
Insights into Membrane Damage by α-Helical and β-Sheet Peptides


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Design of -Helical Peptides
2.2. Simulation Setup
2.3. Experiments
2.3.1. Peptide Synthesis
2.3.2. Peptide Purification and Characterization
2.3.3. Preparation of Large Unilamellar Vesicles (Liposomes)
2.3.4. Phosphorus Concentration Determination
2.3.5. Preparation of Peptide and Liposomes for Leakage Experiments
2.3.6. Circular Dichroism (CD)
2.3.7. Fourier Transform Infrared Spectroscopy (FT-IR)
2.3.8. Cryo-Electron Microscopy (Cryo-EM)
3. Results
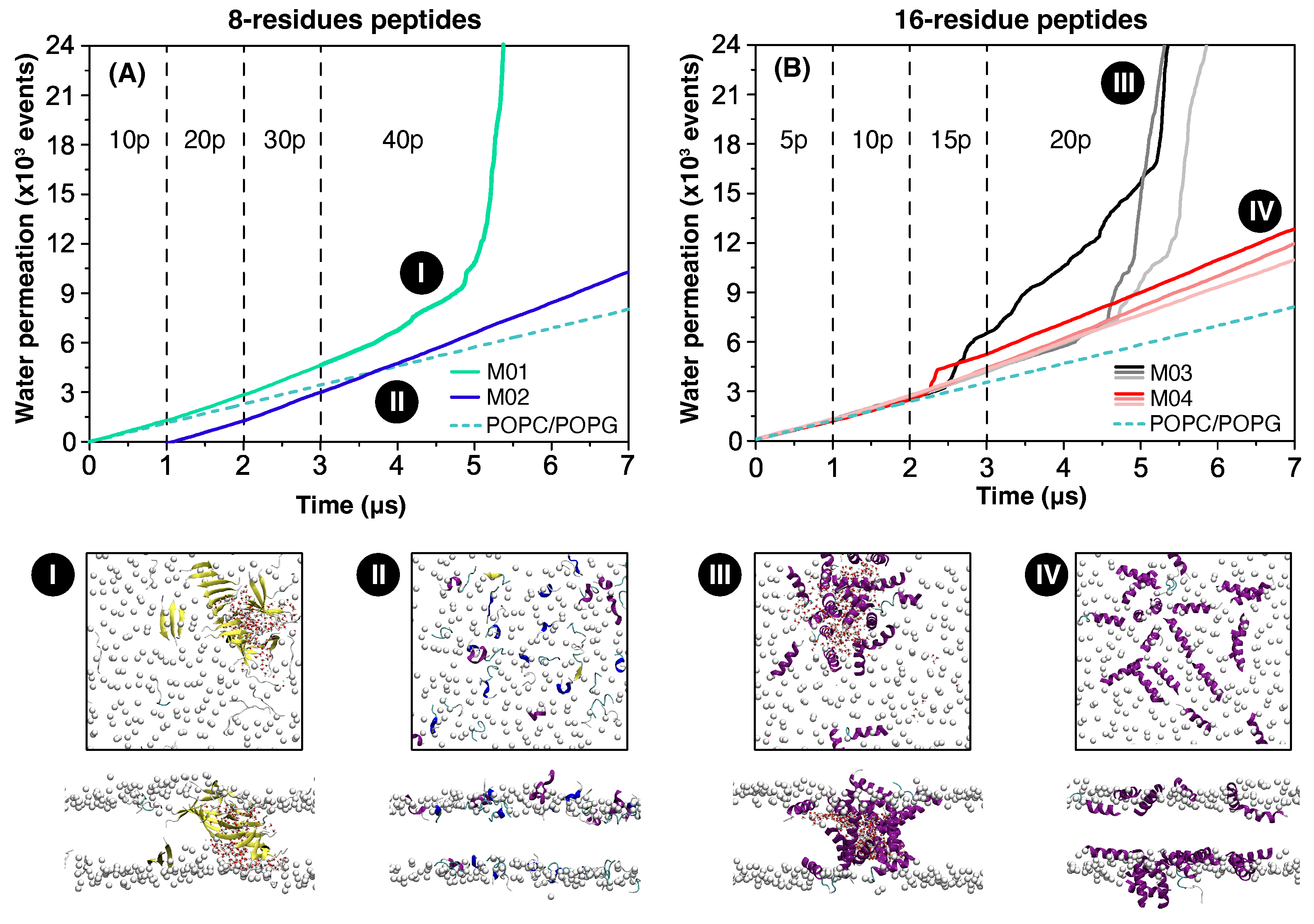
3.1. Water Permeation
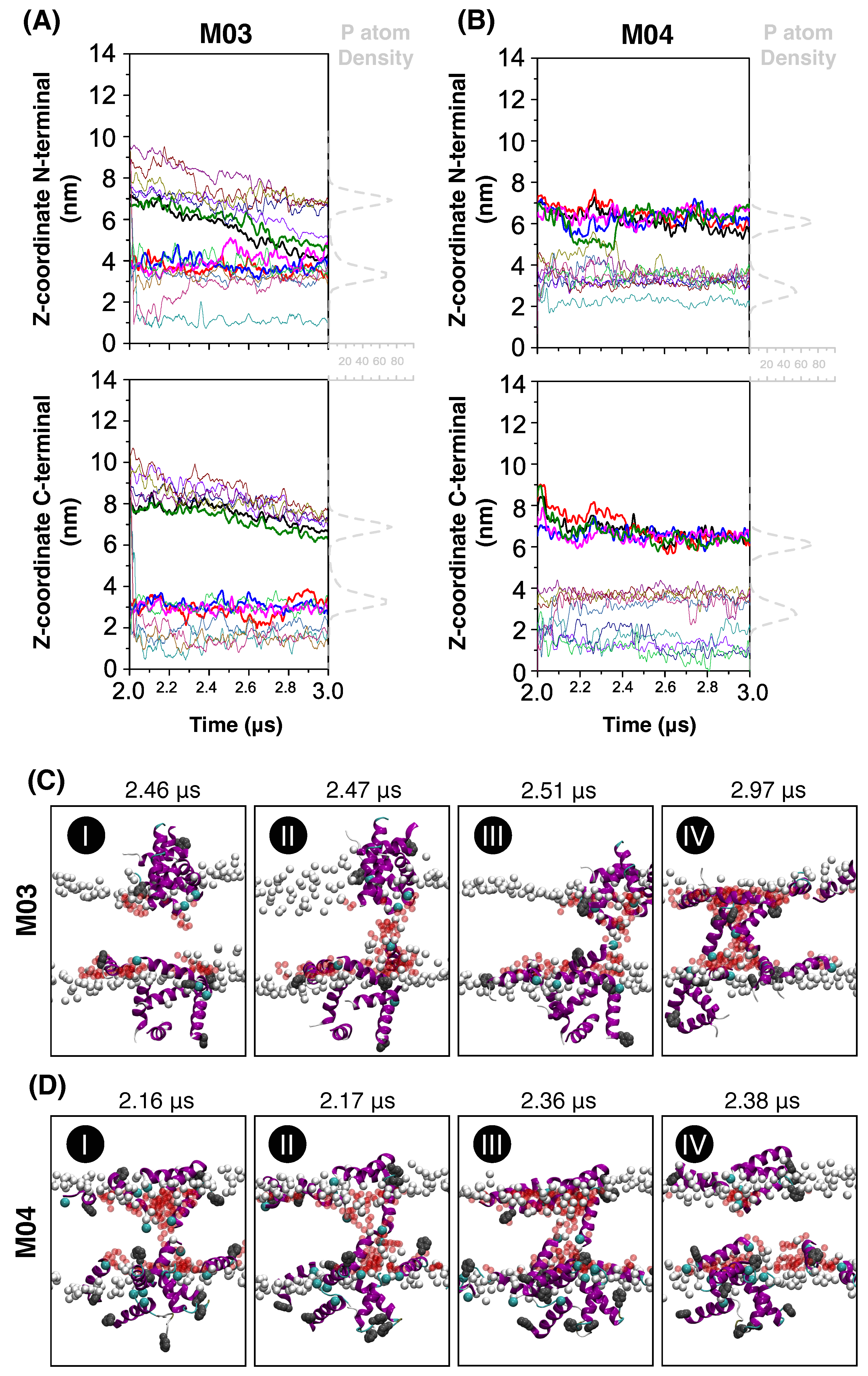
3.2. Mechanism of Membrane Damage
3.3. Experimental Investigation
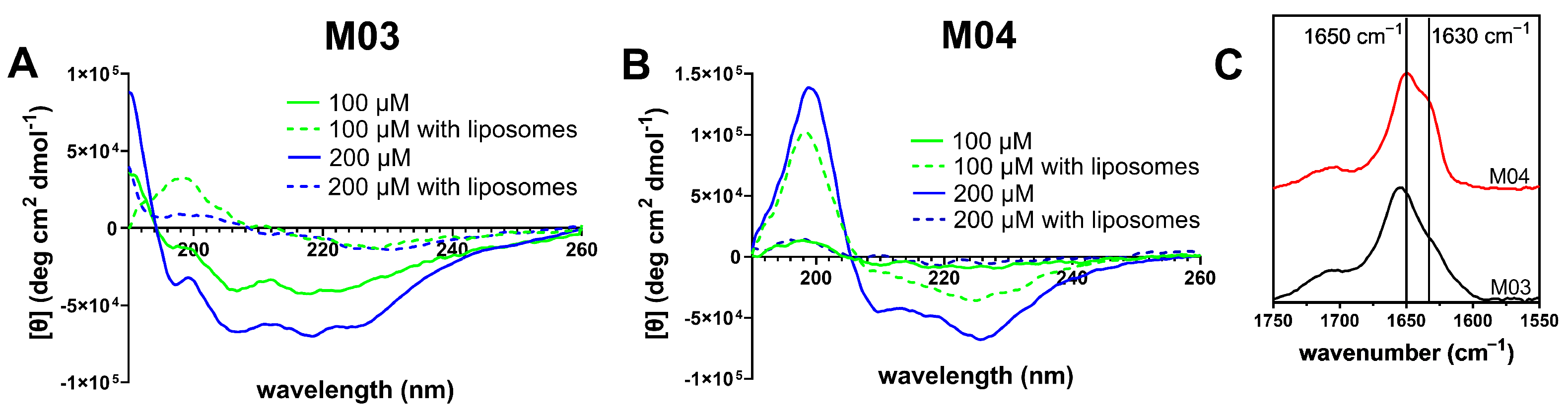
3.3.1. Secondary Structure
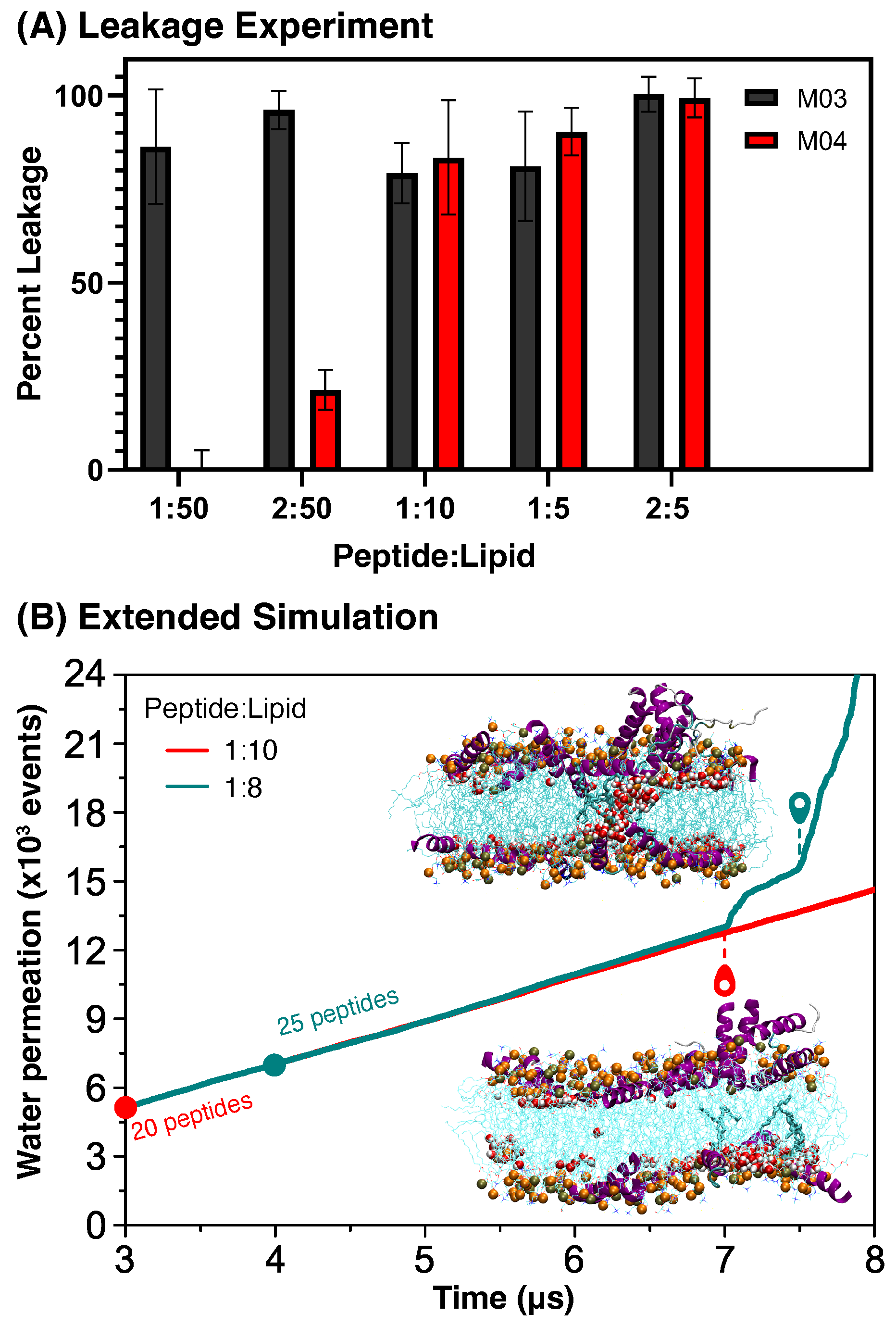
3.3.2. Leakage Assays
3.3.3. High Concentration
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.C.; Gnutt, D.; Gao, M.; Wärmländer, S.K.T.S.; Jarvet, J.; Gräslund, A.; Winter, R.; Ebbinghaus, S.; Strodel, B. Effects of in vivo conditions on amyloid aggregation. Chem. Soc. Rev. 2019, 48, 3946–3996. [Google Scholar] [CrossRef] [PubMed]
- Strikwerda-Brown, C.; Hobbs, D.A.; Gonneaud, J.; St-Onge, F.; Binette, A.P.; Ozlen, H.; Provost, K.; Soucy, J.P.; Buckley, R.F.; Benzinger, T.L.S.; et al. Association of Elevated Amyloid and Tau Positron Emission Tomography Signal With Near-Term Development of Alzheimer Disease Symptoms in Older Adults Without Cognitive Impairment. JAMA Neurol. 2022, 79, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer Disease. Nat. Rev. Dis. Prim. 2021, 7, 33. [Google Scholar] [CrossRef]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Ageitos, J.M.; Sánchez-Pérez, A.; Calo-Mata, P.; Villa, T.G. Antimicrobial peptides (AMPs): Ancient compounds that represent novel weapons in the fight against bacteria. Biochem. Pharmacol. 2017, 133, 117–138. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Kunda, N.K. Antimicrobial peptides as novel therapeutics for non-small cell lung cancer. Drug Discov. Today 2020, 25, 238–247. [Google Scholar] [CrossRef]
- Papo, N.; Shai, Y. Host defense peptides as new weapons in cancer treatment. Cell. Mol. Life Sci. CMLS 2005, 62, 784–790. [Google Scholar] [CrossRef]
- Deslouches, B.; Di, Y.P. Antimicrobial peptides with selective antitumor mechanisms: Prospect for anticancer applications. Oncotarget 2017, 8, 46635–46651. [Google Scholar] [CrossRef]
- Eckert, R.; Qi, F.; Yarbrough, D.K.; He, J.; Anderson, M.H.; Shi, W. Adding Selectivity to Antimicrobial Peptides: Rational Design of a Multidomain Peptide against Pseudomonas spp. Antimicrob. Agents Chemother. 2006, 50, 1480–1488. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Melo, M.C.; Berglund, N.; Khan, A.; de la Fuente-Nunez, C.; Ulmschneider, J.P.; Ulmschneider, M.B. Understanding and modelling the interactions of peptides with membranes: From partitioning to self-assembly. Curr. Opin. Struct. Biol. 2020, 61, 160–166. [Google Scholar] [CrossRef]
- Lipkin, R.; Lazaridis, T. Computational studies of peptide-induced membrane pore formation. Philos. Trans. R. Soc. Biol. Sci. 2017, 372, 20160219. [Google Scholar] [CrossRef] [PubMed]
- Deb, R.; Torres, M.D.T.; Boudný, M.; Koběrská, M.; Cappiello, F.; Popper, M.; Dvořáková Bendová, K.; Drabinová, M.; Hanáčková, A.; Jeannot, K.; et al. Computational Design of Pore-Forming Peptides with Potent Antimicrobial and Anticancer Activities. J. Med. Chem. 2024, 67, 14040–14061. [Google Scholar] [CrossRef]
- Kabelka, I.; Vácha, R. Advances in Molecular Understanding of α-Helical Membrane-Active Peptides. Accounts Chem. Res. 2021, 54, 2196–2204. [Google Scholar] [CrossRef]
- Wang, G.; Zietz, C.M.; Mudgapalli, A.; Wang, S.; Wang, Z. The evolution of the antimicrobial peptide database over 18 years: Milestones and new features. Protein Sci. 2022, 31, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.; Fu, H.; Ma, X. Design, optimization, and nanotechnology of antimicrobial peptides: From exploration to applications. Nano Today 2021, 39, 101229. [Google Scholar] [CrossRef]
- Bin Hafeez, A.; Jiang, X.; Bergen, P.J.; Zhu, Y. Antimicrobial Peptides: An Update on Classifications and Databases. Int. J. Mol. Sci. 2021, 22, 11691. [Google Scholar] [CrossRef]
- Willdigg, J.R.; Helmann, J.D. Mini Review: Bacterial Membrane Composition and Its Modulation in Response to Stress. Front. Mol. Biosci. 2021, 8, 634438. [Google Scholar] [CrossRef]
- Yang, Y.; Jalali, S.; Nilsson, B.L.; Dias, C.L. Binding Mechanisms of Amyloid-like Peptides to Lipid Bilayers and Effects of Divalent Cations. ACS Chem. Neurosci. 2021, 12, 2027–2035. [Google Scholar] [CrossRef]
- Wiradharma, N.; Khoe, U.; Hauser, C.A.E.; Seow, S.V.; Zhang, S.; Yang, Y.Y. Synthetic cationic amphiphilic α-helical peptides as antimicrobial agents. Biomaterials 2011, 32, 2204–2212. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Hu, J.; Zeng, P.; Pan, F.; Yaseen, M.; Xu, H.; Lu, J.R. Molecular mechanisms of anticancer action and cell selectivity of short α-helical peptides. Biomaterials 2014, 35, 1552–1561. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Yang, C.; Chen, Y.; Wang, F.; Mu, Q.; Zhang, J.; Li, Z.; Pan, F.; Xu, H.; Lu, J.R. Surface Physical Activity and Hydrophobicity of Designed Helical Peptide Amphiphiles Control Their Bioactivity and Cell Selectivity. ACS Appl. Mater. Interfaces 2016, 8, 26501–26510. [Google Scholar] [CrossRef]
- Wu, H.; Ong, Z.Y.; Liu, S.; Li, Y.; Wiradharma, N.; Yang, Y.Y.; Ying, J.Y. Synthetic β-sheet forming peptide amphiphiles for treatment of fungal keratitis. Biomaterials 2015, 43, 44–49. [Google Scholar] [CrossRef]
- Jalali, S.; Yang, Y.; Mahmoudinobar, F.; Singh, S.M.; Nilsson, B.L.; Dias, C. Using all-atom simulations in explicit solvent to study aggregation of amphipathic peptides into amyloid-like fibrils. J. Mol. Liq. 2022, 347, 118283. [Google Scholar] [CrossRef]
- Marini, D.M.; Hwang, W.; Lauffenburger, D.A.; Zhang, S.; Kamm, R.D. Left-Handed Helical Ribbon Intermediates in the Self-Assembly of a β-Sheet Peptide. Nano Lett. 2002, 2, 295–299. [Google Scholar] [CrossRef]
- Lee, N.R.; Bowerman, C.J.; Nilsson, B.L. Effects of Varied Sequence Pattern on the Self-Assembly of Amphipathic Peptides. Biomacromolecules 2013, 14, 3267–3277. [Google Scholar] [CrossRef] [PubMed]
- Rangubpit, W.; Sungted, S.; Wong-Ekkabut, J.; Distaffen, H.E.; Nilsson, B.L.; Dias, C.L. Pore Formation by Amyloid-like Peptides: Effects of the Nonpolar–Polar Sequence Pattern. ACS Chem. Neurosci. 2024, 15, 3354–3362. [Google Scholar] [CrossRef]
- Yang, Y.; Distaffen, H.; Jalali, S.; Nieuwkoop, A.J.; Nilsson, B.L.; Dias, C.L. Atomic Insights into Amyloid-Induced Membrane Damage. ACS Chem. Neurosci. 2022, 13, 2766–2777. [Google Scholar] [CrossRef]
- Soscia, S.J.; Kirby, J.E.; Washicosky, K.J.; Tucker, S.M.; Ingelsson, M.; Hyman, B.; Burton, M.A.; Goldstein, L.E.; Duong, S.; Tanzi, R.E.; et al. The Alzheimer’s Disease-Associated Amyloid β-Protein Is an Antimicrobial Peptide. PLoS ONE 2010, 5, e9505. [Google Scholar] [CrossRef]
- Viles, J.H. Imaging Amyloid-β Membrane Interactions: Ion-Channel Pores and Lipid-Bilayer Permeability in Alzheimer’s Disease. Angew. Chem. Int. Ed. 2023, 62, e202215785. [Google Scholar] [CrossRef] [PubMed]
- Ulmschneider, J.P.; Ulmschneider, M.B. Molecular Dynamics Simulations Are Redefining Our View of Peptides Interacting with Biological Membranes. Accounts Chem. Res. 2018, 51, 1106–1116. [Google Scholar] [CrossRef]
- Sciacca, M.F.M.; Milardi, D.; Messina, G.M.L.; Marletta, G.; Brender, J.R.; Ramamoorthy, A.; La Rosa, C. Cations as Switches of Amyloid-Mediated Membrane Disruption Mechanisms: Calcium and IAPP. Biophys. J. 2013, 104, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Sciacca, M.F.M.; Monaco, I.; Rosa, C.L.; Milardi, D. The active role of Ca2+ ions in Aβ-mediated membrane damage. Chem. Commun. 2018, 54, 3629–3631. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, E.; Schweigardt, F.; Wadhwani, P.; Bürck, J.; Reichert, J.; Cravo, H.L.P.; Burger, L.; Ulrich, A.S. Phosphate-dependent aggregation of [KL]n peptides affects their membranolytic activity. Sci. Rep. 2020, 10, 12300. [Google Scholar] [CrossRef]
- Schweigardt, F.; Strandberg, E.; Wadhwani, P.; Reichert, J.; Bürck, J.; Cravo, H.L.P.; Burger, L.; Ulrich, A.S. Membranolytic Mechanism of Amphiphilic Antimicrobial β-Stranded [KL]n Peptides. Biomedicines 2022, 10, 2071. [Google Scholar] [CrossRef]
- Gagnon, M.C.; Strandberg, E.; Grau-Campistany, A.; Wadhwani, P.; Reichert, J.; Bürck, J.; Rabanal, F.; Auger, M.; Paquin, J.F.; Ulrich, A.S. Influence of the Length and Charge on the Activity of α-Helical Amphipathic Antimicrobial Peptides. Biochemistry 2017, 56, 1680–1695. [Google Scholar] [CrossRef]
- Hollmann, A.; Martínez, M.; Noguera, M.E.; Augusto, M.T.; Disalvo, A.; Santos, N.C.; Semorile, L.; Maffía, P.C. Role of amphipathicity and hydrophobicity in the balance between hemolysis and peptide–membrane interactions of three related antimicrobial peptides. Colloids Surfaces Biointerfaces 2016, 141, 528–536. [Google Scholar] [CrossRef]
- Zhang, J.; Chu, A.; Ouyang, X.; Li, B.; Yang, P.; Ba, Z.; Yang, Y.; Mao, W.; Zhong, C.; Gou, S.; et al. Rationally designed highly amphipathic antimicrobial peptides demonstrating superior bacterial selectivity relative to the corresponding α-helix peptide. Eur. J. Med. Chem. 2025, 286, 117310. [Google Scholar] [CrossRef]
- Raheem, N.; Kumar, P.; Lee, E.; Cheng, J.T.J.; Hancock, R.E.W.; Straus, S.K. Insights into the mechanism of action of two analogues of aurein 2.2. Biochim. Biophys. Acta (BBA)-Biomembr. 2020, 1862, 183262. [Google Scholar] [CrossRef]
- Takechi-Haraya, Y.; Ohgita, T.; Kotani, M.; Kono, H.; Saito, C.; Tamagaki-Asahina, H.; Nishitsuji, K.; Uchimura, K.; Sato, T.; Kawano, R.; et al. Effect of hydrophobic moment on membrane interaction and cell penetration of apolipoprotein E-derived arginine-rich amphipathic α-helical peptides. Sci. Rep. 2022, 12, 4959. [Google Scholar] [CrossRef]
- Mahdi, S.; Mechler, A. The role of C-terminal amidation in the mechanism of action of the antimicrobial peptide aurein 1.2. Eurobiotech J. 2020, 4, 25. [Google Scholar] [CrossRef]
- Strandberg, E.; Tiltak, D.; Ieronimo, M.; Kanithasen, N.; Wadhwani, P.; Ulrich, A.S. Influence of C-terminal amidation on the antimicrobial and hemolytic activities of cationic α-helical peptides. Pure Appl. Chem. 2007, 79, 717–728. [Google Scholar] [CrossRef]
- Chen, N.; Jiang, C. Antimicrobial peptides: Structure, mechanism, and modification. Eur. J. Med. Chem. 2023, 255, 115377. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Separovic, F.; Sani, M.A. C-Terminus Amidation Influences Biological Activity and Membrane Interaction of Maculatin 1.1. Biophys. J. 2021, 120, 142a–143a. [Google Scholar] [CrossRef]
- Maloy, W.L.; Kari, U.P. Structure–activity studies on magainins and other host defense peptides. Biopolym. Orig. Res. Biomol. 1995, 37, 105–122. [Google Scholar] [CrossRef]
- Gautier, R.; Douguet, D.; Antonny, B.; Drin, G. HELIQUEST: A web server to screen sequences with specific α-helical properties. Bioinformatics 2008, 24, 2101–2102. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Lindahl, E.; Abraham, M.J.; Hess, B.; van der Spoel, D. GROMACS 2020:User and Reference Manual; Version 2020; Zenodo: Geneva, Switzerland, 2020. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Roberts, B.R.; Lind, M.; Wagen, A.Z.; Rembach, A.; Frugier, T.; Li, Q.X.; Ryan, T.M.; McLean, C.A.; Doecke, J.D.; Rowe, C.C.; et al. Biochemically-defined pools of amyloid-β in sporadic Alzheimer’s disease: Correlation with amyloid PET. Brain 2017, 140, 1486–1498. [Google Scholar] [CrossRef]
- Gravina, S.A.; Ho, L.; Eckman, C.B.; Long, K.E.; Otvos, L.; Younkin, L.H.; Suzuki, N.; Younkin, S.G. Amyloid β Protein (Aβ) in Alzheimeri’s Disease Brain: Biochemical and immunocytochemical analysis with antibodies specific for forms ending at Aβ40 OR Aβ42(43) (*). J. Biol. Chem. 1995, 270, 7013–7016. [Google Scholar] [CrossRef]
- Näslund, J.; Haroutunian, V.; Mohs, R.; Davis, K.L.; Davies, P.; Greengard, P.; Buxbaum, J.D. Correlation Between Elevated Levels of Amyloid β-Peptide in the Brain and Cognitive Decline. JAMA 2000, 283, 1571–1577. [Google Scholar] [CrossRef]
- Urbanc, B. Cross-Linked Amyloid β-Protein Oligomers: A Missing Link in Alzheimer’s Disease Pathology? J. Phys. Chem. B 2021, 125, 1307–1316. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Derreumaux, P. An S-Shaped Aβ42 Cross-β Hexamer Embedded into a Lipid Bilayer Reveals Membrane Disruption and Permeability. ACS Chem. Neurosci. 2023, 14, 936–946. [Google Scholar] [CrossRef]
- Matthes, D.; Groot, B.L.D. Molecular dynamics simulations reveal the importance of amyloid-beta oligomer β-sheet edge conformations in membrane permeabilization. J. Biol. Chem. 2023, 299, 103034. [Google Scholar] [CrossRef]
- Wang, K.; Cai, W. Aggregation, structure and water permeability of membrane-embedded helical Aβ oligomers. Phys. Chem. Chem. Phys. 2024, 26, 5128–5140. [Google Scholar] [CrossRef] [PubMed]
- Hardiagon, A.; Murail, S.; Huang, L.B.; van der Lee, A.; Sterpone, F.; Barboiu, M.; Baaden, M. Molecular dynamics simulations reveal statistics and microscopic mechanisms of water permeation in membrane-embedded artificial water channel nanoconstructs. J. Chem. Phys. 2021, 154, 184102. [Google Scholar] [CrossRef] [PubMed]
- Wimley, W.C. Determining the Effects of Membrane-Interacting Peptides on Membrane Integrity. In Cell-Penetrating Peptides: Methods and Protocols; Methods in Molecular Biology; Langel, Ed.; Springer: New York, NY, USA, 2015; pp. 89–106. [Google Scholar] [CrossRef]
- Wang, F.; Gnewou, O.; Wang, S.; Osinski, T.; Zuo, X.; Egelman, E.H.; Conticello, V.P. Deterministic chaos in the self-assembly of β sheet nanotubes from an amphipathic oligopeptide. Matter 2021, 4, 3217–3231. [Google Scholar] [CrossRef]
- Cerpa, R.; Cohen, F.E.; Kuntz, I.D. Conformational switching in designed peptides: The helix/sheet transition. Fold. Des. 1996, 1, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Ismail, A.A.; Mantsch, H.H. Salt bridge induced changes in the secondary structure of ionic polypeptides. Biopolymers 1992, 32, 1181–1186. [Google Scholar] [CrossRef]
- Przybycien, T.M.; Bailey, J.E. Secondary structure perturbations in salt-induced protein precipitates. Biochim. Biophys. Acta (BBA)-Protein Struct. Mol. Enzymol. 1991, 1076, 103–111. [Google Scholar] [CrossRef]
- Smorodina, E.; Kav, B.; Fatafta, H.; Strodel, B. Effects of ion type and concentration on the structure and aggregation of the amyloid peptide A. Proteins: Struct. Funct. Bioinform. 2023. [Google Scholar] [CrossRef]
- Rogers, D.M.; Jasim, S.B.; Dyer, N.T.; Auvray, F.; Réfrégiers, M.; Hirst, J.D. Electronic Circular Dichroism Spectroscopy of Proteins. Chem 2019, 5, 2751–2774. [Google Scholar] [CrossRef]
- Wimley, W.C.; Hristova, K.; Ladokhin, A.S.; Silvestro, L.; Axelsen, P.H.; White, S.H. Folding of β-sheet membrane proteins: A hydrophobic hexapeptide model. J. Mol. Biol. 1998, 277, 1091–1110. [Google Scholar] [CrossRef]
- Wimley, W.C.; White, S.H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 1996, 3, 842–848. [Google Scholar] [CrossRef]
- Gupta, K.; Jang, H.; Harlen, K.; Puri, A.; Nussinov, R.; Schneider, J.P.; Blumenthal, R. Mechanism of Membrane Permeation Induced by Synthetic β-Hairpin Peptides. Biophys. J. 2013, 105, 2093–2103. [Google Scholar] [CrossRef] [PubMed]
- Kleinschmidt, J.H. Structural Characterization of Membrane Proteins and Peptides by FTIR and ATR-FTIR Spectroscopy. In Lipid-Protein Interactions: Methods and Protocols; Humana Press: Totowa, NJ, USA, 2013; pp. 177–218. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rangubpit, W.; Distaffen, H.E.; Nilsson, B.L.; Dias, C.L. Insights into Membrane Damage by α-Helical and β-Sheet Peptides. Biomolecules 2025, 15, 973. https://doi.org/10.3390/biom15070973
Rangubpit W, Distaffen HE, Nilsson BL, Dias CL. Insights into Membrane Damage by α-Helical and β-Sheet Peptides. Biomolecules. 2025; 15(7):973. https://doi.org/10.3390/biom15070973
Chicago/Turabian StyleRangubpit, Warin, Hannah E. Distaffen, Bradley L. Nilsson, and Cristiano L. Dias. 2025. "Insights into Membrane Damage by α-Helical and β-Sheet Peptides" Biomolecules 15, no. 7: 973. https://doi.org/10.3390/biom15070973
APA StyleRangubpit, W., Distaffen, H. E., Nilsson, B. L., & Dias, C. L. (2025). Insights into Membrane Damage by α-Helical and β-Sheet Peptides. Biomolecules, 15(7), 973. https://doi.org/10.3390/biom15070973