.1. Size Distribution
3.3.2. Protein Folding
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| HDPs | host defense peptides |
| LPS | lipopolysaccharides |
| SPPS | solid-phase peptide synthesis |
| PTMs | post-translational modifications |
| GRAS | generally regarded as safe |
| BNBD1 | bovine neutrophil β-defensin 1 |
| BNBD3 | bovine neutrophil β-defensin 3 |
| Bac5 | bactenecin 5 |
| BMAP27 | bovine myeloid antibacterial peptide 27 |
| MSSA | methicillin-sensitive Staphylococcus aureus |
| BHI | brain heart infusion |
| LB | Luria-Bertani |
| IMAC | immobilized metal affinity chromatography |
| MHB | Mueller Hinton Broth |
| MIC | minimum inhibitory concentration |
| DLS | dynamic light scattering |
| ATR-FTIR | attenuated total reflectance–Fourier-transform infrared spectroscopy |
References
- Ikuta, K.S.; Swetschinski, L.R.; Robles Aguilar, G.; Sharara, F.; Mestrovic, T.; Gray, A.P.; Davis Weaver, N.; Wool, E.E.; Han, C.; Gershberg Hayoon, A.; et al. Global Mortality Associated with 33 Bacterial Pathogens in 2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet 2022, 400, 2221–2248. [Google Scholar] [CrossRef] [PubMed]
- Naghavi, M.; Mestrovic, T.; Gray, A.; Gershberg Hayoon, A.; Swetschinski, L.R.; Robles Aguilar, G.; Davis Weaver, N.; Ikuta, K.S.; Chung, E.; Wool, E.E.; et al. Global Burden Associated with 85 Pathogens in 2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet Infect. Dis. 2024, 24, 868–895. [Google Scholar] [CrossRef] [PubMed]
- Cella, E.; Giovanetti, M.; Benedetti, F.; Scarpa, F.; Johnston, C.; Borsetti, A.; Ceccarelli, G.; Azarian, T.; Zella, D.; Ciccozzi, M. Joining Forces against Antibiotic Resistance: The One Health Solution. Pathogens 2023, 12, 1074. [Google Scholar] [CrossRef] [PubMed]
- Robi, D.T.; Mossie, T.; Temteme, S. A Comprehensive Review of the Common Bacterial Infections in Dairy Calves and Advanced Strategies for Health Management. Vet. Med. Res. Rep. 2024, 15, 1–14. [Google Scholar] [CrossRef]
- Serra-Burriel, M.; Keys, M.; Campillo-Artero, C.; Agodi, A.; Barchitta, M.; Gikas, A.; Palos, C.; López-Casasnovas, G. Impact of Multi-Drug Resistant Bacteria on Economic and Clinical Outcomes of Healthcare-Associated Infections in Adults: Systematic Review and Meta-Analysis. PLoS ONE 2020, 15, e0227139. [Google Scholar] [CrossRef]
- Cloeckaert, A.; Kuchler, K. Grand Challenges in Infectious Diseases: Are We Prepared for Worst-Case Scenarios? Front. Microbiol. 2020, 11, 613383. [Google Scholar] [CrossRef]
- Arnold, K.E.; Laing, G.; McMahon, B.J.; Fanning, S.; Stekel, D.J.; Pahl, O.; Coyne, L.; Latham, S.M.; McIntyre, K.M. The Need for One Health Systems-Thinking Approaches to Understand Multiscale Dissemination of Antimicrobial Resistance. Lancet Planet. Heal. 2024, 8, e124–e133. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell. Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef]
- Kang, H.K.; Kim, C.; Seo, C.H.; Park, Y. The Therapeutic Applications of Antimicrobial Peptides (AMPs): A Patent Review. J. Microbiol. 2017, 55, 1–12. [Google Scholar] [CrossRef]
- Van Dijk, A.; Hedegaard, C.J.; Haagsman, H.P.; Heegaard, P.M.H. The Potential for Immunoglobulins and Host Defense Peptides (HDPs) to Reduce the Use of Antibiotics in Animal Production. Vet. Res. 2018, 49, 68. [Google Scholar] [CrossRef]
- Mookherjee, N.; Anderson, M.A.; Haagsman, H.P.; Davidson, D.J. Antimicrobial Host Defence Peptides: Functions and Clinical Potential. Nat. Rev. Drug Discov. 2020, 19, 311–332. [Google Scholar] [CrossRef] [PubMed]
- Drayton, M.; Deisinger, J.P.; Ludwig, K.C.; Raheem, N.; Müller, A.; Schneider, T.; Straus, S.K. Host Defense Peptides: Dual Antimicrobial and Immunomodulatory Action. Int. J. Mol. Sci. 2021, 22, 11172. [Google Scholar] [CrossRef] [PubMed]
- Graf, M.; Mardirossian, M.; Nguyen, F.; Seefeldt, A.C.; Guichard, G.; Scocchi, M.; Innis, C.A.; Wilson, D.N. Proline-Rich Antimicrobial Peptides Targeting Protein Synthesis. Nat. Prod. Rep. 2017, 34, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.H.; Shah, P.; Chen, Y.W.; Chen, C.S. Systematic Analysis of Intracellular-Targeting Antimicrobial Peptides, Bactenecin 7, Hybrid of Pleurocidin and Dermaseptin, Proline-Arginine-Rich Peptide, and Lactoferricin b, by Using Escherichia Coli Proteome Microarrays. Mol. Cell. Proteom. 2016, 15, 1837–1847. [Google Scholar] [CrossRef]
- Raheem, N.; Straus, S.K. Mechanisms of Action for Antimicrobial Peptides With Antibacterial and Antibiofilm Functions. Front. Microbiol. 2019, 10, 2866. [Google Scholar] [CrossRef]
- Martin, L.; van Meegern, A.; Doemming, S.; Schuerholz, T. Antimicrobial Peptides in Human Sepsis. Front. Immunol. 2015, 6, 404. [Google Scholar] [CrossRef]
- Rosenfeld, Y.; Shai, Y. Lipopolysaccharide (Endotoxin)-Host Defense Antibacterial Peptides Interactions: Role in Bacterial Resistance and Prevention of Sepsis. Biochim. Biophys. Acta—Biomembr. 2006, 1758, 1513–1522. [Google Scholar] [CrossRef]
- Martell, E.M.; González-Garcia, M.; Ständker, L.; Otero-González, A.J. Host Defense Peptides as Immunomodulators: The Other Side of the Coin. Peptides 2021, 146, 170644. [Google Scholar] [CrossRef]
- Cai, J.; Li, X.; Du, H.; Jiang, C.; Xu, S.; Cao, Y. Immunomodulatory Significance of Natural Peptides in Mammalians: Promising Agents for Medical Application. Immunobiology 2020, 225, 151936. [Google Scholar] [CrossRef]
- van der Does, A.M.; Hiemstra, P.S.; Mookherjee, N. Antimicrobial Host Defence Peptides: Immunomodulatory Functions and Translational Prospects. In Antimicrobial Peptides. Advances in Experimental Medicine and Biology; Matsuzaki, K., Ed.; Springer: Singapore, 2019; Volume 1117, pp. 149–171. [Google Scholar] [CrossRef]
- Gill, I.; López-Fandiño, R.; Jorba, X.; Vulfson, E.N. Biologically Active Peptides and Enzymatic Approaches to Their Production. Enzym. Microb. Technol. 1996, 18, 162–183. [Google Scholar] [CrossRef]
- Münzker, L.; Oddo, A.; Hansen, P.R. Chemical Synthesis of Antimicrobial Peptides. Methods Mol. Biol. 2017, 1548, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Kent, S.B.H. Total Chemical Synthesis of Proteins. Chem. Soc. Rev. 2009, 38, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Zompra, A.A.; Galanis, A.S.; Werbitzky, O.; Albericio, F. Manufacturing Peptides as Active Pharmaceutical Ingredients. Future Med. Chem. 2009, 1, 361–377. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.L.; Tofteng, A.P.; Malik, L.; Jensen, K.J. Microwave Heating in Solid-Phase Peptide Synthesis. Chem. Soc. Rev. 2012, 41, 1826–1844. [Google Scholar] [CrossRef]
- Skalska, J.; Andrade, V.M.; Cena, G.L.; Harvey, P.J.; Gaspar, D.M.D.; Mello, É.O.; Henriques, S.T.; Valle, J.; Gomes, V.M.; Conceição, K.; et al. Synthesis, Structure, and Activity of the Antifungal Plant Defensin Pv D 1. J. Med. Chem. 2020, 63, 9391–9402. [Google Scholar] [CrossRef]
- Gaglione, R.; Pane, K.; Dell’Olmo, E.; Cafaro, V.; Pizzo, E.; Olivieri, G.; Notomista, E.; Arciello, A. Cost-Effective Production of Recombinant Peptides in Escherichia coli. N. Biotechnol. 2019, 51, 39–48. [Google Scholar] [CrossRef]
- Jayakrishnan, A.; Wan Rosli, W.R.; Tahir, A.R.M.; Razak, F.S.A.; Kee, P.E.; Ng, H.S.; Chew, Y.-L.; Lee, S.-K.; Ramasamy, M.; Tan, C.S.; et al. Evolving Paradigms of Recombinant Protein Production in Pharmaceutical Industry: A Rigorous Review. Sci 2024, 6, 9. [Google Scholar] [CrossRef]
- Boto, A.; De La Lastra, J.M.P.; González, C.C. The Road from Host-Defense Peptides to a New Generation of Antimicrobial Drugs. Molecules 2018, 23, 311. [Google Scholar] [CrossRef]
- Hilchie, A.L.; Wuerth, K.; Hancock, R.E.W. Immune Modulation by Multifaceted Cationic Host Defense (Antimicrobial) Peptides. Nat. Chem. Biol. 2013, 9, 761–768. [Google Scholar] [CrossRef]
- Kang, J.; Zhao, D.; Lyu, Y.; Tian, L.; Yin, X.; Yang, L.; Teng, K.; Zhou, X. Antimycobacterial Activity of Pichia Pastoris-Derived Mature Bovine Neutrophil β-Defensins 5. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 1823–1834. [Google Scholar] [CrossRef]
- Choi, H.J.; Seo, M.J.; Lee, J.C.; Cheigh, C.I.; Park, H.; Ahn, C.; Pyun, Y.R. Heterologous Expression of Human β-Defensin-1 in Bacteriocin-Producing Lactococcus Lactis. J. Microbiol. Biotechnol. 2005, 15, 330–336. [Google Scholar]
- Roca-Pinilla, R.; Lisowski, L.; Arís, A.; Garcia-Fruitós, E. The Future of Recombinant Host Defense Peptides. Microb. Cell Fact. 2022, 21, 267. [Google Scholar] [CrossRef] [PubMed]
- Deo, S.; Turton, K.L.; Kainth, T.; Kumar, A.; Wieden, H.-J. Strategies for Improving Antimicrobial Peptide Production. Biotechnol. Adv. 2022, 59, 107968. [Google Scholar] [CrossRef]
- Clement, H.; Flores, V.; Diego-Garcia, E.; Corrales-Garcia, L.; Villegas, E.; Corzo, G. A Comparison between the Recombinant Expression and Chemical Synthesis of a Short Cysteine-Rich Insecticidal Spider Peptide. J. Venom. Anim. Toxins Incl. Trop. Dis. 2015, 21, 19. [Google Scholar] [CrossRef]
- Silva, O.N.; Mulder, K.C.; Barbosa, A.A.; Otero-Gonzalez, A.J.; Lopez-Abarrategui, C.; Rezende, T.M.B.; Dias, S.C.; Franco, O.L. Exploring the Pharmacological Potential of Promiscuous Host-Defense Peptides: From Natural Screenings to Biotechnological Applications. Front. Microbiol. 2011, 2, 232. [Google Scholar] [CrossRef]
- Bommarius, B.; Jenssen, H.; Elliott, M.; Kindrachuk, J.; Pasupuleti, M.; Gieren, H.; Jaeger, K.-E.; Hancock, R.E.W.; Kalman, D. Cost-Effective Expression and Purification of Antimicrobial and Host Defense Peptides in Escherichia Coli. Peptides 2010, 31, 1957–1965. [Google Scholar] [CrossRef]
- Li, Y. Recombinant Production of Antimicrobial Peptides in Escherichia Coli: A Review. Protein Expr. Purif. 2011, 80, 260–267. [Google Scholar] [CrossRef]
- Perez-Perez, D.A.; Villanueva-Ramirez, T.d.J.; Hernandez-Pedraza, A.E.; Casillas-Vega, N.G.; Gonzalez-Barranco, P.; Zarate, X. The Small Metal-Binding Protein Smbp Simplifies the Recombinant Expression and Purification of the Antimicrobial Peptide Ll-37. Antibiotics 2021, 10, 1271. [Google Scholar] [CrossRef]
- Panteleev, P.V.; Bolosov, I.A.; Kalashnikov, A.À.; Kokryakov, V.N.; Shamova, O.V.; Emelianova, A.A.; Balandin, S.V.; Ovchinnikova, T.V. Combined Antibacterial Effects of Goat Cathelicidins with Different Mechanisms of Action. Front. Microbiol. 2018, 9, 2983. [Google Scholar] [CrossRef]
- Wright, O.; Yoshimi, T.; Tunnacliffe, A. Recombinant Production of Cathelicidin-Derived Antimicrobial Peptides in Escherichia Coli Using an Inducible Autocleaving Enzyme Tag. N. Biotechnol. 2012, 29, 352–358. [Google Scholar] [CrossRef]
- van der Merwe, J.; Prysliak, T.; Gerdts, V.; Perez-Casal, J. Protein Chimeras Containing the Mycoplasma Bovis GAPDH Protein and Bovine Host-Defence Peptides Retain the Properties of the Individual Components. Microb. Pathog. 2011, 50, 269–277. [Google Scholar] [CrossRef] [PubMed]
- López-Cano, A.; Martínez-Miguel, M.; Guasch, J.; Ratera, I.; Arís, A.; Garcia-Fruitós, E. Exploring the Impact of the Recombinant Escherichia Coli Strain on Defensins Antimicrobial Activity: BL21 versus Origami Strain. Microb. Cell Fact. 2022, 21, 77. [Google Scholar] [CrossRef] [PubMed]
- Xin, A.; Zhao, Y.; Yu, H.; Shi, H.; Liu, H.; Diao, H.; Zhang, Y. Soluble Fusion Expression, Characterization and Localization of Human β-Defensin 6. Mol. Med. Rep. 2014, 9, 149–155. [Google Scholar] [CrossRef]
- Wu, J.; Wang, C.; He, H.; Hu, G.; Yang, H.; Gao, Y.; Zhong, J. Molecular Analysis and Recombinant Expression of Bovine Neutrophil β-Defensin 12 and Its Antimicrobial Activity. Mol. Biol. Rep. 2011, 38, 429–436. [Google Scholar] [CrossRef]
- Ojima-Kato, T. Advances in Recombinant Protein Production in Microorganisms and Functional Peptide Tags. Biosci. Biotechnol. Biochem. 2025, 89, 1–10. [Google Scholar] [CrossRef]
- Morello, E.; Bermúdez-Humarán, L.G.; Llull, D.; Solé, V.; Miraglio, N.; Langella, P.; Poquet, I. Lactococcus Lactis, an Efficient Cell Factory for Recombinant Protein Production and Secretion. J. Mol. Microbiol. Biotechnol. 2008, 14, 48–58. [Google Scholar] [CrossRef]
- Cano-Garrido, O.; Rueda, F.L.; Sànchez-García, L.; Ruiz-Ávila, L.; Bosser, R.; Villaverde, A.; García-Fruitós, E. Expanding the Recombinant Protein Quality in Lactococcus Lactis. Microb. Cell Fact. 2014, 13, 167. [Google Scholar] [CrossRef]
- Zhang, H.; Dong, M.; Xu, H.; Li, H.; Zheng, A.; Sun, G.; Jin, W. Recombinant Lactococcus Lactis Expressing Human LL-37 Prevents Deaths from Viral Infections in Piglets and Chicken. Probiotics Antimicrob. Proteins 2024, 16, 2150–2160. [Google Scholar] [CrossRef]
- Baltà-Foix, R.; Garcia-Fruitós, E.; Arís, A. Time to Consider Ruling out Inclusion Bodies Denaturing Protocols for Spontaneous Solubilization of Biologically Active Proteins. Sci. Rep. 2024, 14, 26061. [Google Scholar] [CrossRef]
- Wang, T.; Wang, Z.; Mi, J.; Wang, W.; Li, K.; Qi, X.; Gao, Y.; Gao, L.; Liu, C.; Zhang, Y.; et al. Recombinant Avian β-Defensin Produced by Food-Grade Lactococcus as a Novel and Potent Immunological Enhancer Adjuvant for Avian Vaccine. Probiotics Antimicrob. Proteins 2021, 13, 1833. [Google Scholar] [CrossRef]
- Harder, J.; Bartels, J.; Christophers, E.; Schröder, J.-M. Isolation and Characterization of Human B-Defensin-3, a Novel Human Inducible Peptide Antibiotic. J. Biol. Chem. 2001, 276, 5707–5713. [Google Scholar] [CrossRef] [PubMed]
- Morin, K.M.; Arcidiacono, S.; Beckwitt, R.; Mello, C.M. Recombinant Expression of Indolicidin Concatamers in Escherichia Coli. Appl. Microbiol. Biotechnol. 2006, 70, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Perinelli, D.R.; Cespi, M.; Lorusso, N.; Palmieri, G.F.; Bonacucina, G.; Blasi, P. Surfactant Self-Assembling and Critical Micelle Concentration: One Approach Fits All? Langmuir 2020, 36, 5745–5753. [Google Scholar] [CrossRef] [PubMed]
- Bello-Madruga, R.; Sandín, D.; Valle, J.; Gómez, J.; Comas, L.; Larrosa, M.N.; González-López, J.J.; Jiménez, M.Á.; Andreu, D.; Torrent, M. Mining the Heparinome for Cryptic Antimicrobial Peptides That Selectively Kill Gram-Negative Bacteria. Mol. Syst. Biol. 2025, 21, 889–910. [Google Scholar] [CrossRef]
- Sreerama, N.; Woody, R.W. Estimation of Protein Secondary Structure from Circular Dichroism Spectra: Comparison of CONTIN, SELCON, and CDSSTR Methods with an Expanded Reference Set. Anal. Biochem. 2000, 287, 252–260. [Google Scholar] [CrossRef]
- Pribic, R.; Vanstokkum, I.H.M.; Chapman, D.; Haris, P.I.; Bloemendal, M. Protein Secondary Structure from Fourier Transform Infrared and/or Circular Dichroism Spectra. Anal. Biochem. 1993, 214, 366–378. [Google Scholar] [CrossRef]
- Tatulian, S.A. Structural Characterization of Membrane Proteins and Peptides by FTIR and ATR-FTIR Spectroscopy. Methods Mol. Biol. 2013, 974, 177–218. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook. Springer Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar] [CrossRef]
- Zeth, K.; Sancho-Vaello, E. Structural Plasticity of LL-37 Indicates Elaborate Functional Adaptation Mechanisms to Bacterial Target Structures. Int. J. Mol. Sci. 2021, 22, 5200. [Google Scholar] [CrossRef]
- Erickson, H.P. Size and Shape of Protein Molecules at the Nanometer Level Determined by Sedimentation, Gel Filtration, and Electron Microscopy. Biol. Proced. Online 2009, 11, 32–51. [Google Scholar] [CrossRef]
- Susi, H.; Byler, D.M. Resolution-Enhanced Fourier Transform Infrared Spectroscopy of Enzymes. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1986; Volume 130, pp. 290–311. [Google Scholar] [CrossRef]
- Goormaghtigh, E.; Cabiaux, V.; Ruysschaert, J. Secondary Structure and Dosage of Soluble and Membrane Proteins by Attenuated Total Reflection Fourier-transform Infrared Spectroscopy on Hydrated Films. Eur. J. Biochem. 1990, 193, 409–420. [Google Scholar] [CrossRef]
- Jackson, M.; Mantsch, H.H. The Use and Misuse of FTIR Spectroscopy in the Determination of Protein Structure. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 95–120. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Li, Q.; Zhang, P. Soy Protein Isolate and Glycerol Hydrogen Bonding Using Two-Dimensional Correlation (2D-COS) Attenuated Total Reflection Fourier Transform Infrared (ATR FT-IR) Spectroscopy. Appl. Spectrosc. 2017, 71, 2437–2445. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.M.; Jasim, S.B.; Dyer, N.T.; Auvray, F.; Réfrégiers, M.; Hirst, J.D. Electronic Circular Dichroism Spectroscopy of Proteins. Chem 2019, 5, 2751–2774. [Google Scholar] [CrossRef]
- Bello-Madruga, R.; Valle, J.; Jiménez, M.Á.; Torrent, M.; Montero-Alejo, V.; Andreu, D. The C-Terminus of Panusin, a Lobster β-Defensin, Is Crucial for Optimal Antimicrobial Activity and Serum Stability. Pharmaceutics 2023, 15, 1777. [Google Scholar] [CrossRef]
- Yang, M.; Zhang, C.; Zhang, X.; Zhang, M.Z.; Rottinghaus, G.E.; Zhang, S. Structure-Function Analysis of Avian β-Defensin-6 and β-Defensin-12: Role of Charge and Disulfide Bridges. BMC Microbiol. 2016, 16, 210. [Google Scholar] [CrossRef]
- Zhao, L.; Yang, M.; Zhang, M.; Zhang, S. Expression, Purification, and in Vitro Comparative Characterization of Avian Beta-Defensin-2,-6, and-12. Avian Dis. 2014, 58, 541–549. [Google Scholar] [CrossRef]
- Bonucci, A.; Balducci, E.; Pistolesi, S.; Pogni, R. The Defensin–Lipid Interaction: Insights on the Binding States of the Human Antimicrobial Peptide HNP-1 to Model Bacterial Membranes. Biochim. Biophys. Acta—Biomembr. 2013, 1828, 758–764. [Google Scholar] [CrossRef]
- Sani, M.-A.; Separovic, F. How Membrane-Active Peptides Get into Lipid Membranes. Acc. Chem. Res. 2016, 49, 1130–1138. [Google Scholar] [CrossRef]
- Benincasa, M.; Lagatolla, C.; Dolzani, L.; Milan, A.; Pacor, S.; Liut, G.; Tossi, A.; Cescutti, P.; Rizzo, R. Biofilms from Klebsiella Pneumoniae: Matrix Polysaccharide Structure and Interactions with Antimicrobial Peptides. Microorganisms 2016, 4, 26. [Google Scholar] [CrossRef]
- Xia, R.; Xiao, H.; Xu, M.; Hou, L.; Han, Y.; Zhou, Z. Insight into the Inhibitory Activity and Mechanism of Bovine Cathelicidin BMAP 27 against Salmonella Typhimurium. Microb. Pathog. 2024, 187, 106540. [Google Scholar] [CrossRef]
- Podda, E.; Benincasa, M.; Pacor, S.; Micali, F.; Mattiuzzo, M.; Gennaro, R.; Scocchi, M. Dual Mode of Action of Bac7, a Proline-Rich Antibacterial Peptide. Biochim. Biophys. Acta—Gen. Subj. 2006, 1760, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Sancho-Vaello, E.; Gil-Carton, D.; François, P.; Bonetti, E.J.; Kreir, M.; Pothula, K.R.; Kleinekathöfer, U.; Zeth, K. The Structure of the Antimicrobial Human Cathelicidin LL-37 Shows Oligomerization and Channel Formation in the Presence of Membrane Mimics. Sci. Rep. 2020, 10, 17356. [Google Scholar] [CrossRef] [PubMed]
- Sancho-Vaello, E.; François, P.; Bonetti, E.J.; Lilie, H.; Finger, S.; Gil-Ortiz, F.; Gil-Carton, D.; Zeth, K. Structural Remodeling and Oligomerization of Human Cathelicidin on Membranes Suggest Fibril-like Structures as Active Species. Sci. Rep. 2017, 7, 15371. [Google Scholar] [CrossRef] [PubMed]
- Wommack, A.J.; Robson, S.A.; Wanniarachchi, Y.A.; Wan, A.; Turner, C.J.; Wagner, G.; Nolan, E.M. NMR Solution Structure and Condition-Dependent Oligomerization of the Antimicrobial Peptide Human Defensin 5. Biochemistry 2012, 51, 9624–9637. [Google Scholar] [CrossRef]
- Chairatana, P.; Chiang, I.L.; Nolan, E.M. Human α-Defensin 6 Self-Assembly Prevents Adhesion and Suppresses Virulence Traits of Candida Albicans. Biochemistry 2017, 56, 1033–1041. [Google Scholar] [CrossRef]
- Unzueta, U.; Ferrer-Miralles, N.; Cedano, J.; Zikung, X.; Pesarrodona, M.; Saccardo, P.; García-Fruitós, E.; Domingo-Espín, J.; Kumar, P.; Gupta, K.C.; et al. Non-Amyloidogenic Peptide Tags for the Regulatable Self-Assembling of Protein-Only Nanoparticles. Biomaterials 2012, 33, 8714–8722. [Google Scholar] [CrossRef]
- Serna, N.; Sánchez, J.M.; Unzueta, U.; Sánchez-Garcia, L.; Sánchez-Chardi, A.; Mangues, R.; Vázquez, E.; Villaverde, A. Recruiting Potent Membrane Penetrability in Tumor Cell-Targeted Protein-Only Nanoparticles. Nanotechnology 2019, 30, 115101. [Google Scholar] [CrossRef]
- López-Laguna, H.; Unzueta, U.; Conchillo-Solé, O.; Sánchez-Chardi, A.; Pesarrodona, M.; Cano-Garrido, O.; Voltà, E.; Sánchez-García, L.; Serna, N.; Saccardo, P.; et al. Assembly of Histidine-Rich Protein Materials Controlled through Divalent Cations. Acta Biomater. 2019, 83, 257–264. [Google Scholar] [CrossRef]
- Céspedes, M.V.; Unzueta, U.; Tatkiewicz, W.; Sánchez-Chardi, A.; Conchillo-Solé, O.; Álamo, P.; Xu, Z.; Casanova, I.; Corchero, J.L.; Pesarrodona, M.; et al. In Vivo Architectonic Stability of Fully de Novo Designed Protein-Only Nanoparticles. ACS Nano 2014, 8, 4166–4176. [Google Scholar] [CrossRef]
- Serna, N.; Sánchez-García, L.; Sánchez-Chardi, A.; Unzueta, U.; Roldán, M.; Mangues, R.; Vázquez, E.; Villaverde, A. Protein-Only, Antimicrobial Peptide-Containing Recombinant Nanoparticles with Inherent Built-in Antibacterial Activity. Acta Biomater. 2017, 60, 256–263. [Google Scholar] [CrossRef]
- Boniotto, M.; Antcheva, N.; Zelezetsky, I.; Tossi, A.; Palumbo, V.; Verga Falzacappa, M.V.; Sgubin, S.; Braida, L.; Amoroso, A.; Crovella, S. A Study of Host Defence Peptide β-Defensin 3 in Primates. Biochem. J. 2003, 374, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Hoover, D.M.; Yang, D.; Boulègue, C.; Santamaria, F.; Oppenheim, J.J.; Lubkowski, J.; Lu, W. Engineering Disulfide Bridges to Dissect Antimicrobial and Chemotactic Activities of Human β-Defensin 3. Proc. Natl. Acad. Sci. USA 2003, 100, 8880–8885. [Google Scholar] [CrossRef] [PubMed]
- Diao, H.; Guo, C.; Lin, D.; Zhang, Y. Intein-Mediated Expression Is an Effective Approach in the Study of β-Defensins. Biochem. Biophys. Res. Commun. 2007, 357, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhou, L.; Li, J.; Suresh, A.; Verma, C.; Foo, Y.H.; Yap, E.P.H.; Tan, D.T.H.; Beuerman, R.W. Linear Analogues of Human β-Defensin 3: Concepts for Design of Antimicrobial Peptides with Reduced Cytotoxicity to Mammalian Cells. ChemBioChem 2008, 9, 964–973. [Google Scholar] [CrossRef]
- Morgera, F.; Antcheva, N.; Pacor, S.; Quaroni, L.; Berti, F.; Vaccari, L.; Tossi, A. Structuring and Interactions of Human β-Defensins 2 and 3 with Model Membranes. J. Pept. Sci. 2008, 14, 518–523. [Google Scholar] [CrossRef]
- Antcheva, N.; Boniotto, M.; Zelezetsky, I.; Pacor, S.; Falzacappa, M.V.V.; Crovella, S.; Tossi, A. Effects of Positively Selected Sequence Variations in Human and Macaca Fascicularis β-Defensins 2 on Antimicrobial Activity. Antimicrob. Agents Chemother. 2004, 48, 685–688. [Google Scholar] [CrossRef]
- Mandal, M.; Jagannadham, M.V.; Nagaraj, R. Antibacterial Activities and Conformations of Bovine β-Defensin BNBD-12 and Analogs:Structural and Disulfide Bridge Requirements for Activity. Peptides 2002, 23, 413–418. [Google Scholar] [CrossRef]
- Krishnakumari, V.; Sharadadevi, A.; Singh, S.; Nagaraj, R. Single Disulfide and Linear Analogues Corresponding to the Carboxy-Terminal Segment of Bovine β-Defensin-2: Effects of Introducing the β-Hairpin Nucleating Sequence D-Pro-Gly on Antibacterial Activity and Biophysical Properties. Biochemistry 2003, 42, 9307–9315. [Google Scholar] [CrossRef]
- Klüver, E.; Schulz-Maronde, S.; Scheid, S.; Meyer, B.; Forssmann, W.G.; Adermann, K. Structure-Activity Relation of Human β-Defensin 3: Influence of Disulfide Bonds and Cysteine Substitution on Antimicrobial Activity and Cytotoxicity. Biochemistry 2005, 44, 9804–9816. [Google Scholar] [CrossRef]
- Bello-Madruga, R.; Torrent Burgas, M. The Limits of Prediction: Why Intrinsically Disordered Regions Challenge Our Understanding of Antimicrobial Peptides. Comput. Struct. Biotechnol. J. 2024, 23, 972–981. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate Structure Prediction of Biomolecular Interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Name | Sequence |
|---|---|---|
| β-Defensins | BNBD1 | DFASC1HTNGGIC2LPNRC3PGHMIQIGIC4FRPRVKC5C6RSW |
| BNBD3 | QGVRNHVTC1RINRGFC2VPIRC3PGRTRQIGTC4FGPRIKC5C6RSW | |
| Cathelicidins | Bac5 | RFRPPIRRPPIRPPFYPPFRPPIRPPIFPPIRPPFRPPLGPFP |
| BMAP27 | GRFKRFRKKFKKLFKKLSPVIPLLHLG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saubi, C.; Carratalá, J.V.; Bello-Madruga, R.; López-Cano, A.; Navarro, S.; Arís, A.; Garcia-Fruitós, E. Evaluating Host Defense Peptides: A Comparative Analysis of Synthetic Peptides and Recombinant Concatemers. Biomolecules 2025, 15, 980. https://doi.org/10.3390/biom15070980
Saubi C, Carratalá JV, Bello-Madruga R, López-Cano A, Navarro S, Arís A, Garcia-Fruitós E. Evaluating Host Defense Peptides: A Comparative Analysis of Synthetic Peptides and Recombinant Concatemers. Biomolecules. 2025; 15(7):980. https://doi.org/10.3390/biom15070980
Chicago/Turabian StyleSaubi, Cristina, José Vicente Carratalá, Roberto Bello-Madruga, Adrià López-Cano, Susanna Navarro, Anna Arís, and Elena Garcia-Fruitós. 2025. "Evaluating Host Defense Peptides: A Comparative Analysis of Synthetic Peptides and Recombinant Concatemers" Biomolecules 15, no. 7: 980. https://doi.org/10.3390/biom15070980
APA StyleSaubi, C., Carratalá, J. V., Bello-Madruga, R., López-Cano, A., Navarro, S., Arís, A., & Garcia-Fruitós, E. (2025). Evaluating Host Defense Peptides: A Comparative Analysis of Synthetic Peptides and Recombinant Concatemers. Biomolecules, 15(7), 980. https://doi.org/10.3390/biom15070980