
Targeting Mitochondrial Quality Control for the Treatment of Triple-Negative Breast Cancer: From Molecular Mechanisms to Precision Therapy

 , ,
, ,
Abstract
1. Introduction
2. Triple-Negative Breast Cancer
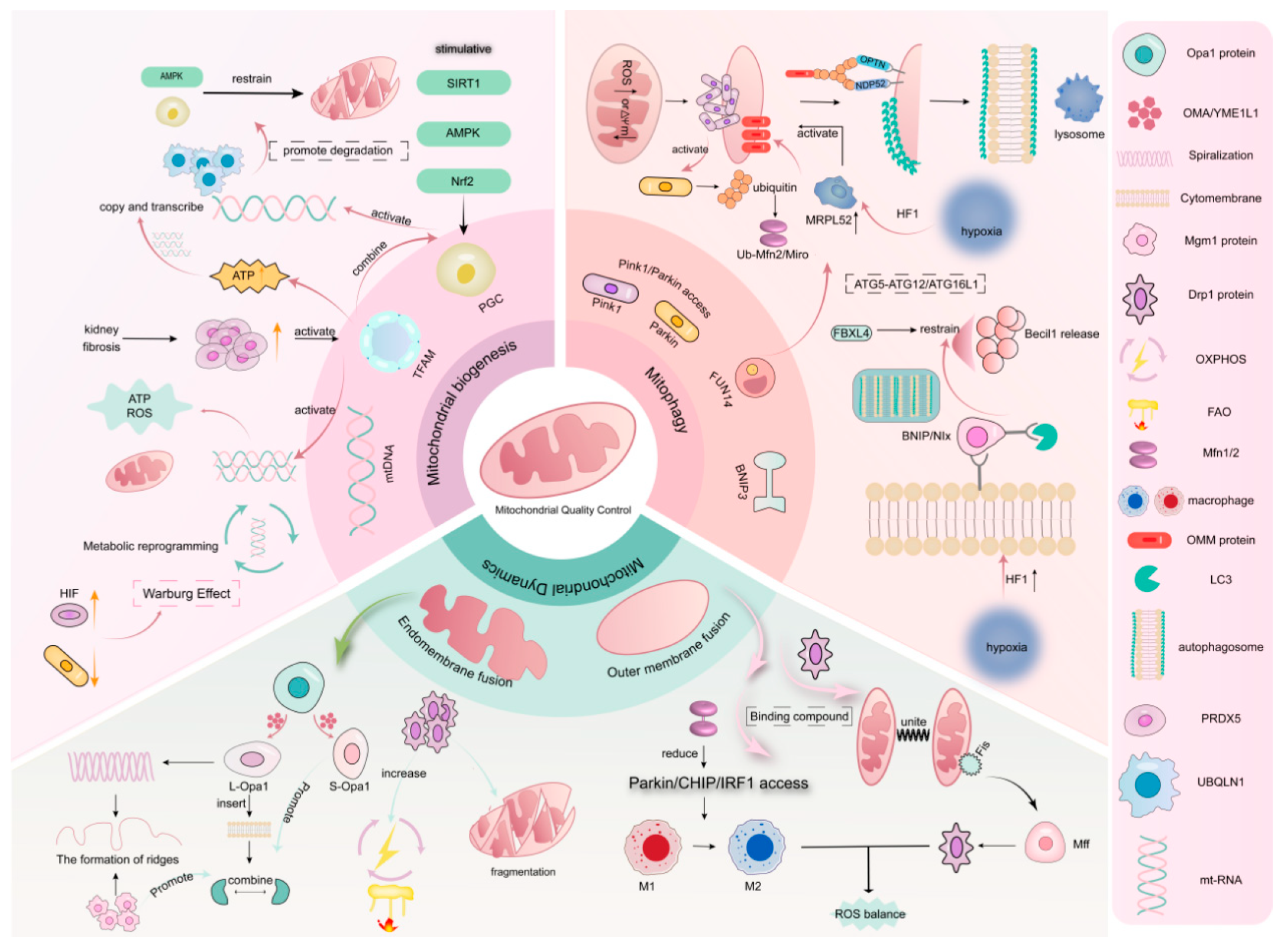
3. Mitochondrial Quality Control
3.1. Mitochondrial Biogenesis
3.2. Mitophagy
3.2.1. PINK1/Parkin Signaling Pathway
3.2.2. BNIP Signaling Pathway
3.3. Mitochondrial Dynamics
3.3.1. Mitochondrial Fusion
3.3.2. Mitochondrial Division
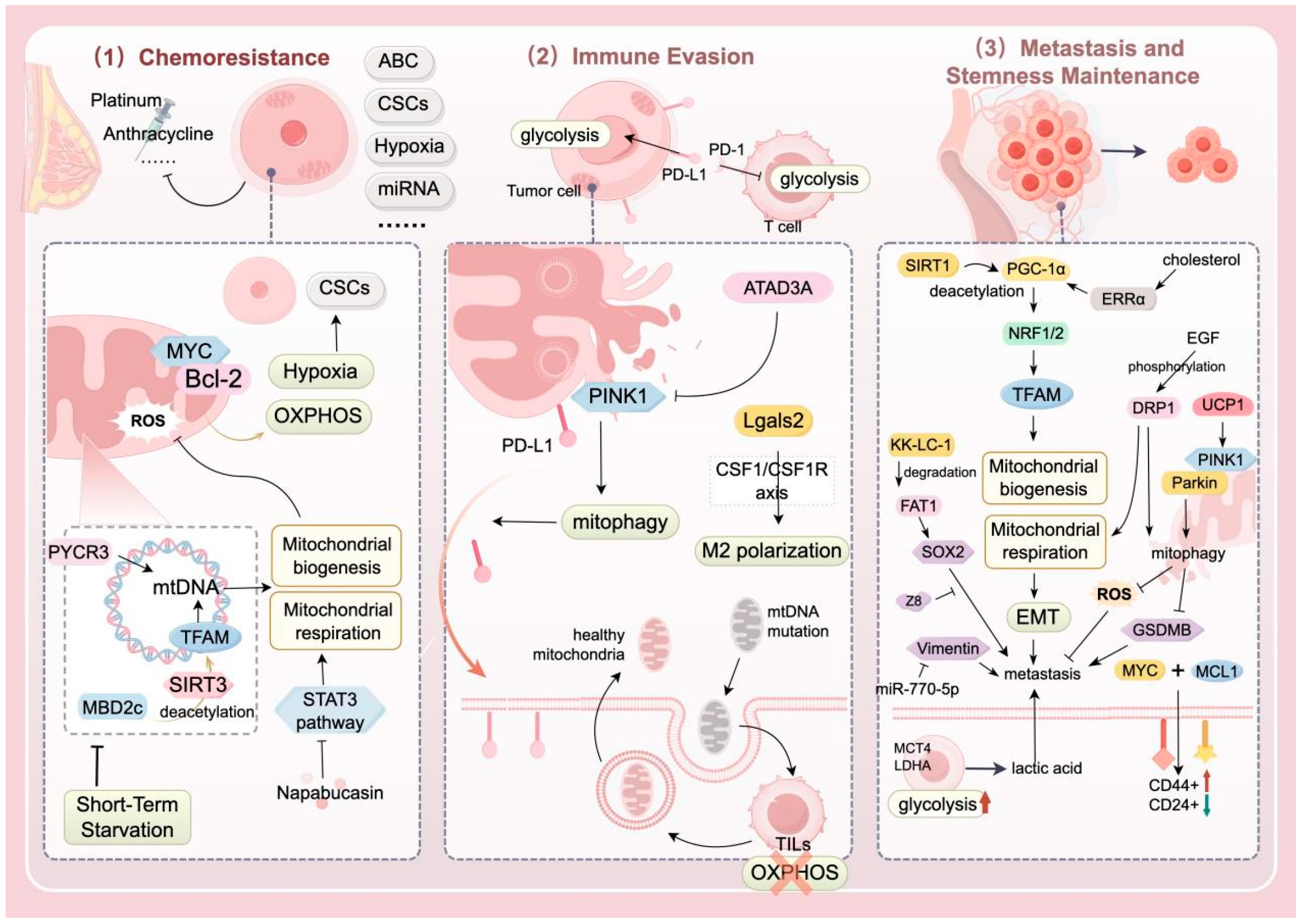
4. Relationship Between MQC and TNBC
4.1. Impact of MQC Abnormalities on the Progression and Prognosis of TNBC
4.1.1. Chemotherapy Resistance
4.1.2. Immune Evasion
4.1.3. Metastasis and Stemness Maintenance

{kind=link}
{kind=link}
{kind=link}
| Mechanism | Function | Author | References |
|---|---|---|---|
| Chemotherapy resistance | Drug efflux; relatively quiescent CSCs; hypoxic microenvironment; evasion of apoptosis; unique miRNA expression profile; intratumoral heterogeneity in TNBC. | Nedeljković M. | [5] |
| Mitochondria-driven metabolic reprogramming promotes tumor survival and disease progression, and cancer stem cells in TNBC tend to favor OXPHOS metabolism. | Missiroli S. | [10,78,79] | |
| MYC and MCL are enriched in drug-resistant TNBC cells, and they enhance mitochondrial OXPHOS, activate the hypoxia pathway, and mediate drug-resistant characteristics. | Lee K.M. | [10] | |
| MBD2c activates TFAM through SIRT3-mediated deacetylation, thereby upregulating the transcription of mtDNA and mitochondrial respiration. Meanwhile, inhibiting MBD2c can significantly enhance the sensitivity to cisplatin. | Hao Y. | [80] | |
| Immune evasion | Cells treated with metformin and 2-deoxy-D-glucose show mitochondria with normal morphology and improved quality under electron microscopy. Glycosylated PD-L1 in these cells is significantly inhibited, and they exhibit potent anti-tumor efficacy. | Repas J. | [85] |
| In paclitaxel-resistant TNBC cells, the expression of ATAD3A is upregulated, inhibiting mitophagy and leading to a decrease in mitochondrial PD-L1, which enhances immune evasion. | Xie X.Q. | [86] | |
| Cancer cells in TNBC induce metabolic reprogramming and functional exhaustion of T cells through a bidirectional mitochondrial transfer mechanism. | Ikeda H. | [87] | |
| Lgals2 can regulate the CSF1/CSF1R axis to induce M2 polarization of macrophages, promoting mitochondrial transfer and immune evasion. | Ji P. | [88] | |
| Metastasis and stemness maintenance | EMT reduces E-cadherin, and the abnormal methylation of the E-cadherin promoter affects the regulation of tumor cell motility and invasiveness. | Noyan S. | [91] |
| Using the antimicrobial peptide merecidin to increase the expression of miR-770-5p can negatively regulate vimentin, thereby delaying the metastasis and proliferation of breast cancer cells. | Ma F. | [92] | |
| SIRT1 controls the outcome of EMT by regulating mitochondrial biogenesis and energy metabolism. | Zhang J. | [9] | |
| PGC-1α is deacetylated and activated by SIRT1, and it binds with the factors NRF-1 and NRF-2 to regulate the expression of genes such as TFAM and TFB1M, affecting the process of mitochondrial biogenesis. When PGC-1α is highly expressed, it drives OXPHOS, thereby promoting the metastasis of TNBC. | Fan S. | [93] | |
| TNBC metabolism is characterized by a preference for glycolysis and low OXPHOS activity, a shift associated with tumor metabolic reprogramming and the tumor microenvironment. | Wang Z. | [94] | |
| Low GLUT1/MCT4 in tumor cells, high MCT4 in stromal cells, and high expression of LDHA play a key role in the reverse Warburg effect, which is of great significance for the reversal of TNBC metastasis. | Cheng S.Y. | [95] | |
| Reducing the expression of MYC and MCL1 via siRNA increases the formation of ALDH+ cells. | Lee K.M. | [10] | |
| ALDH+ cells, as potential targets for CSC-directed therapy in TNBC, can intervene in the development process of TNBC through various mechanisms. | Liu C. | [98] | |
| KK-LC-1 is highly expressed in breast tissue and is significantly associated with the migration, invasion, and scratch healing abilities of TNBC cells. | Zhu X. | [99] | |
| KK-LC-1 promotes the ubiquitination and degradation of FAT1, activates the transcription of SOX2 and ALDH1A1, thereby enhancing the self-renewal and invasive capacity of ALDH+ stem cells. | Bu J. | [100] | |
| EGF phosphorylates DRP1, and high expression of DRP1 is positively correlated with the proportion of ALDH+ cells, OXPHOS, and FAO levels, providing energy for CSCs. | Weiner-Gorzel K. | [101] | |
| Activated DRP1 induces the mitochondrial fission process, promoting the stemness and metastasis of TNBC through metabolic reprogramming and ROS regulation. Meanwhile, the increase in DRP1 is accompanied by upregulation of SOD2, which can clear excessive ROS and maintain the survival of CSCs. | Shome R. | [102,103] | |
| The mitochondrial fission process leads to increased ROS levels, stabilizes HIF, and simultaneously activates the NF-κB and HIF-1α pathways, upregulating stemness genes such as OCT4 and NANOG, thereby promoting transcription. | Altea-Manzano P. | [104] | |
| Hypoxia induces the upregulation of HIF-2α expression, thereby promoting miR-141 to target and bind to MALAT1, inhibiting its mediation of cell autophagy, degradation, and metastasis. | Xu F. | [105] |
4.2. Therapeutic Strategies for TNBC Targeting Mitochondrial Quality Control
4.2.1. Available Drug Regimens
4.2.2. Traditional Chinese Medicine and Novel Therapies
4.2.3. Therapeutic Targets of TNBC by Targeting MQC
| Research Strategy | Mechanism of Action | Drug/Target | Research Model | Key Findings |
|---|---|---|---|---|
| Regulation of mitochondrial metabolism | Activates the AMPK-ACC-FAO signaling pathway and inhibits the Src kinase pathway [106] | Metformin + Dasatinib | TNBC mouse model | Suppresses tumor growth and metastasis |
| Reduces mitochondrial complex I activity, inducing mitochondrial dysfunction [111] | α-Curcumin | TNBC cell lines | Induces cancer cell death | |
| Inhibition of the STAT3 signaling pathway and impairment of mitochondrial function (such as reducing the activity of complex I and ATP levels) [81] | Napabucasin | TNBC cell model | Significantly enhance the sensitivity to paclitaxel | |
| Mitochondrial dynamics regulation | Inhibits MYH9-regulated mitochondrial fission [109] | Isotoosendanin (ITSN) | TNBC metastasis model | Inhibits tumor cell metastasis |
| Inhibits mitochondrial fission, promotes fusion, causing dynamic abnormalities [107] | BET inhibitors | TNBC cellular models | Promotes tumor cell apoptosis | |
| Inhibit OPA1-mediated mitochondrial fusion [59] | OPA1 inhibitor (MYLS22) | Chemotherapy-resistant TNBC model | Inhibit the regrowth of residual tumors | |
| Nanodrug delivery system | Disrupts mitochondrial function, inhibits autophagy, enhances antigen presentation [108] | KLA-5-FU/PTX liposomes | Human breast cancer mouse model | Exhibits significant antitumor activity |
| Photodynamic therapy combined with mitochondrial dysfunction [116] | Doxy/Ce6 nanodrugs | Tumor models | Enhances photodynamic efficacy and promotes immune recognition | |
| Induces mitochondrial dysfunction, ROS accumulation, and photothermal therapy [117] | Au@Zn/CeO engineered bacteria | TNBC stem cell-enriched models | Eliminates CSCs | |
| Mitochondrial biogenesis targeting | Inhibits the PGC-1α/ERRα axis, reducing mitochondrial biogenesis [93] | Shikonin (Lithospermum extract) | TNBC cell metastasis model | Suppresses ATP production and tumor metastasis |
| Mitophagic regulation | Increase the expression of PINK1 and Parkin proteins, while impairing the pro-invasive effect of the pyroptosis protein GSDMB on TNBC [126] | UCP1 | TNBC mouse model | Has a positive effect on inhibiting the proliferation and metastasis of TNBC |
| Phosphorylates Cav-1 to restrict mitophagy, leading to damaged mitochondrial accumulation [120] | Cavity protein-1 (Cav-1) phosphorylation | TNBC cell model | Increases mtROS and suppresses cancer cell survival | |
| Induce mitochondrial dysfunction and mitophagy, promoting ROS accumulation | GSE | Human breast cancer mouse model | Inhibit TNBC cell growth | |
| Prognostic models and gene targets | Mitochondrial autophagy-related genes (MRGs) expression correlates with prognosis [121] | MRPS5, PYCR1, C20orf27 (9 MRGs) | TNBC patient cohort | High MRG expression significantly reduces patient survival rates |
| Increase mtDNA copy number and mitochondrial respiration to promote drug resistance [124] | PYCR3 | TNBC cell metastasis model | Knockdown of PYCR3 can reverse the acquired resistance of TNBC to the chemotherapeutic drug doxorubicin. |
5. Summary and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.U.; Claus, E.; Sohl, J.; Razzak, A.R.; Arnaout, A.; Winer, E.P. Sites of distant recurrence and clinical outcomes in patients with metastatic triple-negative breast cancer: High incidence of central nervous system metastases. Cancer 2008, 113, 2638–2645. [Google Scholar] [CrossRef]
- Qian, J.; Ma, Y.; Tahaney, W.M.; Moyer, C.L.; Lanier, A.; Hill, J.; Coleman, D.; Koupaei, N.; Hilsenbeck, S.G.; Savage, M.I.; et al. The novel phosphatase NUDT5 is a critical regulator of triple-negative breast cancer growth. Breast Cancer Res. 2024, 26, 23. [Google Scholar] [CrossRef]
- Nedeljkovic, M.; Damjanovic, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer-How We Can Rise to the Challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef] [PubMed]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; van de Vijver, K.K.; de Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Mandal, S.K.; Arya, S.; Samanta, S.K.; Kumar, A.; Panchpuri, M.; Abdel-Gawad, H.; Dwibedi, V.; Das, N.; Bose, S.; et al. Therapeutic approach for triple-negative breast Cancer through poly (ADP-ribose) Polymerase-1 inhibitors: Current update. Bioorganic Chem. 2025, 163, 108558. [Google Scholar] [CrossRef]
- Barchiesi, G.; Roberto, M.; Verrico, M.; Vici, P.; Tomao, S.; Tomao, F. Emerging Role of PARP Inhibitors in Metastatic Triple Negative Breast Cancer. Current Scenario and Future Perspectives. Front. Oncol. 2021, 11, 769280. [Google Scholar] [CrossRef]
- Zhang, J.; Peng, J.; Kong, D.; Wang, X.; Wang, Z.; Liu, J.; Yu, W.; Wu, H.; Long, Z.; Zhang, W.; et al. Silent information regulator 1 suppresses epithelial-to-mesenchymal transition in lung cancer cells via its regulation of mitochondria status. Life Sci. 2021, 280, 119716. [Google Scholar] [CrossRef]
- Lee, K.M.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sanchez, V.; Sanders, M.E.; et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017, 26, 633–647.e7. [Google Scholar] [CrossRef]
- Schmid, P.; Rugo, H.S.; Adams, S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): Updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 44–59. [Google Scholar] [CrossRef]
- Jie, H.; Ma, W.; Huang, C. Diagnosis, Prognosis, and Treatment of Triple-Negative Breast Cancer: A Review. Breast Cancer 2025, 17, 265–274. [Google Scholar] [CrossRef]
- Jiang, Y.Z.; Ma, D.; Suo, C.; Shi, J.; Xue, M.; Hu, X.; Xiao, Y.; Yu, K.D.; Liu, Y.R.; Yu, Y.; et al. Genomic and Transcriptomic Landscape of Triple-Negative Breast Cancers: Subtypes and Treatment Strategies. Cancer Cell 2019, 35, 428–440.e5. [Google Scholar] [CrossRef] [PubMed]
- Vtorushin, S.; Dulesova, A.; Krakhmal, N. Luminal androgen receptor (LAR) subtype of triple-negative breast cancer: Molecular, morphological, and clinical features. J. Zhejiang Univ. Sci. B 2022, 23, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Popovic, L.S.; Matovina-Brko, G.; Popovic, M.; Punie, K.; Cvetanovic, A.; Lambertini, M. Targeting triple-negative breast cancer: A clinical perspective. Oncol. Res. 2023, 31, 221–238. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef] [PubMed]
- Manjunath, M.; Choudhary, B. Triple-negative breast cancer: A run-through of features, classification and current therapies. Oncol. Lett. 2021, 22, 512. [Google Scholar] [CrossRef]
- Goldhirsch, A.; Winer, E.P.; Coates, A.S.; Gelber, R.D.; Piccart-Gebhart, M.; Thürlimann, B.; Senn, H.J. Personalizing the treatment of women with early breast cancer: Highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann. Oncol. 2013, 24, 2206–2223. [Google Scholar] [CrossRef]
- Agostinetto, E.; Losurdo, A.; Nader-Marta, G.; Santoro, A.; Punie, K.; Barroso, R.; Popovic, L.; Solinas, C.; Kok, M.; de Azambuja, E.; et al. Progress and pitfalls in the use of immunotherapy for patients with triple negative breast cancer. Expert Opin. Investig. Drugs 2022, 31, 567–591. [Google Scholar] [CrossRef]
- Mizumoto, S.; Inubushi, S.; Miki, M.; Nakamura, H.; Baba, M.; Yamashita, Y.; Yamamoto, M.; Inoue, S.; Tanino, H.; Kunihisa, T. Target-Oriented Classification of Triple-negative Breast Cancer. Anticancer. Res. 2023, 43, 5067–5072. [Google Scholar] [CrossRef]
- Jiang, L.; You, C.; Xiao, Y.; Wang, H.; Su, G.H.; Xia, B.Q.; Zheng, R.C.; Zhang, D.D.; Jiang, Y.Z.; Gu, Y.J.; et al. Radiogenomic analysis reveals tumor heterogeneity of triple-negative breast cancer. Cell Rep. Med. 2022, 3, 100694. [Google Scholar] [CrossRef]
- Futamura, M.; Tokumaru, Y.; Takabe, K.; Arakawa, H.; Asano, Y.; Mori, R.; Mase, J.; Nakakami, A.; Yoshida, K. MIEAP, a p53-downstream gene, is associated with suppression of breast cancer cell proliferation and better survival. Am. J. Cancer Res. 2021, 11, 6060–6073. [Google Scholar]
- Liao, Y.; Octaviani, S.; Tian, Z.; Wang, S.R.; Huang, C.; Huang, J. Mitochondrial quality control in hematopoietic stem cells: Mechanisms, implications, and therapeutic opportunities. Stem Cell Res. Ther. 2025, 16, 180. [Google Scholar] [CrossRef]
- Liu, H.; Zhen, C.; Xie, J.; Luo, Z.; Zeng, L.; Zhao, G.; Lu, S.; Zhuang, H.; Fan, H.; Li, X.; et al. TFAM is an autophagy receptor that limits inflammation by binding to cytoplasmic mitochondrial DNA. Nat. Cell Biol. 2024, 26, 878–891. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.H.; Tan, R.Z.; Lin, J.Y.; Li, T.; Jia, J.; Wu, L.H.; Wang, R.; He, Y.H.; Su, H.W.; Li, P.; et al. Chaihuang Yishen Granule ameliorates mitochondrial homeostasis by upregulating PRDX5/TFAM axis to inhibit renal fibrosis in CKD. Phytomedicine 2025, 139, 156426. [Google Scholar] [CrossRef]
- Guo, B.; Zheng, C.; Cao, J.; Luo, F.; Li, H.; Hu, S.; Lee, S.M.; Yang, X.; Zhang, G.; Zhang, Z.; et al. Tetramethylpyrazine nitrone exerts neuroprotection via activation of PGC-1alpha/Nrf2 pathway in parkinson’s disease models. J. Adv. Res. 2024, 64, 195–211. [Google Scholar] [CrossRef]
- Li, B.; Yu, K.; Zhou, X.; Sun, J.; Qi, L.; Li, W.; Yang, T.; Li, W.; Wang, N.; Gu, X.; et al. Increased TSPO alleviates neuropathic pain by preventing pyroptosis via the AMPK-PGC-1α pathway. J. Headache Pain 2025, 26, 16. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chen, X.; Zhao, Q.; Li, G.; Zhang, H.; Ma, Z.; Yu, H.; Zeng, Q.; Zhang, H.; Xu, D. Berberine improves cardiac insufficiency through AMPK/PGC-1α signaling-mediated mitochondrial homeostasis and apoptosis in HFpEF mice. Int. Immunopharmacol. 2025, 155, 114613. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ji, L.; Ruan, Y.; Wan, Z.; Lin, Z.; Xia, S.; Tao, L.; Zheng, J.; Cai, L.; Wang, Y.; et al. UBQLN1 mediates sorafenib resistance through regulating mitochondrial biogenesis and ROS homeostasis by targeting PGC1β in hepatocellular carcinoma. Signal Transduct. Target. Ther. 2021, 6, 190. [Google Scholar] [CrossRef]
- Liu, S.; Li, Y.; Yuan, M.; Song, Q.; Liu, M. Correlation between the Warburg effect and progression of triple-negative breast cancer. Front. Oncol. 2022, 12, 1060495. [Google Scholar] [CrossRef]
- Fang, Y.; Zhang, Q.; Lv, C.; Guo, Y.; He, Y.; Guo, P.; Wei, Z.; Xia, Y.; Dai, Y. Mitochondrial fusion induced by transforming growth factor-β1 serves as a switch that governs the metabolic reprogramming during differentiation of regulatory T cells. Redox Biol. 2023, 62, 102709. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.L.; Dai, Y.F.; You, M.X.; Li, C.H. Choline dehydrogenase interacts with SQSTM1 to activate mitophagy and promote coelomocyte survival in Apostichopus japonicus following Vibrio splendidus infection. Zool. Res. 2023, 44, 905–918. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Sun, Z.; Tong, G.; Zeng, L.; He, W.; Chen, X.; Zhen, C.; Chen, P.; Tan, N.; He, P. FUNDC1 alleviates doxorubicin-induced cardiotoxicity by restoring mitochondrial-endoplasmic reticulum contacts and blocked autophagic flux. Theranostics 2024, 14, 3719–3738. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Agarwal, E.; Bertolini, I.; Seo, J.H.; Caino, M.C.; Ghosh, J.C.; Kossenkov, A.V.; Liu, Q.; Tang, H.Y.; Goldman, A.R.; et al. The mitophagy effector FUNDC1 controls mitochondrial reprogramming and cellular plasticity in cancer cells. Sci. Signal. 2020, 13, eaaz8240. [Google Scholar] [CrossRef]
- Zheng, B.; Wang, Y.; Zhou, B.; Qian, F.; Liu, D.; Ye, D.; Zhou, X.; Fang, L. Urolithin A inhibits breast cancer progression via activating TFEB-mediated mitophagy in tumor macrophages. J. Adv. Res. 2025, 69, 125–138. [Google Scholar] [CrossRef]
- Ma, K.Y.; Fokkens, M.R.; Reggiori, F.; Mari, M.; Verbeek, D.S. Parkinson’s disease-associated VPS35 mutant reduces mitochondrial membrane potential and impairs PINK1/Parkin-mediated mitophagy. Transl. Neurodegener. 2021, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chan, S.W.; Zhao, H.; Miu, K.K.; Chan, W.Y. Outlook of PINK1/Parkin signaling in molecular etiology of Parkinson’s disease, with insights into Pink1 knockout models. Zool. Res. 2023, 44, 559–576. [Google Scholar] [CrossRef]
- Liu, Y.; Yao, C.; Sheng, B.; Zhi, S.; Chen, X.; Ding, P.; Zhang, J.; Tao, Z.; Li, W.; Zhuang, Z.; et al. Inhibition of USP30 Promotes Mitophagy by Regulating Ubiquitination of MFN2 by Parkin to Attenuate Early Brain Injury After SAH. Transl. Stroke Res. 2025, 16, 448–466. [Google Scholar] [CrossRef]
- Lee, K.J.; Mann, E.; Wright, G.; Piett, C.G.; Nagel, Z.D.; Gassman, N.R. Exploiting DNA repair defects in triple negative breast cancer to improve cell killing. Ther. Adv. Med. Oncol. 2020, 12, 1758835920958354. [Google Scholar] [CrossRef]
- Li, X.; Wang, M.; Li, S.; Chen, Y.; Wang, M.; Wu, Z.; Sun, X.; Yao, L.; Dong, H.; Song, Y.; et al. HIF-1-induced mitochondrial ribosome protein L52: A mechanism for breast cancer cellular adaptation and metastatic initiation in response to hypoxia. Theranostics 2021, 11, 7337–7359. [Google Scholar] [CrossRef]
- Xia, M.; Li, C.; Chen, J.; Wu, C.; Zhang, J.; Hong, H.; Jiang, J.; Xu, G.; Qian, Z.; Cui, Z. Activation of FANCC attenuates mitochondrial ROS-driven necroptosis by targeting TBK1-dependent mitophagy in astrocytes after spinal cord injury. Theranostics 2025, 15, 4188–4211. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Liu, Y.; Zhang, Q.; Xu, H.; Fang, Y.; Chen, X.; Fu, J.; Yuan, Y.; Li, Y.; Yuan, L.; et al. Human menstrual blood-derived stem cells reverse sorafenib resistance in hepatocellular carcinoma cells through the hyperactivation of mitophagy. Stem Cell Res. Ther. 2023, 14, 58. [Google Scholar] [CrossRef]
- Zhu, H.L.; Shi, X.T.; Xu, X.F.; Zhou, G.X.; Xiong, Y.W.; Yi, S.J.; Liu, W.B.; Dai, L.M.; Cao, X.L.; Xu, D.X.; et al. Melatonin protects against environmental stress-induced fetal growth restriction via suppressing ROS-mediated GCN2/ATF4/BNIP3-dependent mitophagy in placental trophoblasts. Redox Biol. 2021, 40, 101854. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, J.; Sun, Q.; Xue, Z.; Tang, Z.; Liu, W.; Liu, J.; Miao, B.; Su, N.; He, Y.; et al. Calnexin promotes glioblastoma progression by inducing protective mitophagy through the MEK/ERK/BNIP3 pathway. Theranostics 2025, 15, 2624–2648. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.J.; Wang, Z.Y.; Xu, L.; Chen, X.H.; Li, X.X.; Liao, W.T.; Ma, H.K.; Jiang, M.D.; Xu, T.T.; Xu, J.; et al. HIF-1α-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol. 2020, 36, 101671. [Google Scholar] [CrossRef]
- Chiu, H.W.; Yeh, Y.L.; Wang, Y.C.; Huang, W.J.; Ho, S.Y.; Lin, P.; Wang, Y.J. Combination of the novel histone deacetylase inhibitor YCW1 and radiation induces autophagic cell death through the downregulation of BNIP3 in triple-negative breast cancer cells in vitro and in an orthotopic mouse model. Mol. Cancer 2016, 15, 46. [Google Scholar] [CrossRef]
- Kuhikar, R.; Khan, N.; Philip, J.; Melinkeri, S.; Kale, V.; Limaye, L. Transforming growth factor β1 accelerates and enhances in vitro red blood cell formation from hematopoietic stem cells by stimulating mitophagy. Stem Cell Res. Ther. 2020, 11, 71. [Google Scholar] [CrossRef]
- Sulkshane, P.; Ram, J.; Thakur, A.; Reis, N.; Kleifeld, O.; Glickman, M.H. Ubiquitination and receptor-mediated mitophagy converge to eliminate oxidation-damaged mitochondria during hypoxia. Redox Biol. 2021, 45, 102047. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Urbe, S. Diverse routes to mitophagy governed by ubiquitylation and mitochondrial import. Trends Cell Biol. 2025, 35, 527–538. [Google Scholar] [CrossRef]
- Zacharioudakis, E.; Gavathiotis, E. Mitochondrial dynamics proteins as emerging drug targets. Trends Pharmacol. Sci. 2023, 44, 112–127. [Google Scholar] [CrossRef]
- Ban, T.; Ishihara, T.; Kohno, H.; Saita, S.; Ichimura, A.; Maenaka, K.; Oka, T.; Mihara, K.; Ishihara, N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol. 2017, 19, 856–863. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Liu, W.; Ding, L.; Zhang, X.; Wang, B.; Tong, Z.; Yue, X.; Li, C.; Xu, L.; et al. TIM-4 orchestrates mitochondrial homeostasis to promote lung cancer progression via ANXA2/PI3K/AKT/OPA1 axis. Cell Death Dis. 2023, 14, 141. [Google Scholar] [CrossRef]
- Ban, T.; Kohno, H.; Ishihara, T.; Ishihara, N. Relationship between OPA1 and cardiolipin in mitochondrial inner-membrane fusion. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.L.; Meng, S.; Chen, Y.; Feng, J.X.; Gu, D.D.; Yu, B.; Li, Y.J.; Yang, J.Y.; Liao, S.; Chan, D.C.; et al. MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature 2017, 542, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Li, J.; Chen, F.; Han, Y.; Chen, D.; Hu, H. Engineering nanoparticles boost TNBC therapy by CD24 blockade and mitochondrial dynamics regulation. J. Control. Release 2023, 355, 211–227. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. 2020, 15, 235–259. [Google Scholar] [CrossRef]
- Yan, L.; Qi, Y.; Ricketson, D.; Li, L.; Subramanian, K.; Zhao, J.; Yu, C.; Wu, L.; Sarsam, R.; Wong, M.; et al. Structural analysis of a trimeric assembly of the mitochondrial dynamin-like GTPase Mgm1. Proc. Natl. Acad. Sci. USA 2020, 117, 4061–4070. [Google Scholar] [CrossRef]
- Baek, M.L.; Lee, J.; Pendleton, K.E.; Berner, M.J.; Goff, E.B.; Tan, L.; Martinez, S.A.; Mahmud, I.; Wang, T.; Meyer, M.D.; et al. Mitochondrial structure and function adaptation in residual triple negative breast cancer cells surviving chemotherapy treatment. Oncogene 2023, 42, 1117–1131. [Google Scholar] [CrossRef]
- Ji, W.K.; Chakrabarti, R.; Fan, X.; Schoenfeld, L.; Strack, S.; Higgs, H.N. Receptor-mediated Drp1 oligomerization on endoplasmic reticulum. J. Cell Biol. 2017, 216, 4123–4139. [Google Scholar] [CrossRef]
- Kashatus, J.A.; Nascimento, A.; Myers, L.J.; Sher, A.; Byrne, F.L.; Hoehn, K.L.; Counter, C.M.; Kashatus, D.F. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol. Cell 2015, 57, 537–551. [Google Scholar] [CrossRef]
- Humphries, B.A.; Cutter, A.C.; Buschhaus, J.M.; Chen, Y.C.; Qyli, T.; Palagama, D.S.W.; Eckley, S.; Robison, T.H.; Bevoor, A.; Chiang, B.; et al. Enhanced mitochondrial fission suppresses signaling and metastasis in triple-negative breast cancer. Breast Cancer Res. 2020, 22, 60. [Google Scholar] [CrossRef]
- Jin, J.Y.; Wei, X.X.; Zhi, X.L.; Wang, X.H.; Meng, D. Drp1-dependent mitochondrial fission in cardiovascular disease. Acta Pharmacol. Sin. 2021, 42, 655–664. [Google Scholar] [CrossRef]
- Garrido-Bazan, V.; Pardo, J.P.; Aguirre, J. DnmA and FisA Mediate Mitochondria and Peroxisome Fission, and Regulate Mitochondrial Function, ROS Production and Development in Aspergillus nidulans. Front. Microbiol. 2020, 11, 837. [Google Scholar] [CrossRef]
- Liu, R.; Chan, D.C. The mitochondrial fission receptor Mff selectively recruits oligomerized Drp1. Mol. Biol. Cell 2015, 26, 4466–4477. [Google Scholar] [CrossRef]
- Yu, S.; Cao, Z.; Cai, F.; Yao, Y.; Chang, X.; Wang, X.; Zhuang, H.; Hua, Z.C. ADT-OH exhibits anti-metastatic activity on triple-negative breast cancer by combinatorial targeting of autophagy and mitochondrial fission. Cell Death Dis. 2024, 15, 463. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, Y.; Guo, Y.; Shi, X.; Chen, X.; Feng, W.; Wu, L.L.; Zhang, J.; Yu, S.; Wang, Y.; et al. An Overview: The Diversified Role of Mitochondria in Cancer Metabolism. Int. J. Biol. Sci. 2023, 19, 897–915. [Google Scholar] [CrossRef] [PubMed]
- Arnold, P.K.; Jackson, B.T.; Paras, K.I.; Brunner, J.S.; Hart, M.L.; Newsom, O.J.; Alibeckoff, S.P.; Endress, J.; Drill, E.; Sullivan, L.B.; et al. A non-canonical tricarboxylic acid cycle underlies cellular identity. Nature 2022, 603, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B.; Cheng, G.; Hardy, M.; You, M. OXPHOS-targeting drugs in oncology: New perspectives. Expert. Opin. Ther. Targets 2023, 27, 939–952. [Google Scholar] [CrossRef]
- Walkon, L.L.; Strubbe-Rivera, J.O.; Bazil, J.N. Calcium Overload and Mitochondrial Metabolism. Biomolecules 2022, 12, 1891. [Google Scholar] [CrossRef]
- Li, J.; Jia, Y.C.; Ding, Y.X.; Bai, J.; Cao, F.; Li, F. The crosstalk between ferroptosis and mitochondrial dynamic regulatory networks. Int. J. Biol. Sci. 2023, 19, 2756–2771. [Google Scholar] [CrossRef] [PubMed]
- Hall-Younger, E.; Tait, S.W. Mitochondria and cell death signalling. Curr. Opin. Cell Biol. 2025, 94, 102510. [Google Scholar] [CrossRef]
- Obidiro, O.; Battogtokh, G.; Akala, E.O. Triple Negative Breast Cancer Treatment Options and Limitations: Future Outlook. Pharmaceutics 2023, 15, 1796. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.A.; Gubbins, L.; Sharma, S.; Tully, R.; Guang, M.H.; Weiner-Gorzel, K.; McCaffrey, J.; Harrison, M.; Furlong, F.; Kell, M.; et al. The fate of chemoresistance in triple negative breast cancer (TNBC). BBA Clin. 2015, 3, 257–275. [Google Scholar] [CrossRef]
- Mosca, L.; Ilari, A.; Fazi, F.; Assaraf, Y.G.; Colotti, G. Taxanes in cancer treatment: Activity, chemoresistance and its overcoming. Drug Resist. Updat. 2021, 54, 100742. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Missiroli, S.; Perrone, M.; Genovese, I.; Pinton, P.; Giorgi, C. Cancer metabolism and mitochondria: Finding novel mechanisms to fight tumours. EBioMedicine 2020, 59, 102943. [Google Scholar] [CrossRef]
- Li, F.; Wang, Y.; Zeller, K.I.; Potter, J.J.; Wonsey, D.R.; O’Donnell, K.A.; Kim, J.W.; Yustein, J.T.; Lee, L.A.; Dang, C.V. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell. Biol. 2005, 25, 6225–6234. [Google Scholar] [CrossRef]
- Hao, Y.; Zhou, Z.; Liu, R.; Shen, S.; Liu, H.; Zhou, Y.; Sun, Y.; Mao, Q.; Zhang, T.; Li, S.T.; et al. Mitochondria-localized MBD2c facilitates mtDNA transcription and drug resistance. Nat. Chem. Biol. 2024, 21, 926–938. [Google Scholar] [CrossRef]
- Yuan, L.; Zhu, Y.; Guan, G.; Liu, M. Napabucasin targets resistant triple negative breast cancer through suppressing STAT3 and mitochondrial function. Cancer Chemother. Pharmacol. 2025, 95, 51. [Google Scholar] [CrossRef]
- Maeda, T.; Hiraki, M.; Jin, C.; Rajabi, H.; Tagde, A.; Alam, M.; Bouillez, A.; Hu, X.; Suzuki, Y.; Miyo, M.; et al. MUC1-C Induces PD-L1 and Immune Evasion in Triple-Negative Breast Cancer. Cancer Res. 2018, 78, 205–215. [Google Scholar] [CrossRef]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Qin, G.; Wang, X.; Ye, S.; Li, Y.; Chen, M.; Wang, S.; Qin, T.; Zhang, C.; Li, Y.; Long, Q.; et al. NPM1 upregulates the transcription of PD-L1 and suppresses T cell activity in triple-negative breast cancer. Nat. Commun. 2020, 11, 1669. [Google Scholar] [CrossRef] [PubMed]
- Repas, J.; Zupin, M.; Vodlan, M.; Veranic, P.; Gole, B.; Potocnik, U.; Pavlin, M. Dual Effect of Combined Metformin and 2-Deoxy-D-Glucose Treatment on Mitochondrial Biogenesis and PD-L1 Expression in Triple-Negative Breast Cancer Cells. Cancers 2022, 14, 1343. [Google Scholar] [CrossRef]
- Xie, X.Q.; Yang, Y.; Wang, Q.; Liu, H.F.; Fang, X.Y.; Li, C.L.; Jiang, Y.Z.; Wang, S.; Zhao, H.Y.; Miao, J.Y.; et al. Targeting ATAD3A-PINK1-mitophagy axis overcomes chemoimmunotherapy resistance by redirecting PD-L1 to mitochondria. Cell Res. 2023, 33, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Kawase, K.; Nishi, T.; Watanabe, T.; Takenaga, K.; Inozume, T.; Ishino, T.; Aki, S.; Lin, J.; Kawashima, S.; et al. Immune evasion through mitochondrial transfer in the tumour microenvironment. Nature 2025, 638, 225–236. [Google Scholar] [CrossRef]
- Ji, P.; Gong, Y.; Jin, M.L.; Wu, H.L.; Guo, L.W.; Pei, Y.C.; Chai, W.J.; Jiang, Y.Z.; Liu, Y.; Ma, X.Y.; et al. In vivo multidimensional CRISPR screens identify Lgals2 as an immunotherapy target in triple-negative breast cancer. Sci. Adv. 2022, 8, eabl8247. [Google Scholar] [CrossRef]
- Varzaru, V.B.; Vlad, T.; Popescu, R.; Vlad, C.S.; Moatar, A.E.; Cobec, I.M. Triple-Negative Breast Cancer: Molecular Particularities Still a Challenge. Diagnostics 2024, 14, 1875. [Google Scholar] [CrossRef]
- Passaniti, A.; Kim, M.S.; Polster, B.M.; Shapiro, P. Targeting mitochondrial metabolism for metastatic cancer therapy. Mol. Carcinog. 2022, 61, 827–838. [Google Scholar] [CrossRef]
- Noyan, S.; Ozketen, A.A.; Gurdal, H.; Dedeoglu, B.G. miR-770-5p regulates EMT and invasion in TNBC cells by targeting DNMT3A. Cell Signal 2021, 83, 109996. [Google Scholar] [CrossRef]
- Ma, F.; Song, J.; He, M.; Wang, X. The Antimicrobial Peptide Merecidin Inhibit the Metastasis of Triple-Negative Breast Cancer by Obstructing EMT via miR-30d-5p/Vimentin. Technol. Cancer Res. Treat. 2024, 23, 15330338241281310. [Google Scholar] [CrossRef]
- Fan, S.; Yan, X.; Hu, X.; Liu, X.; Zhao, S.; Zhang, Y.; Zhou, X.; Shen, X.; Qi, Q.; Chen, Y. Shikonin blocks CAF-induced TNBC metastasis by suppressing mitochondrial biogenesis through GSK-3β/NEDD4-1 mediated phosphorylation-dependent degradation of PGC-1α. J. Exp. Clin. Cancer Res. 2024, 43, 180. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, Q.; Dong, C. Metabolic reprogramming in triple-negative breast cancer. Cancer Biol. Med. 2020, 17, 44–59. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.Y.; Yang, Y.C.; Ting, K.L.; Wen, S.Y.; Viswanadha, V.P.; Huang, C.Y.; Kuo, W.W. Lactate dehydrogenase downregulation mediates the inhibitory effect of diallyl trisulfide on proliferation, metastasis, and invasion in triple-negative breast cancer. Environ. Toxicol. 2017, 32, 1390–1398. [Google Scholar] [CrossRef] [PubMed]
- Hua, Z.; White, J.; Zhou, J. Cancer stem cells in TNBC. Semin. Cancer Biol. 2022, 82, 26–34. [Google Scholar] [CrossRef]
- Castaneda, M.; den Hollander, P.; Werden, S.; Ramirez-Pena, E.; Vasaikar, S.V.; Kuburich, N.A.; Gould, C.; Soundararajan, R.; Mani, S.A. beta-Catenin Drives the FOXC2-Mediated Epithelial-Mesenchymal Transition and Acquisition of Stem Cell Properties. Cancers 2025, 17, 1114. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Qiang, J.; Deng, Q.; Xia, J.; Deng, L.; Zhou, L.; Wang, D.; He, X.; Liu, Y.; Zhao, B.; et al. ALDH1A1 Activity in Tumor-Initiating Cells Remodels Myeloid-Derived Suppressor Cells to Promote Breast Cancer Progression. Cancer Res. 2021, 81, 5919–5934. [Google Scholar] [CrossRef]
- Zhu, X.; Bu, J.; Zhu, T.; Jiang, Y. Targeting KK-LC-1 inhibits malignant biological behaviors of triple-negative breast cancer. J. Transl. Med. 2023, 21, 184. [Google Scholar] [CrossRef]
- Bu, J.; Zhang, Y.; Wu, S.; Li, H.; Sun, L.; Liu, Y.; Zhu, X.; Qiao, X.; Ma, Q.; Liu, C.; et al. KK-LC-1 as a therapeutic target to eliminate ALDH+ stem cells in triple negative breast cancer. Nat. Commun. 2023, 14, 2602. [Google Scholar] [CrossRef]
- Weiner-Gorzel, K.; Murphy, M. Mitochondrial dynamics, a new therapeutic target for Triple Negative Breast Cancer. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188518. [Google Scholar] [CrossRef] [PubMed]
- Shome, R.; Ghosh, S.S. Tweaking EMT and MDR dynamics to constrain triple-negative breast cancer invasiveness by EGFR and Wnt/β-catenin signaling regulation. Cell. Oncol 2021, 44, 405–422. [Google Scholar] [CrossRef]
- Huang, S.Q.; Cao, K.X.; Wang, C.L.; Chen, P.L.; Chen, Y.X.; Zhang, Y.T.; Yu, S.H.; Bai, Z.X.; Guo, S.; Liao, M.X.; et al. Decreasing mitochondrial fission ameliorates HIF-1α-dependent pathological retinal angiogenesis. Acta Pharmacol. Sin. 2024, 45, 1438–1450. [Google Scholar] [CrossRef] [PubMed]
- Altea-Manzano, P.; Doglioni, G.; Liu, Y.; Cuadros, A.M.; Nolan, E.; Fernandez-Garcia, J.; Wu, Q.; Planque, M.; Laue, K.J.; Cidre-Aranaz, F.; et al. A palmitate-rich metastatic niche enables metastasis growth via p65 acetylation resulting in pro-metastatic NF-κB signaling. Nat. Cancer 2023, 4, 344–364. [Google Scholar] [CrossRef]
- Xu, F.; Hu, Y.; Gao, J.; Wang, J.; Xie, Y.; Sun, F.; Wang, L.; Miyamoto, A.; Xia, O.; Zhang, C. HIF-1α/Malat1/miR-141 Axis Activates Autophagy to Increase Proliferation, Migration, and Invasion in Triple-negative Breast Cancer. Curr. Cancer Drug Targets 2023, 23, 363–378. [Google Scholar] [CrossRef]
- Park, J.H.; Jung, K.H.; Jia, D.; Yang, S.; Attri, K.S.; Ahn, S.; Murthy, D.; Samanta, T.; Dutta, D.; Ghidey, M.; et al. Biguanides antithetically regulate tumor properties by the dose-dependent mitochondrial reprogramming-driven c-Src pathway. Cell Rep. Med. 2025, 6, 101941. [Google Scholar] [CrossRef] [PubMed]
- Rossi, T.; Iorio, E.; Chirico, M.; Pisanu, M.E.; Amodio, N.; Cantafio, M.E.G.; Perrotta, I.; Colciaghi, F.; Fiorillo, M.; Gianferrari, A.; et al. BET inhibitors (BETi) influence oxidative phosphorylation metabolism by affecting mitochondrial dynamics leading to alterations in apoptotic pathways in triple-negative breast cancer (TNBC) cells. Cell Prolif. 2024, 57, e13730. [Google Scholar] [CrossRef]
- Chen, T.; Chen, H.; Jiang, Y.; Yan, Q.; Zheng, S.; Wu, M. Co-Delivery of 5-Fluorouracil and Paclitaxel in Mitochondria-Targeted KLA-Modified Liposomes to Improve Triple-Negative Breast Cancer Treatment. Pharmaceuticals 2022, 15, 881. [Google Scholar] [CrossRef]
- Zhang, J.N.; Zhang, Z.; Huang, Z.L.; Guo, Q.; Wu, Z.Q.; Ke, C.; Lu, B.; Wang, Z.T.; Ji, L.L. Isotoosendanin inhibits triple-negative breast cancer metastasis by reducing mitochondrial fission and lamellipodia formation regulated by the Smad2/3-GOT2-MYH9 signaling axis. Acta Pharmacol. Sin. 2024, 45, 2672–2683. [Google Scholar] [CrossRef]
- Tanner, L.; Haynes, R.K.; Wiesner, L. Accumulation of TB-Active Compounds in Murine Organs Relevant to Infection by Mycobacterium tuberculosis. Front. Pharmacol. 2020, 11, 724. [Google Scholar] [CrossRef]
- Cruz-Gregorio, A.; Aranda-Rivera, A.K.; Aparicio-Trejo, O.E.; Medina-Campos, O.N.; Sciutto, E.; Fragoso, G.; Pedraza-Chaverri, J. α-Mangostin induces oxidative damage, mitochondrial dysfunction, and apoptosis in a triple-negative breast cancer model. Phytother. Res. 2023, 37, 3394–3407. [Google Scholar] [CrossRef]
- Wang, H.; Wei, L.; Mao, D.; Che, X.; Ye, X.; Liu, Y.; Chen, Y. Combination of oxymatrine (Om) and astragaloside IV (As) enhances the infiltration and function of TILs in triple-negative breast cancer (TNBC). Int. Immunopharmacol. 2023, 125, 111026. [Google Scholar] [CrossRef] [PubMed]
- Falcone, I.G.; Rushing, B.R. Untargeted Metabolomics Reveals Acylcarnitines as Major Metabolic Targets of Resveratrol in Breast Cancer Cells. Metabolites 2025, 15, 250. [Google Scholar] [CrossRef]
- Hong, Y.; Fan, D. Ginsenoside Rk1 induces cell cycle arrest and apoptosis in MDA-MB-231 triple negative breast cancer cells. Toxicology 2019, 418, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, J.; Wang, C.; Wang, T.; Zeng, Y.; Li, X.; Zuo, Y.; Chen, H.; Zhang, C.; Cao, Y.; et al. Aptamer-functionalized triptolide with release controllability as a promising targeted therapy against triple-negative breast cancer. J. Exp. Clin. Cancer Res. 2024, 43, 207. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Li, X.; Li, B.; Yang, S.; Qin, J.; Han, S.; Ren, J.; Shuai, X. Nanodrug Inducing Autophagy Inhibition and Mitochondria Dysfunction for Potentiating Tumor Photo-Immunotherapy. Small 2023, 19, e2300280. [Google Scholar] [CrossRef]
- Zhang, W.; Jiang, Y.; Liu, L.; Shen, H.; Huang, X.; Zheng, W.; Chu, Z.; Wang, W.; Guo, Y.; Qian, H. Implantable Microneedles Loaded with Nanoparticles Surface Engineered Escherichia coli for Efficient Eradication of Triple-Negative Breast Cancer Stem Cells. Nano Lett. 2025, 25, 2041–2051. [Google Scholar] [CrossRef]
- Duan, H.; Liu, Y.; Gao, Z.; Huang, W. Recent advances in drug delivery systems for targeting cancer stem cells. Acta Pharm. Sin. B 2021, 11, 55–70. [Google Scholar] [CrossRef]
- Ghanbari, F.; Mader, S.; Philip, A. Cholesterol as an Endogenous Ligand of ERRα Promotes ERRα-Mediated Cellular Proliferation and Metabolic Target Gene Expression in Breast Cancer Cells. Cells 2020, 9, 1765. [Google Scholar] [CrossRef]
- Jiang, Y.; Krantz, S.; Qin, X.; Li, S.; Gunasekara, H.; Kim, Y.M.; Zimnicka, A.; Bae, M.; Ma, K.; Toth, P.T.; et al. Caveolin-1 controls mitochondrial damage and ROS production by regulating fission-fusion dynamics and mitophagy. Redox Biol. 2022, 52, 102304. [Google Scholar] [CrossRef]
- Ding, P.; Pei, S.; Qu, Z.; Yang, Y.; Liu, Q.; Kong, X.; Wang, Z.; Wang, J.; Fang, Y. Single-cell sequencing unveils mitophagy-related prognostic model for triple-negative breast cancer. Front. Immunol. 2024, 15, 1489444. [Google Scholar] [CrossRef] [PubMed]
- Pateras, I.S.; Williams, C.; Gianniou, D.D.; Margetis, A.T.; Avgeris, M.; Rousakis, P.; Legaki, A.I.; Mirtschink, P.; Zhang, W.; Panoutsopoulou, K.; et al. Short term starvation potentiates the efficacy of chemotherapy in triple negative breast cancer via metabolic reprogramming. J. Transl. Med. 2023, 21, 169. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, F.; Huang, S.; Liu, L. PYCR3 modulates mtDNA copy number to drive proliferation and doxorubicin resistance in triple-negative breast cancer. Int. J. Biochem. Cell Biol. 2024, 171, 106581. [Google Scholar] [CrossRef]
- Kawashima, M.; Bensaad, K.; Zois, C.E.; Barberis, A.; Bridges, E.; Wigfield, S.; Lagerholm, C.; Dmitriev, R.I.; Tokiwa, M.; Toi, M.; et al. Correction to: Disruption of hypoxia-inducible fatty acid binding protein 7 induces beige fat-like differentiation and thermogenesis in breast cancer cells. Cancer Metab. 2020, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Bertholet, A.M.; Kirichok, Y. UCP1: A transporter for H+ and fatty acid anions. Biochimie 2017, 134, 28–34. [Google Scholar] [CrossRef]
- Xia, J.; Chu, C.; Li, W.; Chen, H.; Xie, W.; Cheng, R.; Hu, K.; Li, X. Mitochondrial Protein UCP1 Inhibits the Malignant Behaviors of Triple-negative Breast Cancer through Activation of Mitophagy and Pyroptosis. Int. J. Biol. Sci. 2022, 18, 2949–2961. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pei, W.; Dai, L.; Li, M.; Cao, S.; Xiao, Y.; Yang, Y.; Ma, M.; Deng, M.; Mo, Y.; Liu, M. Targeting Mitochondrial Quality Control for the Treatment of Triple-Negative Breast Cancer: From Molecular Mechanisms to Precision Therapy. Biomolecules 2025, 15, 970. https://doi.org/10.3390/biom15070970
Pei W, Dai L, Li M, Cao S, Xiao Y, Yang Y, Ma M, Deng M, Mo Y, Liu M. Targeting Mitochondrial Quality Control for the Treatment of Triple-Negative Breast Cancer: From Molecular Mechanisms to Precision Therapy. Biomolecules. 2025; 15(7):970. https://doi.org/10.3390/biom15070970
Chicago/Turabian StylePei, Wanjuan, Ling Dai, Mingxiao Li, Sihui Cao, Yili Xiao, Yan Yang, Minghao Ma, Minjie Deng, Yang Mo, and Mi Liu. 2025. "Targeting Mitochondrial Quality Control for the Treatment of Triple-Negative Breast Cancer: From Molecular Mechanisms to Precision Therapy" Biomolecules 15, no. 7: 970. https://doi.org/10.3390/biom15070970
APA StylePei, W., Dai, L., Li, M., Cao, S., Xiao, Y., Yang, Y., Ma, M., Deng, M., Mo, Y., & Liu, M. (2025). Targeting Mitochondrial Quality Control for the Treatment of Triple-Negative Breast Cancer: From Molecular Mechanisms to Precision Therapy. Biomolecules, 15(7), 970. https://doi.org/10.3390/biom15070970