
Endogenous Ribonucleases: Therapeutic Targeting of the Transcriptome Through Oligonucleotide-Triggered RNA Inactivation


Abstract

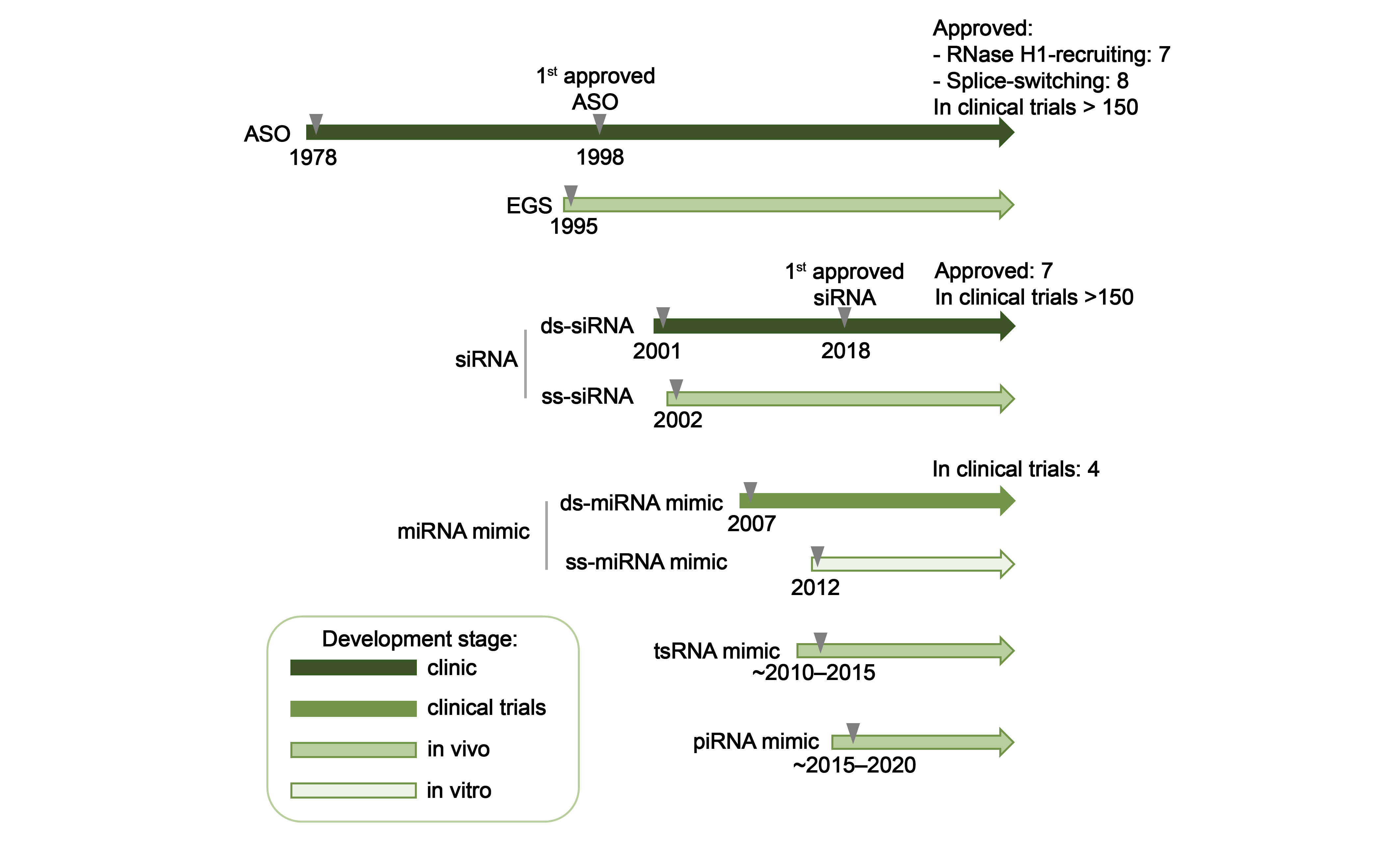
1. Introduction
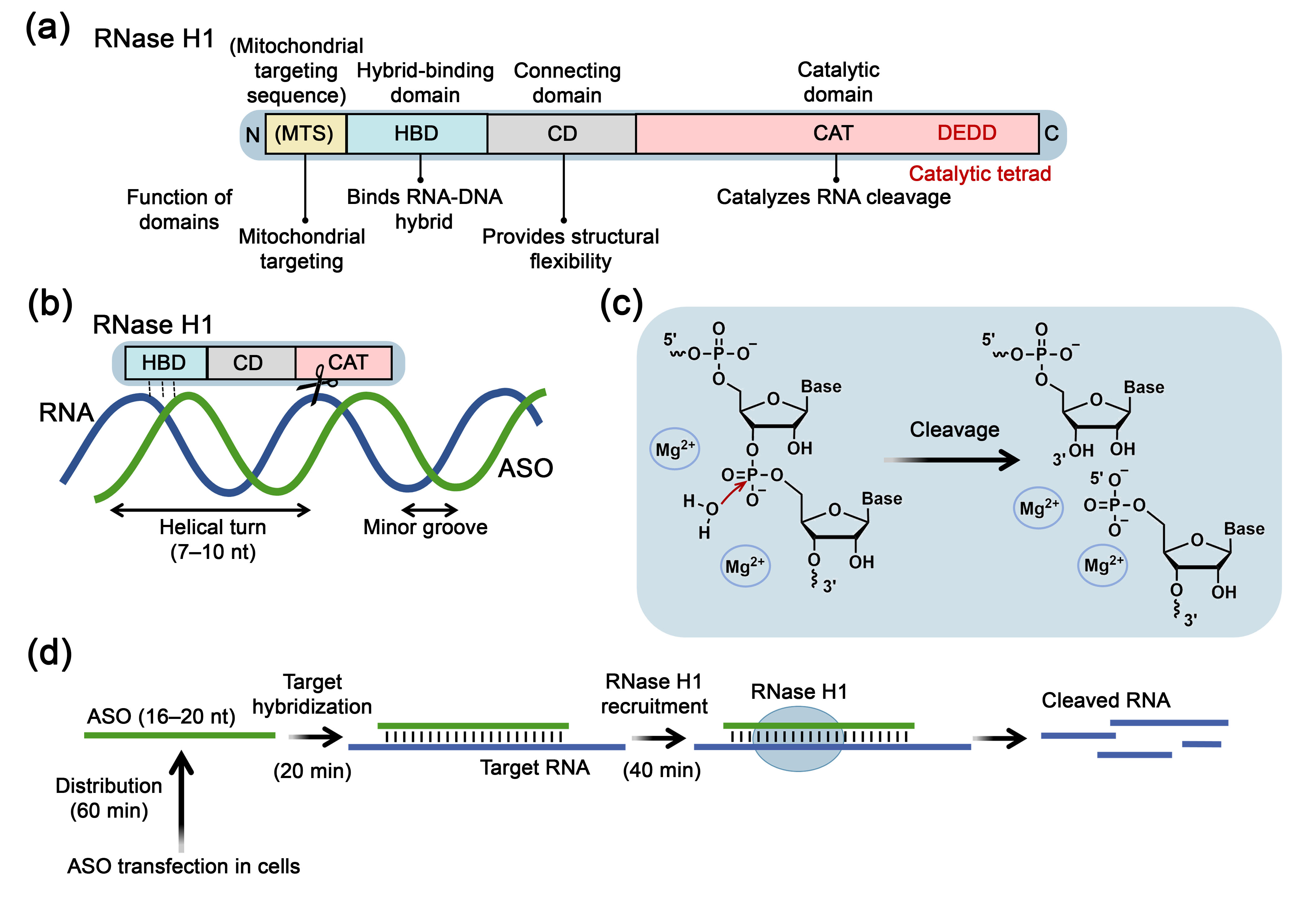
2. RNase H1 as a Key Enzyme Facilitating Biological Activity of Antisense Oligonucleotides
2.1. Structure, Mechanism of Action and Physiological Functions of RNase H1
2.2. RNase H1 as an Enzymatic Effector in Antisense Oligonucleotide-Based Therapeutics
2.2.1. Kinetic Parameters of RNase H1-Mediated Target RNA Degradation
2.2.2. Chemical Modifications of Antisense Oligonucleotides Compatible with RNase H1 Activity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Modification | Nuclease Resistance 1 | Duplex Stability 2 | Studies in Biological Systems | Reference |
|---|---|---|---|---|
| Phosphorothioate (PS) | + | − | Cell culture/in vivo | [67,77] |
| Phosphorodithioate (SPS) | +++ | − | n.s. | [69,70] |
| Phosphonoacetate (AcPO) | +++ | − | n.s. | [74] |
| Thiophosphonoacetate (AcPS) | +++ | − | n.s. | [74] |
| Phosphonoformate (PF) | +++ | + | n.s. | [76] |
| Boranophosphate | ++ | − | n.s. | [71,72,73] |
| Mesyl-(methanesulfonyl)-phosphoramidate (μ) | +++ | − | Cell culture/in vivo | [78,79,80,81,82] |
| 5′-O-methylenephosphonate (5′-MEP) | n.s. | + | n.s. | [75] |
| 5′-hydroxyphosphonate (5′-HP) | n.s. | + | n.s. | [75] |
| Arabinonucleic acid (ANA) | + | − | n.s. | [83,84] |
| 2′-deoxy-2′-fluoro-β-d-arabino nucleic acid (2′-F-ANA or FANA) | ++ | + | Cell culture/in vivo | [84,85,86,87,88,89,90] |
| 2′-deoxy-2′-fluoro-3′-C-hydroxymethyl-β-d-lyxo-configured pyrimidine nucleotides (U/C) | + | + | Cell culture | [91] |
| 2′-β-fluoro-tricyclo nucleotides (2′-F-tc-ANA) | n.s | + | n.s. | [92] |
| 6′-fluoro[4.3.0]bicyclo-nucleotides (6′-F-bc4,3-DNA) | n.s. | − − | n.s. | [93] |
| 6′-difluorinated[4.3.0]bicyclo-nucleotides (6′-diF-bc4,3-DNA) | n.s. | − − | n.s. | [94] |
| 4′-C-aminoethoxy thymidine (AEoT) | +++ | + | Cell culture | [95] |
| 4′-C-2-aminopropoxy thymidine in S- and R-configuration (4′-(S)-2-APoT and 4′-(R)-2-APoT) | +++ | − | n.s. | [96] |
| Cyclohexenyl nucleic acid (CeNA) | +++ | + | n.s. | [97,98] |
| C5-propynyl arabinouridine (araUP) and arabinocytidine (araCP) | +++ | + | n.s. | [99] |
2.2.3. Therapeutic Outcomes of RNase H1-Mediated Antisense Activity
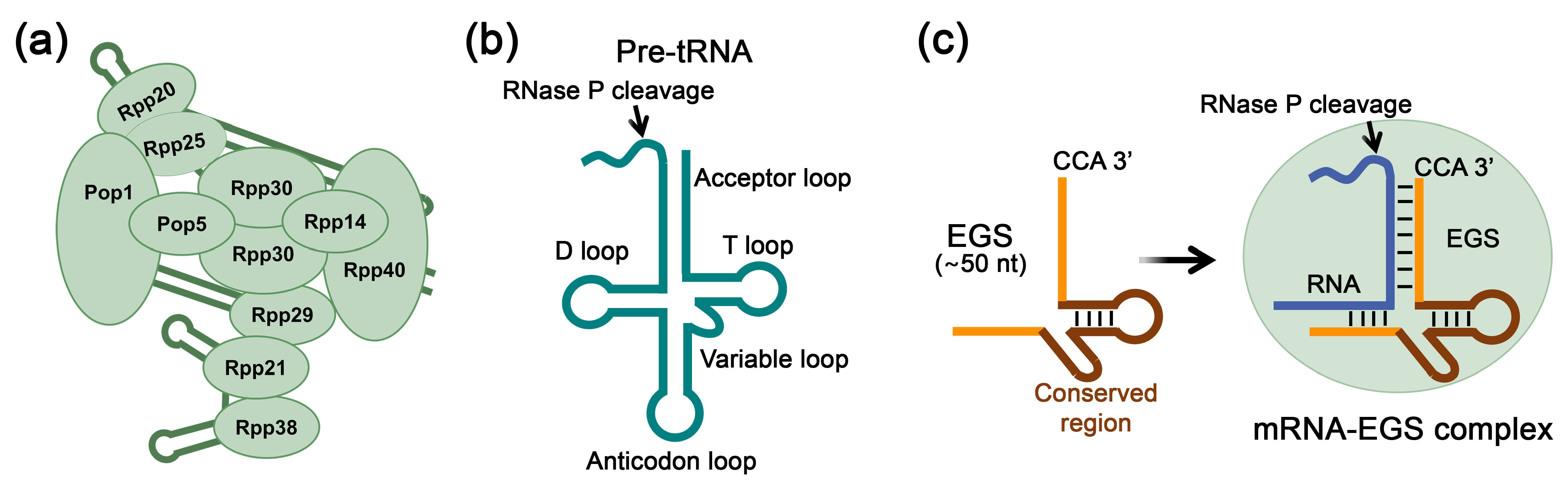
3. RNase P-Mediated RNA Inhibition Using External Guide Sequences
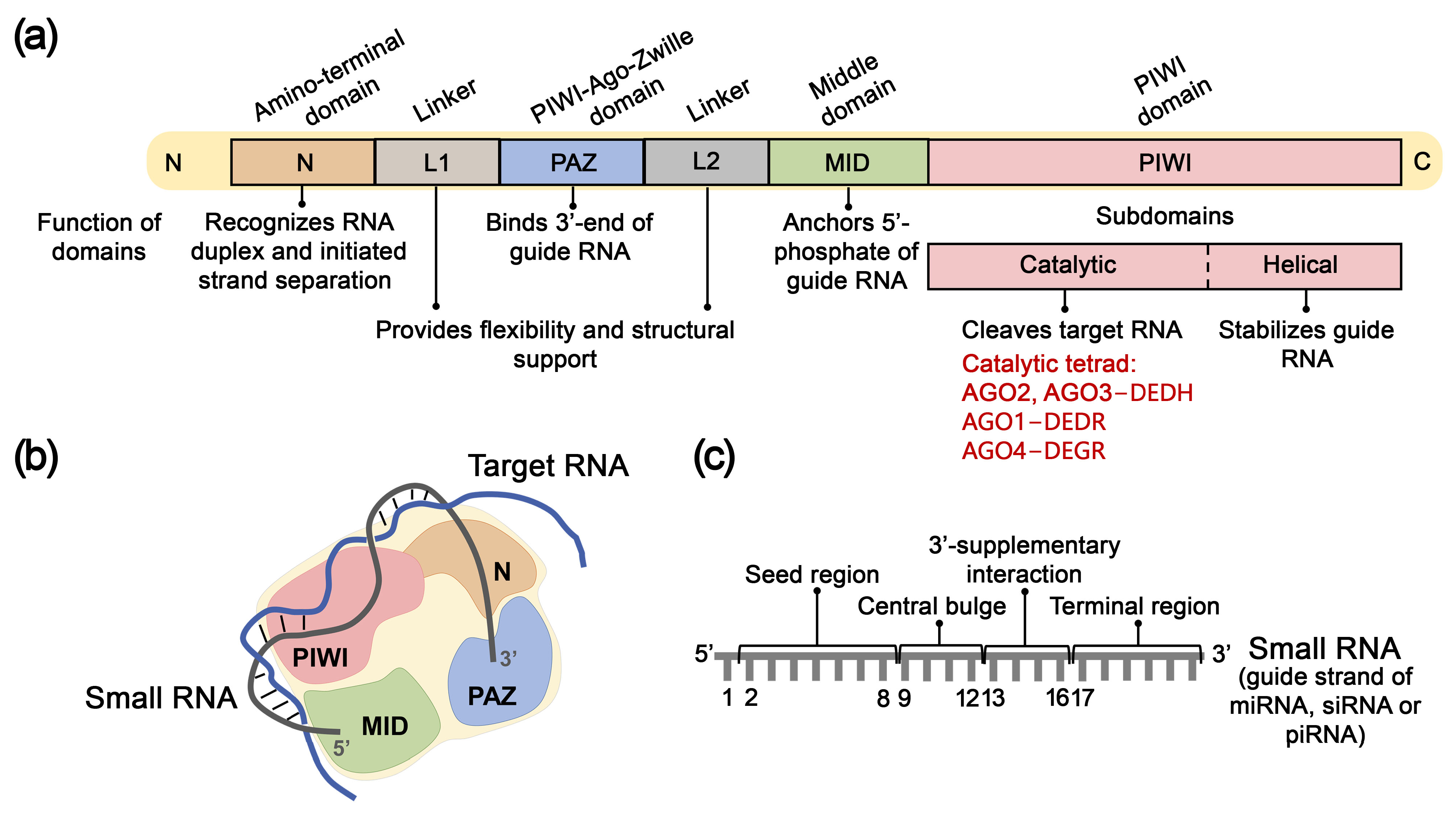
4. Argonaute Family Proteins for Target RNA Inhibition
4.1. Structure, Mechanism of Action, and Key Functions of AGO Proteins
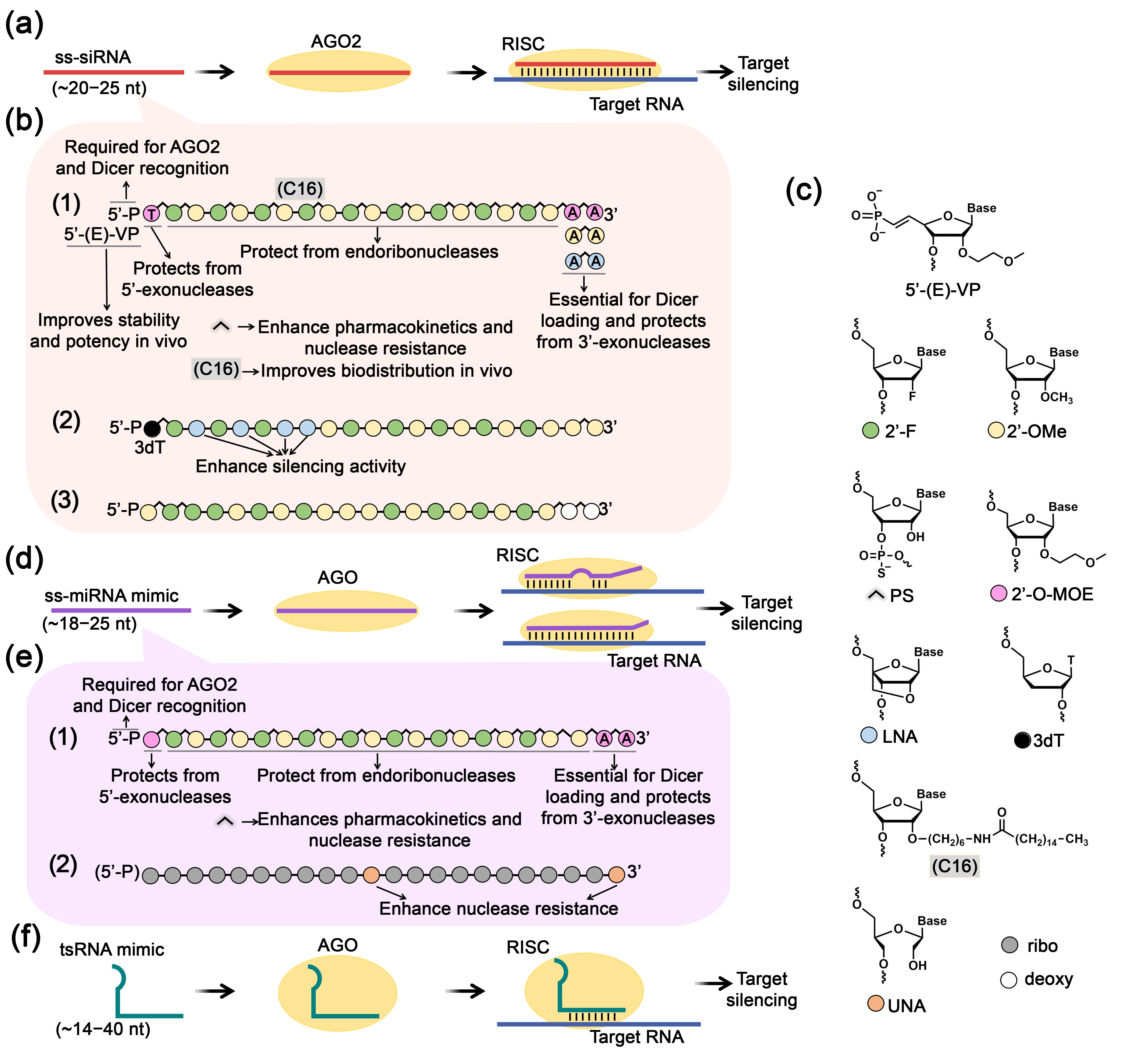
4.2. AGO-Mediated RNA Inhibition Using Single-Stranded Oligonucleotides
4.2.1. Single-Stranded siRNA
4.2.2. Single-Stranded miRNA Mimics
4.2.3. Small tRNA-Derived RNA Mimics
| tsRNA Mimic/Origin | Experimental Model and mRNA Targets (Signaling Pathway) | Cell Culture Effects * | In Vivo Effects * | Refs |
|---|---|---|---|---|
| 5′-tRF (17 nt) tRF-17-79MP9PP/ tRNA-Val-CAG, tRNA-Val-AAC | Breast cancer: MCF-7, BT-549 Target: THBS1 (THBS1/TGC/Smad3) | THBS1 mRNA level ↓ 50–60% THBS1 protein level ↓ Proliferation ↓ 20–30% Colony formation ↓ 40–60% Invasion ↓ 65–70% Migration ↓ 65–75% Cell cycle: G1/S arrest | N.s. | [184] |
| 5′-tRF (18 nt) tRF-5026a/ tRNA-Val-AAC | Gastric cancer: AGS, MGC-803, HGC-27, BGC-823, SGC-7901 Target: PTEN/PI3K/AKT | Protein levels: PI3K ↓ 25–60%, AKT ↓ 25–35%, PTEN ↑ 25–60% Proliferation ↓ 15–30% Colony formation ↓ 40–60% Migration ↓ 40–50% Cell cycle: G0/G1 arrest | SGC-823 and MGC-803 xenograft model: 0.05 µM mimic: tumor growth ↓ 50% 0.1 µM mimic: tumor growth inhibition in 5/6 mice | [185] |
| tRF (15 nt) tRF-Val-CAC-016/ tRNA-Val-CAC | Gastric cancer: NCI-N87, HGC-27 Target: CACNA1d (CACNA1d/MAPK) | CACNA1d mRNA level ↓ 30–45% Protein levels: CyclinD1, CyclinB, c-Myc ↓ Proliferation ↓ 65–80% Colony formation ↓ 70% Cell cycle: S arrest | NCI-N87 xenograft model: Tumor volume ↓ 50% Protein levels: CACNA1d, Ki-67 ↓ | [186] |
| 3′-tRF (17 nt) tRF-3008A/ tRNA-Val | Colorectal cancer: HCT116 Target: FOXK1 (Wnt/β-catenin) | Proliferation ↓ 20% Invasion ↓ 40% Migration ↓ 50% Apoptosis ↑ 60% | In vitro treatment of HCT116 cells with subsequent implantation into mice: Tumor volume ↓ 50% Metastases ↓ Protein levels: Caspase-3, Ki-67, MMP9 ↓ | [187] |
| 3′-tRF (27 nt) tRF-Glu49/ tRNA-Glu-TTC, tRNA-Glu-CTC | Cervical cancer: HeLa Target: FGL1 | FGL1 mRNA level ↓ 60% Proliferation ↓ 25% Invasion ↓ 60% Migration ↓ 50% | N.s. | [188] |
| 3′-tiRNA (41 nt) tRF-41-YDLBRY73W0K5KKOVD/ tRNA-Asn-GTT | Gastric cancer: HGC-27, AGS Target: PAPSS2 | Proliferation ↓ 25% Migration ↓ 50–60% Cell cycle: G0/G1 arrest Apoptosis ↑ 40% | N.s. | [189] |
| 5′-tiRNA (33 nt) tRF-33-P4R8YP9LON4VDP/ tRNA-Gly-GCC | Gastric cancer: HGC-27, AGS Target: STAT3 | Proliferation ↓ 25% Colony formation ↓ 20% Invasion ↓ 30–40% Migration ↓ 30–35% Apoptosis ↑ 1.3–2.5-fold | In vitro treatment of HGC-27 cells with subsequent implantation into mice: 0.05 µM mimic: tumor growth ↓ 60–70% 0.1 µM mimic: tumor growth ↓ 90–97% | [190] |
| 3′-tRF (17 nt) tRF-60:76-Val-AAC-1-M5/ tRNA-Val-AAC | Angiogenesis model: HUVEC Target: Tnfrsf10b, Bcl2l1 | mRNA levels: Tnfrsf10b ↓ 10%, Bcl2l1 ↓ 7% | N.s. | [191] |
| i-tRF (21 nt) tRF-21-NB8PLML3E/ tRNA-Gln-CTG | Cardiac hypertrophy: angiotensin II-stimulated H9c2 cardiomyocytes | Cardiomyocyte hypertrophy ↓ Natriuretic peptides levels: ANP ↓ 40%, BNP ↓ 75% | N.s. | [192] |
| 3′-tRF (17 nt) tRF3-Thr-AGT/ tRNA-Thr-AGT | Cellular acute pancreatitis: AR42J rat pancreatic acinar cells treated with sodium taurocholate Target: ZBP1 (ZBP1/NLRP3) | Proliferation ↑ 60% # Pro-inflammatory cytokines levels: IL-1β ↓ 55–65% and IL-18 ↓ 65% # Proteins levels: NLRP3, ASC, Gasdermin-D ↓ 40–50% #, Caspase-1 ↓ # | N.s. | [193] |
| 3′-tRF (17 nt) tRF-3003a/ tRNA-Cys-GCA | Osteoarthritis: IL-1β-treated TC28/I2 chondrocytes Target: JAK3 (JAK/STAT) | JAK3 mRNA level ↓ 90% IL-6 protein level ↓ 65% | N.s. | [194] |
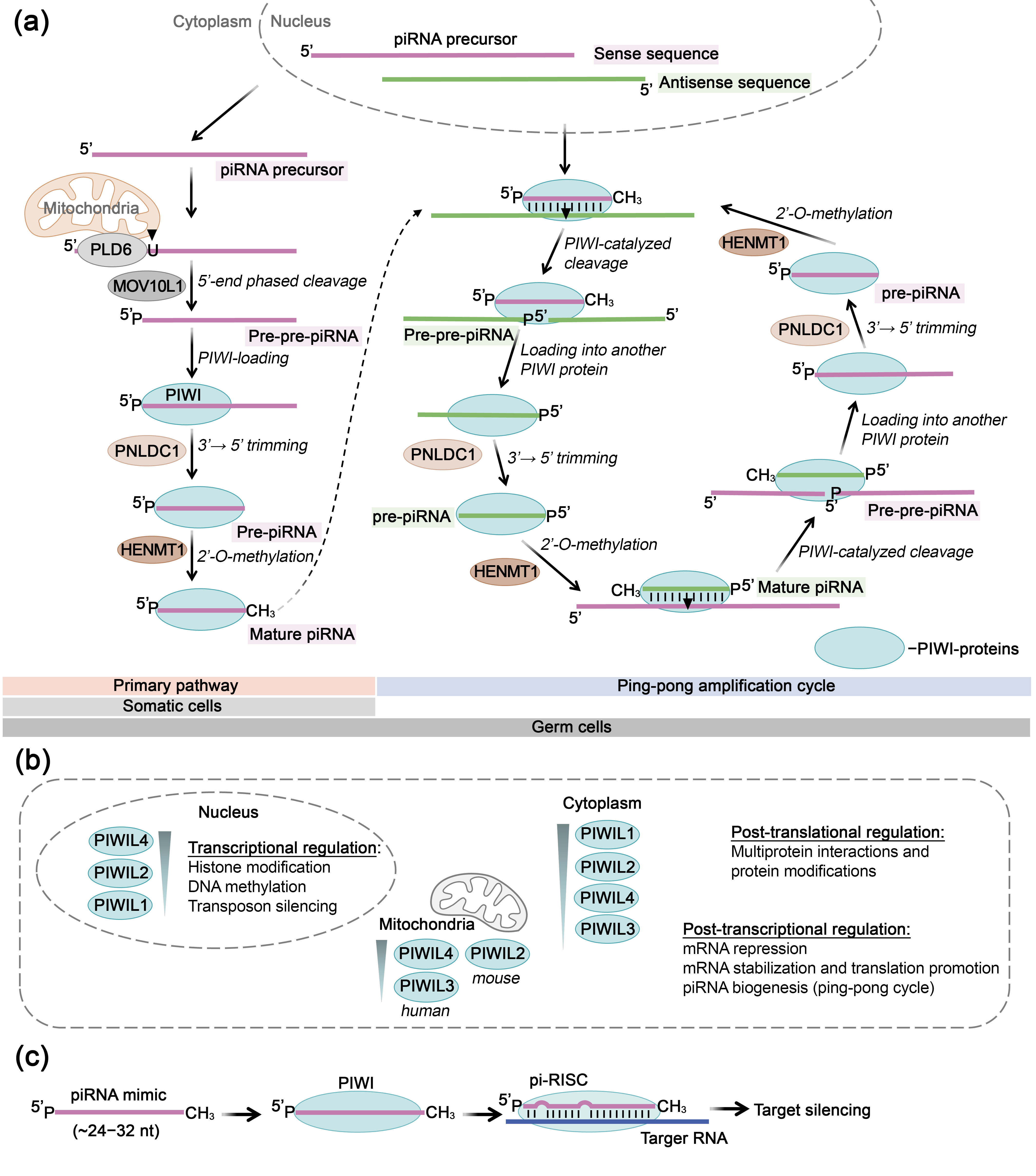
5. PIWI Proteins as Key Mediators of RNA-Inhibitory Action of piRNAs
5.1. Structure, Mechanism of Action, and Primary Functions of PIWI Proteins
5.2. PIWI-Mediated RNA Inhibition Using Synthetic piRNAs
6. Conclusionss
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 2′-F-tc-ANA | 2′-β-fluoro-tricyclo-arabinonucleic acid |
| 2′-F | 2′-fluororibose |
| 2′-F-ANA | 2′-deoxy-2′-fluoro-β-d-arabinonucleic acid |
| 2′-OMe | 2′-O-methyl |
| 2′-O-MOE | 2′-O-methoxyethyl |
| 3′-UTR | 3′-untranslated region |
| 3dT | 3′-deoxythymidine |
| 4′-(S)-2-APoT and 4′-(R)-2-APoT | 4′-C-2-aminopropoxy-modified thymidine in S- and R-configuration |
| 5′-(E)-VP | 5′-(E)-vinylphosphonate |
| 5′-HP | 5′-hydroxyphosphonate |
| 5′-MEP | 5′-O-methylenephosphonate |
| 6′-diF-bc4,3-DNA | 6′-difluorinated[4.3.0]bicyclo-nucleotides |
| 6′-F-bc4,3-DNA | 6′-fluoro[4.3.0]bicyclo-nucleotides |
| AcPO | Phosphonoacetate |
| AcPS | Thiophosphonoacetate |
| ADAM33 | Adam metallopeptidase domain 33 |
| AEoT | 4′-C-aminoethoxy thymidine |
| ALS | Amyotrophic lateral sclerosis |
| ANA | Arabinonucleic acid |
| ANP | Atrial natriuretic peptide |
| ANXA2 | Annexin A2 |
| ApoB-100 | Apolipoprotein B-100 |
| Apo-CIII | Apolipoprotein CIII |
| araUP and araCP | C5-propynyl arabinouridine and arabinocytidine |
| ASO | Antisense oligonucleotide |
| Bcl2l1 | Bcl-2-like protein 1 |
| BNP | B-type natriuretic peptide |
| CACNA1d | Calcium voltage-gated channel subunit alpha1 D |
| CAT | Catalytic domain |
| CCL3 | CC motif chemokine ligand 3 |
| CCR4-NOT | Carbon catabolite repression 4—negative on TATA-less |
| CCR5 | CC-chemokine receptor 5 |
| CD | Connecting domain |
| CeNA | Cyclohexenyl nucleic acid |
| cEt | Constrained ethyl nucleic acids |
| circRNA | Circular RNA |
| CTNNB1 | Catenin beta 1 |
| CTTN | Cortactin |
| DDX21 | DExD-box helicase 21 |
| DNCA/CLD | Neutral cytidine lipid/cationic lipid |
| EFNA5 | Ephrin A5 |
| EGS | External guide sequences |
| eIF4F | Eukaryotic initiation factor 4F |
| FGL1 | Fibrinogen-like protein 1 |
| FOXK1 | Forkhead box protein K1 |
| ftsZ | Filamenting temperature-sensitive mutant Z |
| gyrA | DNA gyrase subunit A |
| HBD | Hybrid-binding domain |
| HBV | Hepatitis B virus |
| HCMV | Human cytomegalovirus |
| HENMT1 | HEN Methyltransferase 1 |
| HIV | Human immunodeficiency virus |
| hnRNP | Heterogeneous nuclear ribonucleoproteins |
| hspA8 | Heat shock protein family a (hsp70) member 8 |
| HSV-1 | Herpes simplex virus type 1 |
| HSV-2 | Herpes simplex virus type 2 |
| HTT | Huntingtin |
| ICP8 | Infected cell protein 8 |
| IRES | Internal ribosome entry site |
| JAK3 | Janus kinase 3 |
| KRAS | Kirsten rat sarcoma virus |
| LDL | Low-density lipoprotein |
| LNA | Locked nucleic acids |
| lncRNA | Long non-coding RNA |
| MALAT1 | Metastasis associated lung adenocarcinoma transcript 1 |
| MCP | Major capsid protein |
| MID | Middle domain |
| miRNA | MicroRNA |
| MOV10L1 | Mov10-like RNA Helicase 1 |
| MTS | Mitochondrial targeting sequence |
| n.s. | Not studied |
| NAT10 | N-acetyltransferase 10 |
| NPC1 | Niemann-pick C1 |
| NPM1 | Nucleophosmin 1 |
| P54nrb protein | 54-kda nuclear RNA-binding protein |
| PAPSS2 | 3′-phosphoadenosine 5′-phosphosulfate synthetase 2 |
| PAZ | PIWI-Argonaute-Zwille |
| PC4 | Positive cofactor 4 |
| PF | Phosphonoformate |
| piRISC | PiRNA-induced gene silencing complex |
| piRNA | PIWI (P element induced wimpy testis)-interacting RNA |
| PIWI | P element induced wimpy testis |
| PLD6 | Phospholipase D Family Member 6 |
| PNLDC1 | PARN Like Ribonuclease Domain Containing Exonuclease 1 |
| POPC | Palmityl-oleyl-phosphatidylcholine |
| pre-tRNA | Precursor transfer RNA |
| PS | Phosphorothioate |
| PTEN | Phosphatase and tensin homolog |
| PURPL | P53 upregulated regulator of p53 levels |
| PVT1 | Plasmacytoma variant translocation 1 |
| RISC | RNA-induced silencing complex |
| RNAi | RNA interference |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| siRNA | Small interfering RNA |
| SOD1 | Superoxide dismutase 1 |
| SPS | Phosphorodithioate |
| ss-siRNA | Single-stranded siRNA |
| TAT | Transactivator of transcription |
| TCF4 | Transcription factor 4 |
| TCP1 | T-Complex 1 protein |
| THBS1 | Thrombospondin 1 |
| tiRNA | tRNA-derived stress-induced RNA |
| TNA | Therapeutic nucleic acid |
| Tnfrsf10b | Tumor necrosis factor receptor superfamily member 10B |
| tRF | Transfer RNA-derived fragment |
| tRNA | Transfer RNA |
| tsRNA | Transfer RNA-derived small RNA |
| TTR | Transthyretin |
| tyRNA | Tiny RNA |
| UNA | Unlocked nucleic acid |
| VARS | Valyl-tRNA synthetase |
| ZBP1 | Z-DNA binding protein 1 |
| μ | Mesyl-(methanesulfonyl)-phosphoramidate |
References
- Conn, V.M.; Chinnaiyan, A.M.; Conn, S.J. Circular RNA in Cancer. Nat. Rev. Cancer 2024, 24, 597–613. [Google Scholar] [CrossRef]
- Goodall, G.J.; Wickramasinghe, V.O. RNA in Cancer. Nat. Rev. Cancer 2021, 21, 22–36. [Google Scholar] [CrossRef]
- Li, Y.; Sun, S. RNA Dysregulation in Neurodegenerative Diseases. EMBO J. 2025, 44, 613–638. [Google Scholar] [CrossRef]
- Sessa, F.; Salerno, M.; Esposito, M.; Cocimano, G.; Pomara, C. MiRNA Dysregulation in Cardiovascular Diseases: Current Opinion and Future Perspectives. Int. J. Mol. Sci. 2023, 24, 5192. [Google Scholar] [CrossRef]
- Kumar, D.; Sahoo, S.S.; Chauss, D.; Kazemian, M.; Afzali, B. Non-Coding RNAs in Immunoregulation and Autoimmunity: Technological Advances and Critical Limitations. J. Autoimmun. 2023, 134, 102982. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Zhang, G.; Lv, X.; Ren, L. Critical Role of Cellular MicroRNAs in Virus Infection: Decades of Progress. Anim. Zoonoses. 2025, In press. [CrossRef]
- Bhatti, G.K.; Khullar, N.; Sidhu, I.S.; Navik, U.S.; Reddy, A.P.; Reddy, P.H.; Bhatti, J.S. Emerging Role of Non-coding RNA in Health and Disease. Metab. Brain Dis. 2021, 36, 1119–1134. [Google Scholar] [CrossRef] [PubMed]
- Harries, L.W. Dysregulated RNA Processing and Metabolism: A New Hallmark of Ageing and Provocation for Cellular Senescence. FEBS J. 2023, 290, 1221–1234. [Google Scholar] [CrossRef]
- Wei, H.; Xu, Y.; Lin, L.; Li, Y.; Zhu, X. A Review on the Role of RNA Methylation in Aging-Related Diseases. Int. J. Biol. Macromol. 2024, 254, 127769. [Google Scholar] [CrossRef]
- Flores-Concha, M.; Oñate, Á.A. Long Non-Coding RNAs in the Regulation of the Immune Response and Trained Immunity. Front. Genet. 2020, 11, 718. [Google Scholar] [CrossRef]
- Jarrous, N.; Liu, F. Human RNase P: Overview of a Ribonuclease of Interrelated Molecular Networks and Gene-Targeting Systems. RNA 2023, 29, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Hyjek, M.; Figiel, M.; Nowotny, M. RNases H: Structure and Mechanism. DNA Repair 2019, 84, 102672. [Google Scholar] [CrossRef]
- Naeem, S.; Zhang, J.; Zhang, Y.; Wang, Y. Nucleic Acid Therapeutics: Past, Present, and Future. Mol. Ther. Nucleic Acids 2025, 36, 102440. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.; Xia, Z.J.; Lo, N.; Daud, K.; He, H.H. Assembling the RNA Therapeutics Toolbox. Med. Rev. 2024, 4, 110–128. [Google Scholar] [CrossRef]
- Liang, X.; Shen, W.; Sun, H.; Migawa, M.T.; Vickers, T.A.; Crooke, S.T. Translation Efficiency of MRNAs Is Increased by Antisense Oligonucleotides Targeting Upstream Open Reading Frames. Nat. Biotechnol. 2016, 34, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Sun, R.; Bui, H.-H.; Crosby, J.R.; Monia, B.P.; Guo, S. Steric Inhibition of 5′ UTR Regulatory Elements Results in Upregulation of Human CFTR. Mol. Ther. 2019, 27, 1749–1757. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Zhan, J.; Zhou, Y. Argonaute Proteins: Structures and Their Endonuclease Activity. Mol. Biol. Rep. 2021, 48, 4837–4849. [Google Scholar] [CrossRef]
- Jiang, M.; Hong, X.; Gao, Y.; Kho, A.T.; Tantisira, K.G.; Li, J. PiRNA Associates with Immune Diseases. Cell Commun. Signal. 2024, 22, 347. [Google Scholar] [CrossRef]
- Zhang, K.; Li, Y.; Huang, Y.; Sun, K. PiRNA in Cardiovascular Disease: Focus on Cardiac Remodeling and Cardiac Protection. J. Cardiovasc. Transl. Res. 2023, 16, 768–777. [Google Scholar] [CrossRef]
- Deng, X.; Liao, T.; Xie, J.; Kang, D.; He, Y.; Sun, Y.; Wang, Z.; Jiang, Y.; Miao, X.; Yan, Y.; et al. The Burgeoning Importance of PIWI-Interacting RNAs in Cancer Progression. Sci. China Life Sci. 2024, 67, 653–662. [Google Scholar] [CrossRef]
- Wu, Z.; Yu, X.; Zhang, S.; He, Y.; Guo, W. Novel Roles of PIWI Proteins and PIWI-Interacting RNAs in Human Health and Diseases. Cell Commun. Signal. 2023, 21, 343. [Google Scholar] [CrossRef] [PubMed]
- Aravin, A.; Gaidatzis, D.; Pfeffer, S.; Lagos-Quintana, M.; Landgraf, P.; Iovino, N.; Morris, P.; Brownstein, M.J.; Kuramochi-Miyagawa, S.; Nakano, T.; et al. A Novel Class of Small RNAs Bind to MILI Protein in Mouse Testes. Nature 2006, 442, 203–207. [Google Scholar] [CrossRef]
- Girard, A.; Sachidanandam, R.; Hannon, G.J.; Carmell, M.A. A Germline-Specific Class of Small RNAs Binds Mammalian Piwi Proteins. Nature 2006, 442, 199–202. [Google Scholar] [CrossRef]
- Kumar, P.; Anaya, J.; Mudunuri, S.B.; Dutta, A. Meta-Analysis of TRNA Derived RNA Fragments Reveals That They Are Evolutionarily Conserved and Associate with AGO Proteins to Recognize Specific RNA Targets. BMC Biol. 2014, 12, 78. [Google Scholar] [CrossRef]
- Liu, Y.; Li, C.; Cui, X.; Li, M.; Liu, S.; Wang, Z. Potentially Diagnostic and Prognostic Roles of PiRNAs/PIWIs in Pancreatic Cancer: A Review. Biochim. Biophys. Acta-Rev. Cancer 2025, 1880, 189286. [Google Scholar] [CrossRef]
- Loubalova, Z.; Konstantinidou, P.; Haase, A.D. Themes and Variations on PiRNA-Guided Transposon Control. Mob. DNA 2023, 14, 10. [Google Scholar] [CrossRef]
- Egli, M.; Manoharan, M. Chemistry, Structure and Function of Approved Oligonucleotide Therapeutics. Nucleic Acids Res. 2023, 51, 2529–2573. [Google Scholar] [CrossRef] [PubMed]
- Stein, H.; Hausen, P. Enzyme from Calf Thymus Degrading the RNA Moiety of DNA-RNA Hybrids: Effect on DNA-Dependent RNA Polymerase. Science 1969, 166, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Malik, H.S. Phylogenetic Analysis of Ribonuclease H Domains Suggests a Late, Chimeric Origin of LTR Retrotransposable Elements and Retroviruses. Genome Res. 2001, 11, 1187–1197. [Google Scholar] [CrossRef]
- Lee, H.; Cho, H.; Kim, J.; Lee, S.; Yoo, J.; Park, D.; Lee, G. RNase H Is an Exo- and Endoribonuclease with Asymmetric Directionality, Depending on the Binding Mode to the Structural Variants of RNA:DNA Hybrids. Nucleic Acids Res. 2022, 50, 1801–1814. [Google Scholar] [CrossRef]
- Pizzi, S.; Sertic, S.; Orcesi, S.; Cereda, C.; Bianchi, M.; Jackson, A.P.; Lazzaro, F.; Plevani, P.; Muzi-Falconi, M. Reduction of HRNase H2 Activity in Aicardi–Goutières Syndrome Cells Leads to Replication Stress and Genome Instability. Hum. Mol. Genet. 2015, 24, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Cerritelli, S.M.; Frolova, E.G.; Feng, C.; Grinberg, A.; Love, P.E.; Crouch, R.J. Failure to Produce Mitochondrial DNA Results in Embryonic Lethality in Rnaseh1 Null Mice. Mol. Cell 2003, 11, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Crow, Y.J.; Leitch, A.; Hayward, B.E.; Garner, A.; Parmar, R.; Griffith, E.; Ali, M.; Semple, C.; Aicardi, J.; Babul-Hirji, R.; et al. Mutations in Genes Encoding Ribonuclease H2 Subunits Cause Aicardi-Goutières Syndrome and Mimic Congenital Viral Brain Infection. Nat. Genet. 2006, 38, 910–916. [Google Scholar] [CrossRef]
- Crow, Y.J.; Chase, D.S.; Lowenstein Schmidt, J.; Szynkiewicz, M.; Forte, G.M.A.; Gornall, H.L.; Oojageer, A.; Anderson, B.; Pizzino, A.; Helman, G.; et al. Characterization of Human Disease Phenotypes Associated with Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med. Genet. Part A 2015, 167, 296–312. [Google Scholar] [CrossRef] [PubMed]
- Pendergraft, W.F.; Means, T.K. AGS, SLE, and RNASEH2 Mutations: Translating Insights into Therapeutic Advances. J. Clin. Investig. 2015, 125, 102–104. [Google Scholar] [CrossRef]
- Günther, C.; Kind, B.; Reijns, M.A.M.; Berndt, N.; Martinez-Bueno, M.; Wolf, C.; Tüngler, V.; Chara, O.; Lee, Y.A.; Hübner, N.; et al. Defective Removal of Ribonucleotides from DNA Promotes Systemic Autoimmunity. J. Clin. Investig. 2015, 125, 413–424. [Google Scholar] [CrossRef]
- Aden, K.; Bartsch, K.; Dahl, J.; Reijns, M.A.M.; Esser, D.; Sheibani-Tezerji, R.; Sinha, A.; Wottawa, F.; Ito, G.; Mishra, N.; et al. Epithelial RNase H2 Maintains Genome Integrity and Prevents Intestinal Tumorigenesis in Mice. Gastroenterology 2019, 156, 145–159.e19. [Google Scholar] [CrossRef]
- Hiller, B.; Hoppe, A.; Haase, C.; Hiller, C.; Schubert, N.; Müller, W.; Reijns, M.A.M.; Jackson, A.P.; Kunkel, T.A.; Wenzel, J.; et al. Ribonucleotide Excision Repair Is Essential to Prevent Squamous Cell Carcinoma of the Skin. Cancer Res. 2018, 78, 5917–5926. [Google Scholar] [CrossRef]
- Cerritelli, S.M.; El Hage, A. RNases H1 and H2: Guardians of the Stability of the Nuclear Genome When Supply of DNTPs Is Limiting for DNA Synthesis. Curr. Genet. 2020, 66, 1073–1084. [Google Scholar] [CrossRef]
- Lima, W.F.; Murray, H.M.; Damle, S.S.; Hart, C.E.; Hung, G.; De Hoyos, C.L.; Liang, X.-H.; Crooke, S.T. Viable RNaseH1 Knockout Mice Show RNaseH1 Is Essential for R Loop Processing, Mitochondrial and Liver Function. Nucleic Acids Res. 2016, 44, 5299–5312. [Google Scholar] [CrossRef]
- Liang, X.-H.; Sun, H.; Nichols, J.G.; Crooke, S.T. RNase H1-Dependent Antisense Oligonucleotides Are Robustly Active in Directing RNA Cleavage in Both the Cytoplasm and the Nucleus. Mol. Ther. 2017, 25, 2075–2092. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Sun, H.; De Hoyos, C.L.; Bailey, J.K.; Liang, X.; Crooke, S.T. Dynamic Nucleoplasmic and Nucleolar Localization of Mammalian RNase H1 in Response to RNAP I Transcriptional R-Loops. Nucleic Acids Res. 2017, 45, 10672–10692. [Google Scholar] [CrossRef]
- Cerritelli, S.M.; Crouch, R.J. Ribonuclease H: The Enzymes in Eukaryotes. FEBS J. 2009, 276, 1494–1505. [Google Scholar] [CrossRef]
- Holmes, J.B.; Akman, G.; Wood, S.R.; Sakhuja, K.; Cerritelli, S.M.; Moss, C.; Bowmaker, M.R.; Jacobs, H.T.; Crouch, R.J.; Holt, I.J. Primer Retention Owing to the Absence of RNase H1 Is Catastrophic for Mitochondrial DNA Replication. Proc. Natl. Acad. Sci. USA 2015, 112, 9334–9339. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Liang, X.-H.; Baker, B.F.; Crooke, R.M. Antisense Technology: A Review. J. Biol. Chem. 2021, 296, 100416. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Liang, X.; Crooke, R.M.; Baker, B.F.; Geary, R.S. Antisense Drug Discovery and Development Technology Considered in a Pharmacological Context. Biochem. Pharmacol. 2020, 189, 114196. [Google Scholar] [CrossRef]
- Nowotny, M.; Cerritelli, S.M.; Ghirlando, R.; Gaidamakov, S.A.; Crouch, R.J.; Yang, W. Specific Recognition of RNA/DNA Hybrid and Enhancement of Human RNase H1 Activity by HBD. EMBO J. 2008, 27, 1172–1181. [Google Scholar] [CrossRef]
- Davis, R.R.; Shaban, N.M.; Perrino, F.W.; Hollis, T. Crystal Structure of RNA-DNA Duplex Provides Insight into Conformational Changes Induced by RNase H Binding. Cell Cycle 2015, 14, 668–673. [Google Scholar] [CrossRef]
- Pang, J.; Guo, Q.; Lu, Z. The Catalytic Mechanism, Metal Dependence, Substrate Specificity, and Biodiversity of Ribonuclease H. Front. Microbiol. 2022, 13, 1034811. [Google Scholar] [CrossRef]
- Samara, N.L.; Yang, W. Cation Trafficking Propels RNA Hydrolysis. Nat. Struct. Mol. Biol. 2018, 25, 715–721. [Google Scholar] [CrossRef]
- Wu, H.; Lima, W.F.; Crooke, S.T. Investigating the Structure of Human RNase H1 by Site-Directed Mutagenesis. J. Biol. Chem. 2001, 276, 23547–23553. [Google Scholar] [CrossRef] [PubMed]
- Goedken, E.R.; Marqusee, S. Co-Crystal of Escherichia Coli RNase HI with Mn 2+ Ions Reveals Two Divalent Metals Bound in the Active Site. J. Biol. Chem. 2001, 276, 7266–7271. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, M.; Yang, W. Stepwise Analyses of Metal Ions in RNase H Catalysis from Substrate Destabilization to Product Release. EMBO J. 2006, 25, 1924–1933. [Google Scholar] [CrossRef]
- Belikova, A.M.; Zarytova, V.F.; Grineva, N.I. Synthesis of Ribonucleosides and Diribonucleoside Phosphates Containing 2-Chloro-Ethylamine and Nitrogen Mustard Residues. Tetrahedron Lett. 1967, 8, 3557–3562. [Google Scholar] [CrossRef]
- Grineva, N.I.; Karpova, G.G. Complementary Addressed Alkylation of Ribosomal RNA with Alkylating Derivatives of Oligonucleotides. Mol. Biol. 1974, 8, 832–844. [Google Scholar] [PubMed]
- Grineva, N.I.; Karpova, G.G.; Kuznetsova, L.M.; Uimitova, T.A.; Venkstern, T.B. Complementarily Addressed Alkylation of Yeast TRNA 1 Val with Chloroethylmethylaminobenzylidene d(PC-G)-A. Proof of the Modification of the Third Nucleotide Located at the 5′-Terminus of the Complete Binding Site of the Reagent. Mol. Biol. 1976, 10, 1260–1271. [Google Scholar]
- Paterson, B.M.; Roberts, B.E.; Kuff, E.L. Structural Gene Identification and Mapping by DNA.MRNA Hybrid-Arrested Cell-Free Translation. Proc. Natl. Acad. Sci. USA 1977, 74, 4370–4374. [Google Scholar] [CrossRef]
- Sang, A.; Zhuo, S.; Bochanis, A.; Manautou, J.E.; Bahal, R.; Zhong, X.; Rasmussen, T.P. Mechanisms of Action of the US Food and Drug Administration-Approved Antisense Oligonucleotide Drugs. BioDrugs 2024, 38, 511–526. [Google Scholar] [CrossRef]
- Collotta, D.; Bertocchi, I.; Chiapello, E.; Collino, M. Antisense Oligonucleotides: A Novel Frontier in Pharmacological Strategy. Front. Pharmacol. 2023, 14, 1304342. [Google Scholar] [CrossRef]
- Vickers, T.A.; Crooke, S.T. The Rates of the Major Steps in the Molecular Mechanism of RNase H1-Dependent Antisense Oligonucleotide Induced Degradation of RNA. Nucleic Acids Res. 2015, 43, 8955–8963. [Google Scholar] [CrossRef]
- Liang, X.; Sun, H.; Shen, W.; Crooke, S.T. Identification and Characterization of Intracellular Proteins That Bind Oligonucleotides with Phosphorothioate Linkages. Nucleic Acids Res. 2015, 43, 2927–2945. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Wang, S.; Vickers, T.A.; Shen, W.; Liang, X. Cellular Uptake and Trafficking of Antisense Oligonucleotides. Nat. Biotechnol. 2017, 35, 230–237. [Google Scholar] [CrossRef]
- Wu, H.; Sun, H.; Liang, X.; Lima, W.F.; Crooke, S.T. Human RNase H1 Is Associated with Protein P32 and Is Involved in Mitochondrial Pre-rRNA Processing. PLoS ONE 2013, 8, e71006. [Google Scholar] [CrossRef]
- Zhang, L.; Bernardo, K.D.; Vickers, T.A.; Tian, J.; Liang, X.; Crooke, S.T. NAT10 and DDX21 Proteins Interact with RNase H1 and Affect the Performance of Phosphorothioate Oligonucleotides. Nucleic Acid Ther. 2022, 32, 280–299. [Google Scholar] [CrossRef]
- Zamaratski, E.; Pradeepkumar, P.I.; Chattopadhyaya, J. A Critical Survey of the Structure-Function of the Antisense Oligo/RNA Heteroduplex as Substrate for RNase H. J. Biochem. Biophys. Methods 2001, 48, 189–208. [Google Scholar] [CrossRef] [PubMed]
- Lima, W.F.; Nichols, J.G.; Wu, H.; Prakash, T.P.; Migawa, M.T.; Wyrzykiewicz, T.K.; Bhat, B.; Crooke, S.T. Structural Requirements at the Catalytic Site of the Heteroduplex Substrate for Human RNase H1 Catalysis. J. Biol. Chem. 2004, 279, 36317–36326. [Google Scholar] [CrossRef]
- Stein, C.A.; Subasinghe, C.; Shinozuka, K.; Cohen, J.S. Physicochemical Properties of Phosphorothioate Oligodeoxynucleotides. Nucleic Acids Res. 1988, 16, 3209–3221. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Vickers, T.A.; Liang, X. Phosphorothioate Modified Oligonucleotide–protein Interactions. Nucleic Acids Res. 2020, 48, 5235–5253. [Google Scholar] [CrossRef]
- Caruthers, M.H.; Beaton, G.; Cummins, L.; Dellinger, D.; Graff, D.; Ma, Y.-X.; Marshall, W.S.; Sasmor, H.; Shankland, P.; Van Wu, J.; et al. Chemical and Biochemical Studies with Dithioate DNA. Nucleosides Nucleotides 1991, 10, 47–59. [Google Scholar] [CrossRef]
- Cummins, L.; Graff, D.; Beaton, G.; Marshall, W.S.; Caruthers, M.H. Biochemical and Physicochemical Properties of Phosphorodithioate DNA. Biochemistry 1996, 35, 8734–8741. [Google Scholar] [CrossRef]
- Sergueev, D.S.; Shaw, B.R. H-Phosphonate Approach for Solid-Phase Synthesis of Oligodeoxyribonucleoside Boranophosphates and Their Characterization. J. Am. Chem. Soc. 1998, 120, 9417–9427. [Google Scholar] [CrossRef]
- Takahashi, Y.; Sato, K.; Wada, T. Solid-Phase Synthesis of Boranophosphate/Phosphorothioate/Phosphate Chimeric Oligonucleotides and Their Potential as Antisense Oligonucleotides. J. Org. Chem. 2022, 87, 3895–3909. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Nukaga, Y.; Wada, T. Solid-Phase Synthesis and Properties of Stereocontrolled Boranophosphate/Phosphate and Phosphorothioate/Phosphate Chimeric Oligouridylates. R. Soc. Open Sci. 2023, 10, 230095. [Google Scholar] [CrossRef]
- Sheehan, D.; Lunstad, B.; Yamada, C.M.; Stell, B.G.; Caruthers, M.H.; Dellinger, D.J. Biochemical Properties of Phosphonoacetate and Thiophosphonoacetate Oligodeoxyribonucleotides. Nucleic Acids Res. 2003, 31, 4109–4118. [Google Scholar] [CrossRef] [PubMed]
- Šípová, H.; Špringer, T.; Rejman, D.; Šimák, O.; Petrová, M.; Novák, P.; Rosenbergová, Š.; Páv, O.; Liboska, R.; Barvík, I.; et al. 5′-O-Methylphosphonate Nucleic Acids—New Modified DNAs That Increase the Escherichia coli RNase H Cleavage Rate of Hybrid Duplexes. Nucleic Acids Res. 2014, 42, 5378–5389. [Google Scholar] [CrossRef] [PubMed]
- Yamada, C.M.; Dellinger, D.J.; Caruthers, M.H. Synthesis and Biochemical Evaluation of Phosphonoformate Oligodeoxyribonucleotides. J. Am. Chem. Soc. 2006, 128, 5251–5261. [Google Scholar] [CrossRef]
- Perry, C.M.; Barman Balfour, J.A. Fomivirsen. Drugs 1999, 57, 375–380. [Google Scholar] [CrossRef]
- Miroshnichenko, S.K.; Patutina, O.A.; Burakova, E.A.; Chelobanov, B.P.; Fokina, A.A.; Vlassov, V.V.; Altman, S.; Zenkova, M.A.; Stetsenko, D.A. Mesyl Phosphoramidate Antisense Oligonucleotides as an Alternative to Phosphorothioates with Improved Biochemical and Biological Properties. Proc. Natl. Acad. Sci. USA 2019, 116, 1229–1234. [Google Scholar] [CrossRef]
- Patutina, O.A.; Gaponova (Miroshnichenko), S.K.; Sen’kova, A.V.; Savin, I.A.; Gladkikh, D.V.; Burakova, E.A.; Fokina, A.A.; Maslov, M.A.; Shmendel’, E.V.; Wood, M.J.A.; et al. Mesyl Phosphoramidate Backbone Modified Antisense Oligonucleotides Targeting MiR-21 with Enhanced in Vivo Therapeutic Potency. Proc. Natl. Acad. Sci. USA 2020, 117, 32370–32379. [Google Scholar] [CrossRef]
- Gaponova, S.; Patutina, O.; Sen’kova, A.; Burakova, E.; Savin, I.; Markov, A.; Shmendel, E.; Maslov, M.; Stetsenko, D.; Vlassov, V.; et al. Single Shot vs. Cocktail: A Comparison of Mono- and Combinative Application of MiRNA-Targeted Mesyl Oligonucleotides for Efficient Antitumor Therapy. Cancers 2022, 14, 4396. [Google Scholar] [CrossRef]
- Anderson, B.A.; Freestone, G.C.; Low, A.; De-Hoyos, C.L.; Drury, W.J., III; Østergaard, M.E.; Migawa, M.T.; Fazio, M.; Wan, W.B.; Berdeja, A.; et al. Towards next Generation Antisense Oligonucleotides: Mesylphosphoramidate Modification Improves Therapeutic Index and Duration of Effect of Gapmer Antisense Oligonucleotides. Nucleic Acids Res. 2021, 49, 9026–9041. [Google Scholar] [CrossRef]
- Zhang, L.; Liang, X.; De Hoyos, C.L.; Migawa, M.; Nichols, J.G.; Freestone, G.; Tian, J.; Seth, P.P.; Crooke, S.T. The Combination of Mesyl-Phosphoramidate Inter-Nucleotide Linkages and 2′- O -Methyl in Selected Positions in the Antisense Oligonucleotide Enhances the Performance of RNaseH1 Active PS-ASOs. Nucleic Acid Ther. 2022, 32, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Noronha, A.M.; Wilds, C.J.; Lok, C.-N.; Viazovkina, K.; Arion, D.; Parniak, M.A.; Damha, M.J. Synthesis and Biophysical Properties of Arabinonucleic Acids (ANA): Circular Dichroic Spectra, Melting Temperatures, and Ribonuclease H Susceptibility of ANA·RNA Hybrid Duplexes. Biochemistry 2000, 39, 7050–7062. [Google Scholar] [CrossRef]
- Damha, M.J.; Wilds, C.J.; Noronha, A.; Brukner, I.; Borkow, G.; Arion, D.; Parniak, M.A. Hybrids of RNA and Arabinonucleic Acids (ANA and 2′F-ANA) Are Substrates of Ribonuclease H. J. Am. Chem. Soc. 1998, 120, 13545. [Google Scholar] [CrossRef]
- Li, F.; Sarkhel, S.; Wilds, C.J.; Wawrzak, Z.; Prakash, T.P.; Manoharan, M.; Egli, M. 2′-Fluoroarabino- and Arabinonucleic Acid Show Different Conformations, Resulting in Deviating RNA Affinities and Processing of Their Heteroduplexes with RNA by RNase H. Biochemistry 2006, 45, 4141–4152. [Google Scholar] [CrossRef] [PubMed]
- Kalota, A.; Karabon, L.; Swider, C.R.; Viazovkina, E.; Elzagheid, M.; Damha, M.J.; Gewirtz, A.M. 2′-Deoxy-2′-Fluoro-Beta-D-Arabinonucleic Acid (2′F-ANA) Modified Oligonucleotides (ON) Effect Highly Efficient, and Persistent, Gene Silencing. Nucleic Acids Res. 2006, 34, 451–461. [Google Scholar] [CrossRef]
- Takahashi, M.; Li, H.; Zhou, J.; Chomchan, P.; Aishwarya, V.; Damha, M.J.; Rossi, J.J. Dual Mechanisms of Action of Self-Delivering, Anti-HIV-1 FANA Oligonucleotides as a Potential New Approach to HIV Therapy. Mol. Ther.-Nucleic Acids 2019, 17, 615–625. [Google Scholar] [CrossRef]
- Pelisch, N.; Rosas Almanza, J.; Stehlik, K.E.; Aperi, B.V.; Kroner, A. Use of a Self-Delivering Anti-CCL3 FANA Oligonucleotide as an Innovative Approach to Target Inflammation after Spinal Cord Injury. eNeuro 2021, 8, ENEURO.0338-20.2021. [Google Scholar] [CrossRef]
- Chorzalska, A.; Morgan, J.; Ahsan, N.; Treaba, D.O.; Olszewski, A.J.; Petersen, M.; Kingston, N.; Cheng, Y.; Lombardo, K.; Schorl, C.; et al. Bone Marrow–specific Loss of ABI1 Induces Myeloproliferative Neoplasm with Features Resembling Human Myelofibrosis. Blood 2018, 132, 2053–2066. [Google Scholar] [CrossRef]
- Akimova, T.; Wang, L.; Bartosh, Z.; Christensen, L.M.; Eruslanov, E.; Singhal, S.; Aishwarya, V.; Hancock, W.W. Antisense Targeting of FOXP3+ Tregs to Boost Anti-Tumor Immunity. Front. Immunol. 2024, 15, 1426657. [Google Scholar] [CrossRef]
- Danielsen, M.B.; Lou, C.; Lisowiec-Wachnicka, J.; Pasternak, A.; Jørgensen, P.T.; Wengel, J. Gapmer Antisense Oligonucleotides Containing 2′, 3′-Dideoxy-2′-Fluoro-3′-C-Hydroxymethyl-β-d-Lyxofuranosyl Nucleotides Display Site-Specific RNase H Cleavage and Induce Gene Silencing. Chem.—A Eur. J. 2020, 26, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Istrate, A.; Katolik, A.; Istrate, A.; Leumann, C.J. 2′β-Fluoro-Tricyclo Nucleic Acids (2′F-tc-ANA): Thermal Duplex Stability, Structural Studies, and RNase H Activation. Chem.—A Eur. J. 2017, 23, 10310–10318. [Google Scholar] [CrossRef]
- Frei, S.; Istrate, A.; Leumann, C.J. 6′-Fluoro[4.3.0]Bicyclo Nucleic Acid: Synthesis, Biophysical Properties and Molecular Dynamics Simulations. Beilstein J. Org. Chem. 2018, 14, 3088–3097. [Google Scholar] [CrossRef]
- Frei, S.; Katolik, A.K.; Leumann, C.J. Synthesis, Biophysical Properties, and RNase H Activity of 6′-Difluoro[4.3.0]Bicyclo-DNA. Beilstein J. Org. Chem. 2019, 15, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Katsuzaki, Y.; Tsukimura, R.; Chandela, A.; Chano, T.; Ueno, Y. 4′-C-Aminoethoxy-Modified DNAs Exhibit Increased Nuclease Resistance, Sustained RNase H Activity, and Inhibition of KRAS Gene Expression. Chem. Biodivers. 2022, 19, e202200125. [Google Scholar] [CrossRef]
- Onishi, S.; Chandela, A.; Ueno, Y. Synthesis and Evaluation of 4′-C-(S) and 4′-C-(R)-2-Aminopropoxy−Thymidine-Modified DNAs for Thermal Stability, RNase H Digestion, and Nuclease Resistance. ChemistrySelect 2024, 9, e202305090. [Google Scholar] [CrossRef]
- Wang, J.; Verbeure, B.; Luyten, I.; Lescrinier, E.; Froeyen, M.; Hendrix, C.; Rosemeyer, H.; Seela, F.; Van Aerschot, A.; Herdewijn, P. Cyclohexene Nucleic Acids (CeNA): Serum Stable Oligonucleotides That Activate RNase H and Increase Duplex Stability with Complementary RNA. J. Am. Chem. Soc. 2000, 122, 8595–8602. [Google Scholar] [CrossRef]
- Verbeure, B. RNase H Mediated Cleavage of RNA by Cyclohexene Nucleic Acid (CeNA). Nucleic Acids Res. 2001, 29, 4941–4947. [Google Scholar] [CrossRef] [PubMed]
- Pontarelli, A.; Wilds, C.J. Oligonucleotides Containing C5-Propynyl Modified Arabinonucleic Acids: Synthesis, Biophysical and Antisense Properties. ChemBioChem 2023, 24, e202300068. [Google Scholar] [CrossRef]
- Stetsenko, D.A. Mesyl Phosphoramidate Oligonucleotides: A New Promising Type of Antisense Agents. In Handbook of Chemical Biology of Nucleic Acids; Springer Nature: Singapore, 2023; pp. 543–583. [Google Scholar]
- Souleimanian, N.; Deleavey, G.F.; Soifer, H.; Wang, S.; Tiemann, K.; Damha, M.J.; Stein, C.A. Antisense 2′-Deoxy, 2′-Fluoroarabino Nucleic Acid (2′F-ANA) Oligonucleotides: In Vitro Gymnotic Silencers of Gene Expression Whose Potency Is Enhanced by Fatty Acids. Mol. Ther.-Nucleic Acids 2012, 1, e43. [Google Scholar] [CrossRef]
- Le, B.T.; Chen, S.; Abramov, M.; Herdewijn, P.; Veedu, R.N. Evaluation of Anhydrohexitol Nucleic Acid, Cyclohexenyl Nucleic Acid and d-Altritol Nucleic Acid-Modified 2′-O-Methyl RNA Mixmer Antisense Oligonucleotides for Exon Skipping in Vitro. Chem. Commun. 2016, 52, 13467–13470. [Google Scholar] [CrossRef] [PubMed]
- Sergeeva, O.; Akhmetova, E.; Dukova, S.; Beloglazkina, E.; Uspenskaya, A.; Machulkin, A.; Stetsenko, D.; Zatsepin, T. Structure-Activity Relationship Study of Mesyl and Busyl Phosphoramidate Antisense Oligonucleotides for Unaided and PSMA-Mediated Uptake into Prostate Cancer Cells. Front. Chem. 2024, 12, 1342178. [Google Scholar] [CrossRef] [PubMed]
- Astaneh, B.; Makhdami, N.; Astaneh, V.; Guyatt, G. The Effect of Mipomersen in the Management of Patients with Familial Hypercholesterolemia: A Systematic Review and Meta-Analysis of Clinical Trials. J. Cardiovasc. Dev. Dis. 2021, 8, 82. [Google Scholar] [CrossRef] [PubMed]
- Gales, L. Tegsedi (Inotersen): An Antisense Oligonucleotide Approved for the Treatment of Adult Patients with Hereditary Transthyretin Amyloidosis. Pharmaceuticals 2019, 12, 78. [Google Scholar] [CrossRef]
- Paik, J.; Duggan, S. Volanesorsen: First Global Approval. Drugs 2019, 79, 1349–1354. [Google Scholar] [CrossRef]
- Nie, T. Eplontersen: First Approval. Drugs 2024, 84, 473–478. [Google Scholar] [CrossRef]
- Meyer, T.; Schumann, P.; Weydt, P.; Petri, S.; Koc, Y.; Spittel, S.; Bernsen, S.; Günther, R.; Weishaupt, J.H.; Dreger, M.; et al. Neurofilament Light-Chain Response during Therapy with Antisense Oligonucleotide Tofersen in SOD1-Related ALS: Treatment Experience in Clinical Practice. Muscle Nerve 2023, 67, 515–521. [Google Scholar] [CrossRef]
- Syed, Y.Y. Olezarsen: First Approval. Drugs 2025, 85, 571–576. [Google Scholar] [CrossRef]
- Laxton, C.; Brady, K.; Moschos, S.; Turnpenny, P.; Rawal, J.; Pryde, D.C.; Sidders, B.; Corbau, R.; Pickford, C.; Murray, E.J. Selection, Optimization, and Pharmacokinetic Properties of a Novel, Potent Antiviral Locked Nucleic Acid-Based Antisense Oligomer Targeting Hepatitis C Virus Internal Ribosome Entry Site. Antimicrob. Agents Chemother. 2011, 55, 3105–3114. [Google Scholar] [CrossRef]
- Chery, J.; Petri, A.; Wagschal, A.; Lim, S.-Y.; Cunningham, J.; Vasudevan, S.; Kauppinen, S.; Näär, A.M. Development of Locked Nucleic Acid Antisense Oligonucleotides Targeting Ebola Viral Proteins and Host Factor Niemann-Pick C1. Nucleic Acid Ther. 2018, 28, 273–284. [Google Scholar] [CrossRef]
- Sadewasser, A.; Dietzel, E.; Michel, S.; Klüver, M.; Helfer, M.; Thelemann, T.; Klar, R.; Eickmann, M.; Becker, S.; Jaschinski, F. Anti-Niemann Pick C1 Single-Stranded Oligonucleotides with Locked Nucleic Acids Potently Reduce Ebola Virus Infection In Vitro. Mol. Ther.-Nucleic Acids 2019, 16, 686–697. [Google Scholar] [CrossRef]
- Okamoto, S.; Echigoya, Y.; Tago, A.; Segawa, T.; Sato, Y.; Itou, T. Antiviral Efficacy of RNase H-Dependent Gapmer Antisense Oligonucleotides against Japanese Encephalitis Virus. Int. J. Mol. Sci. 2023, 24, 14846. [Google Scholar] [CrossRef] [PubMed]
- Lulla, V.; Wandel, M.P.; Bandyra, K.J.; Ulferts, R.; Wu, M.; Dendooven, T.; Yang, X.; Doyle, N.; Oerum, S.; Beale, R.; et al. Targeting the Conserved Stem Loop 2 Motif in the SARS-CoV-2 Genome. J. Virol. 2021, 95, e0066321. [Google Scholar] [CrossRef]
- Qiao, Y.; Wotring, J.W.; Zhang, C.J.; Jiang, X.; Xiao, L.; Watt, A.; Gattis, D.; Scandalis, E.; Freier, S.; Zheng, Y.; et al. Antisense Oligonucleotides to Therapeutically Target SARS-CoV-2 Infection. PLoS ONE 2023, 18, e0281281. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, M.; Saso, W.; Yamamoto, T.; Sato, M.; Takagi, H.; Hasegawa, T.; Kozakura, Y.; Yokoi, H.; Ohashi, H.; Tsuchimoto, K.; et al. Anti-SARS-CoV-2 Gapmer Antisense Oligonucleotides Targeting the Main Protease Region of Viral RNA. Antivir. Res. 2024, 230, 105992. [Google Scholar] [CrossRef]
- Zhu, C.; Lee, J.Y.; Woo, J.Z.; Xu, L.; Nguyenla, X.; Yamashiro, L.H.; Ji, F.; Biering, S.B.; Van Dis, E.; Gonzalez, F.; et al. An Intranasal ASO Therapeutic Targeting SARS-CoV-2. Nat. Commun. 2022, 13, 4503. [Google Scholar] [CrossRef] [PubMed]
- Morelli, E.; Biamonte, L.; Federico, C.; Amodio, N.; Di Martino, M.T.; Gallo Cantafio, M.E.; Manzoni, M.; Scionti, F.; Samur, M.K.; Gullà, A.; et al. Therapeutic Vulnerability of Multiple Myeloma to MIR17PTi, a First-in-Class Inhibitor of Pri-MiR-17-92. Blood 2018, 132, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Morelli, E.; Fulciniti, M.; Samur, M.K.; Ribeiro, C.F.; Wert-Lamas, L.; Henninger, J.E.; Gullà, A.; Aktas-Samur, A.; Todoerti, K.; Talluri, S.; et al. A MIR17HG-Derived Long Noncoding RNA Provides an Essential Chromatin Scaffold for Protein Interaction and Myeloma Growth. Blood 2023, 141, 391–405. [Google Scholar] [CrossRef]
- Løvendorf, M.B.; Holm, A.; Petri, A.; Thrue, C.A.; Uchida, S.; Venø, M.T.; Kauppinen, S. Knockdown of Circular RNAs Using LNA-Modified Antisense Oligonucleotides. Nucleic Acid Ther. 2023, 33, 45–57. [Google Scholar] [CrossRef]
- Nasseri, S.; Sharifi, M.; Mehrzad, V. Effects of Hsa-PiR-32877 Suppression with Antisense LNA GapmeRs on the Proliferation and Apoptosis of Human Acute Myeloid Leukemia Cells. Int. J. Mol. Cell. Med. 2023, 12, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Berhane, T.; Holm, A.; Karstensen, K.T.; Petri, A.; Ilieva, M.S.; Krarup, H.; Vyberg, M.; Løvendorf, M.B.; Kauppinen, S. Knockdown of the Long Noncoding RNA PURPL Induces Apoptosis and Sensitizes Liver Cancer Cells to Doxorubicin. Sci. Rep. 2022, 12, 19502. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Sun, C.; Mahapatra, K.D.; Riihilä, P.; Knuutila, J.; Nissinen, L.; Lapins, J.; Kähäri, V.-M.; Homey, B.; Sonkoly, E.; et al. Long Noncoding RNA Plasmacytoma Variant Translocation 1 Is Overexpressed in Cutaneous Squamous Cell Carcinoma and Exon 2 Is Critical for Its Oncogenicity. Br. J. Dermatol. 2024, 190, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; Stamato, M.A.; Juli, G.; Morelli, E.; Fulciniti, M.; Manzoni, M.; Taiana, E.; Agnelli, L.; Cantafio, M.E.G.; Romeo, E.; et al. Drugging the LncRNA MALAT1 via LNA GapmeR ASO Inhibits Gene Expression of Proteasome Subunits and Triggers Anti-Multiple Myeloma Activity. Leukemia 2018, 32, 1948–1957. [Google Scholar] [CrossRef]
- Adewunmi, O.; Shen, Y.; Zhang, X.H.-F.; Rosen, J.M. Targeted Inhibition of LncRNA Malat1 Alters the Tumor Immune Microenvironment in Preclinical Syngeneic Mouse Models of Triple-Negative Breast Cancer. Cancer Immunol. Res. 2023, 11, 1462–1479. [Google Scholar] [CrossRef]
- Bothwell, A.L.; Altman, S. Partial Purification and Properties of an Endoribonuclease Isolated from Human KB Cells. J. Biol. Chem. 1975, 250, 1451–1459. [Google Scholar] [CrossRef]
- Koski, R.A.; Bothwell, A.L.M.; Altman, S. Identification of a Ribonuclease P-like Activity from Human KB Cells. Cell 1976, 9, 101–116. [Google Scholar] [CrossRef]
- Forster, A.C.; Altman, S. External Guide Sequences for an RNA Enzyme. Science 1990, 249, 783–786. [Google Scholar] [CrossRef]
- Yuan, Y.; Altman, S. Selection of Guide Sequences That Direct Efficient Cleavage of MRNA by Human Ribonuclease P. Science 1994, 263, 1269–1273. [Google Scholar] [CrossRef]
- Altman, S. Antibiotics Present and Future. FEBS Lett. 2014, 588, 1–2. [Google Scholar] [CrossRef]
- Kirsebom, L.A.; Liu, F.; McClain, W.H. The Discovery of a Catalytic RNA within RNase P and Its Legacy. J. Biol. Chem. 2024, 300, 107318. [Google Scholar] [CrossRef] [PubMed]
- Danilin, N.A.; Matveev, A.L.; Tikunova, N.V.; Venyaminova, A.G.; Novopashina, D.S. Conjugates of RNase P-Guiding Oligonucleotides with Oligo(N-Methylpyrrole) as Prospective Antibacterial Agents. Russ. J. Bioorg. Chem. 2021, 47, 469–477. [Google Scholar] [CrossRef]
- Deng, Q.; Liu, Y.; Li, X.; Yan, B.; Sun, X.; Tang, W.; Trang, P.; Yang, Z.; Gong, H.; Wang, Y.; et al. Inhibition of Human Cytomegalovirus Major Capsid Protein Expression and Replication by Ribonuclease P–associated External Guide Sequences. RNA 2019, 25, 645–655. [Google Scholar] [CrossRef]
- Yan, B.; Liu, Y.; Chen, Y.-C.; Liu, F. External Guide Sequence Effectively Suppresses the Gene Expression and Replication of Herpes Simplex Virus 2. Molecules 2024, 29, 2052. [Google Scholar] [CrossRef]
- Liu, Y.; Yan, B.; Zhang, I.; Liu, F. RNase P-Associated External Guide Sequences Inhibit HIV-1 Infection by Shutting Down Human CCR5 Expression. Receptors 2025, 4, 3. [Google Scholar] [CrossRef]
- Höck, J.; Meister, G. The Argonaute Protein Family. Genome Biol. 2008, 9, 210. [Google Scholar] [CrossRef]
- Meister, G. Argonaute Proteins: Functional Insights and Emerging Roles. Nat. Rev. Genet. 2013, 14, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K. Anatomy of Four Human Argonaute Proteins. Nucleic Acids Res. 2022, 50, 6618–6638. [Google Scholar] [CrossRef]
- Quévillon Huberdeau, M.; Zeitler, D.M.; Hauptmann, J.; Bruckmann, A.; Fressigné, L.; Danner, J.; Piquet, S.; Strieder, N.; Engelmann, J.C.; Jannot, G.; et al. Phosphorylation of Argonaute Proteins Affects MRNA Binding and Is Essential for MicroRNA-guided Gene Silencing in Vivo. EMBO J. 2017, 36, 2088–2106. [Google Scholar] [CrossRef]
- Mohamed, A.A.; Wang, P.Y.; Bartel, D.P.; Vos, S.M. The Structural Basis for RNA Slicing by Human Argonaute2. Cell Rep. 2025, 44, 115166. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Du, X.; Muramatsu, S.; Gomez, C.M. An MiRNA-Mediated Therapy for SCA6 Blocks IRES-Driven Translation of the CACNA1A Second Cistron. Sci. Transl. Med. 2016, 8, 347ra94. [Google Scholar] [CrossRef] [PubMed]
- Anzelon, T.A.; Chowdhury, S.; Hughes, S.M.; Xiao, Y.; Lander, G.C.; MacRae, I.J. Structural Basis for PiRNA Targeting. Nature 2021, 597, 285–289. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Tomari, Y.; Zamore, P.D. The RNA-Induced Silencing Complex Is a Mg2+-Dependent Endonuclease. Curr. Biol. 2004, 14, 787–791. [Google Scholar] [CrossRef]
- Liu, J.; Carmell, M.A.; Rivas, F.V.; Marsden, C.G.; Thomson, J.M.; Song, J.-J.; Hammond, S.M.; Joshua-Tor, L.; Hannon, G.J. Argonaute2 Is the Catalytic Engine of Mammalian RNAi. Science 2004, 305, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Meister, G.; Landthaler, M.; Patkaniowska, A.; Dorsett, Y.; Teng, G.; Tuschl, T. Human Argonaute2 Mediates RNA Cleavage Targeted by MiRNAs and SiRNAs. Mol. Cell 2004, 15, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Park, M.S.; Phan, H.-D.; Busch, F.; Hinckley, S.H.; Brackbill, J.A.; Wysocki, V.H.; Nakanishi, K. Human Argonaute3 Has Slicer Activity. Nucleic Acids Res. 2017, 45, 11867–11877. [Google Scholar] [CrossRef]
- Park, M.S.; Sim, G.; Kehling, A.C.; Nakanishi, K. Human Argonaute2 and Argonaute3 Are Catalytically Activated by Different Lengths of Guide RNA. Proc. Natl. Acad. Sci. USA 2020, 117, 28576–28578. [Google Scholar] [CrossRef]
- Nakanishi, K. Are Argonaute-Associated Tiny RNAs Junk, Inferior MiRNAs, or a New Type of Functional RNAs? Front. Mol. Biosci. 2021, 8, 795356. [Google Scholar] [CrossRef]
- Sim, G.; Kehling, A.C.; Park, M.S.; Divoky, C.; Zhang, H.; Malhotra, N.; Secor, J.; Nakanishi, K. Determining the Defining Lengths between Mature MicroRNAs/Small Interfering RNAs and TinyRNAs. Sci. Rep. 2023, 13, 19761. [Google Scholar] [CrossRef]
- Hauptmann, J.; Dueck, A.; Harlander, S.; Pfaff, J.; Merkl, R.; Meister, G. Turning Catalytically Inactive Human Argonaute Proteins into Active Slicer Enzymes. Nat. Struct. Mol. Biol. 2013, 20, 814–817. [Google Scholar] [CrossRef]
- Faehnle, C.R.; Elkayam, E.; Haase, A.D.; Hannon, G.J.; Joshua-Tor, L. The Making of a Slicer: Activation of Human Argonaute-1. Cell Rep. 2013, 3, 1901–1909. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, S.; Qi, H.H.; Chowdhury, D.; Shi, Y.; Novina, C.D. Distinct Passenger Strand and MRNA Cleavage Activities of Human Argonaute Proteins. Nat. Struct. Mol. Biol. 2009, 16, 1259–1266. [Google Scholar] [CrossRef]
- Pong, S.K.; Gullerova, M. Noncanonical Functions of MicroRNA Pathway Enzymes-Drosha, DGCR8, Dicer and Ago Proteins. FEBS Lett. 2018, 592, 2973–2986. [Google Scholar] [CrossRef]
- Wu, J.; Yang, J.; Cho, W.C.; Zheng, Y. Argonaute Proteins: Structural Features, Functions and Emerging Roles. J. Adv. Res. 2020, 24, 317–324. [Google Scholar] [CrossRef]
- Wang, J.; Li, Y. Current Advances in Antiviral RNA Interference in Mammals. FEBS J. 2024, 291, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.-W.; Voinnet, O. Antiviral Immunity Directed by Small RNAs. Cell 2007, 130, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Dexheimer, P.J.; Cochella, L. MicroRNAs: From Mechanism to Organism. Front. Cell Dev. Biol. 2020, 8, 409. [Google Scholar] [CrossRef]
- Isenmann, M.; Stoddart, M.J.; Schmelzeisen, R.; Gross, C.; Della Bella, E.; Rothweiler, R.M. Basic Principles of RNA Interference: Nucleic Acid Types and In Vitro Intracellular Delivery Methods. Micromachines 2023, 14, 1321. [Google Scholar] [CrossRef]
- Sheu-Gruttadauria, J.; MacRae, I.J. Phase Transitions in the Assembly and Function of Human MiRISC. Cell 2018, 173, 946–957. [Google Scholar] [CrossRef]
- Saliminejad, K.; Khorram Khorshid, H.R.; Soleymani Fard, S.; Ghaffari, S.H. An Overview of MicroRNAs: Biology, Functions, Therapeutics, and Analysis Methods. J. Cell. Physiol. 2019, 234, 5451–5465. [Google Scholar] [CrossRef]
- Lima, W.F.; Prakash, T.P.; Murray, H.M.; Kinberger, G.A.; Li, W.; Chappell, A.E.; Li, C.S.; Murray, S.F.; Gaus, H.; Seth, P.P.; et al. Single-Stranded SiRNAs Activate RNAi in Animals. Cell 2012, 150, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Pendergraff, H.; Liu, J.; Kordasiewicz, H.B.; Cleveland, D.W.; Swayze, E.E.; Lima, W.F.; Crooke, S.T.; Prakash, T.P.; Corey, D.R. Single-Stranded RNAs Use RNAi to Potently and Allele-Selectively Inhibit Mutant Huntingtin Expression. Cell 2012, 150, 895–908. [Google Scholar] [CrossRef] [PubMed]
- Matsui, M.; Prakash, T.P.; Corey, D.R. Argonaute 2-Dependent Regulation of Gene Expression by Single-Stranded MiRNA Mimics. Mol. Ther. 2016, 24, 946–955. [Google Scholar] [CrossRef]
- Hu, J.; Liu, J.; Yu, D.; Aiba, Y.; Lee, S.; Pendergraff, H.; Boubaker, J.; Artates, J.W.; Lagier-Tourenne, C.; Lima, W.F.; et al. Exploring the Effect of Sequence Length and Composition on Allele-Selective Inhibition of Human Huntingtin Expression by Single-Stranded Silencing RNAs. Nucleic Acid Ther. 2014, 24, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Prakash, T.P.; Lima, W.F.; Murray, H.M.; Li, W.; Kinberger, G.A.; Chappell, A.E.; Gaus, H.; Seth, P.P.; Bhat, B.; Crooke, S.T.; et al. Identification of Metabolically Stable 5′-Phosphate Analogs That Support Single-Stranded SiRNA Activity. Nucleic Acids Res. 2017, 45, 6994. [Google Scholar] [CrossRef]
- Hu, J.; Rigo, F.; Prakash, T.P.; Corey, D.R. Recognition of C9orf72 Mutant RNA by Single-Stranded Silencing RNAs. Nucleic Acid Ther. 2017, 27, 87–94. [Google Scholar] [CrossRef]
- Pendergraff, H.M.; Debacker, A.J.; Watts, J.K. Single-Stranded Silencing RNAs: Hit Rate and Chemical Modification. Nucleic Acid Ther. 2016, 26, 216–222. [Google Scholar] [CrossRef]
- Hu, J.; Shen, X.; Rigo, F.; Prakash, T.P.; Mootha, V.V.; Corey, D.R. Duplex RNAs and Ss-SiRNAs Block RNA Foci Associated with Fuchs’ Endothelial Corneal Dystrophy. Nucleic Acid Ther. 2019, 29, 73–81. [Google Scholar] [CrossRef]
- Chang, W.; Pei, Y.; Guidry, E.N.; Zewge, D.; Parish, C.A.; Sherer, E.C.; DiMuzio, J.; Zhang, H.; South, V.J.; Strapps, W.R.; et al. Systematic Chemical Modifications of Single Stranded SiRNAs Significantly Improved CTNNB1 MRNA Silencing. Bioorg. Med. Chem. Lett. 2016, 26, 4513–4517. [Google Scholar] [CrossRef]
- Li, Z.; Wang, X.; Zhou, X.; Wang, J.; Guan, Z.; Yang, Z. Optimization in Chemical Modification of Single-Stranded SiRNA Encapsulated by Neutral Cytidinyl/Cationic Lipids. Front. Chem. 2022, 10, 843181. [Google Scholar] [CrossRef]
- Ferino, A.; Miglietta, G.; Picco, R.; Vogel, S.; Wengel, J.; Xodo, L.E. MicroRNA Therapeutics: Design of Single-Stranded MiR-216b Mimics to Target KRAS in Pancreatic Cancer Cells. RNA Biol. 2018, 15, 1273–1285. [Google Scholar] [CrossRef]
- Lima, W.F.; Murray, H.; Nichols, J.G.; Wu, H.; Sun, H.; Prakash, T.P.; Berdeja, A.R.; Gaus, H.J.; Crooke, S.T. Human Dicer Binds Short Single-Strand and Double-Strand RNA with High Affinity and Interacts with Different Regions of the Nucleic Acids. J. Biol. Chem. 2009, 284, 2535–2548. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Sun, X.; Wang, M.; Hou, Y.; Zhan, Y.; Jiang, Y.; Liu, Z.; Cao, X.; Chen, P.; Liu, Z.; et al. A MicroRNA 221– and 222–Mediated Feedback Loop Maintains Constitutive Activation of NFκB and STAT3 in Colorectal Cancer Cells. Gastroenterology 2014, 147, 847–859.e11. [Google Scholar] [CrossRef]
- Castellanos-Gonzalez, A.; Sadiqova, A.; Ortega-Mendez, J.; White, A.C. RNA-Based Therapy for Cryptosporidium Parvum Infection: Proof-of-Concept Studies. Infect. Immun. 2022, 90, e0019622. [Google Scholar] [CrossRef]
- Chen, L.; Wu, Y.; Tang, Q.; Tang, F. Oncogenic-TsRNA: A Novel Diagnostic and Therapeutic Molecule for Cancer Clinic. J. Cancer 2024, 15, 5403–5414. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Rafi, M.; Aldarmaki, M.; ElSiddig, M.; Al Nuaimi, M.; Amiri, K.M.A. TRNA Derived Small RNAs—Small Players with Big Roles. Front. Genet. 2022, 13, 997780. [Google Scholar] [CrossRef]
- Tian, H.; Hu, Z.; Wang, C. The Therapeutic Potential of TRNA-Derived Small RNAs in Neurodegenerative Disorders. Aging Dis. 2022, 13, 389. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, C.-Y.; Fang, B.; Li, B.; Li, Y.-H.; Xia, Q.-Q.; Zhao, Y.; Cheng, X.-L.; Yang, S.-M.; Zhang, M.-H.; et al. The Function and Therapeutic Potential of Transfer RNA-Derived Small RNAs in Cardiovascular Diseases: A Review. Pharmacol. Res. 2024, 206, 107279. [Google Scholar] [CrossRef] [PubMed]
- Saad, A.A.A.; Zhang, K.; Deng, Q.; Zhou, J.; Ge, L.; Wang, H. The Functions and Modifications of TRNA-Derived Small RNAs in Cancer Biology. Cancer Metastasis Rev. 2025, 44, 38. [Google Scholar] [CrossRef]
- Li, G.; Manning, A.C.; Bagi, A.; Yang, X.; Gokulnath, P.; Spanos, M.; Howard, J.; Chan, P.P.; Sweeney, T.; Kitchen, R.; et al. Distinct Stress-Dependent Signatures of Cellular and Extracellular TRNA-Derived Small RNAs. Adv. Sci. 2022, 9, e2200829. [Google Scholar] [CrossRef]
- Wang, Y.; Weng, Q.; Ge, J.; Zhang, X.; Guo, J.; Ye, G. TRNA-Derived Small RNAs: Mechanisms and Potential Roles in Cancers. Genes Dis. 2022, 9, 1431–1442. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shao, Z.; Wu, S. Research Progress on the TsRNA Biogenesis, Function, and Application in Lung Cancer. Non-coding RNA Res. 2025, 10, 63–69. [Google Scholar] [CrossRef]
- Akiyama, Y.; Ivanov, P. TRNA-Derived RNAs: Biogenesis and Roles in Translational Control. Wiley Interdiscip. Rev. RNA 2023, 14, e1805. [Google Scholar] [CrossRef]
- Mo, D.; He, F.; Zheng, J.; Chen, H.; Tang, L.; Yan, F. TRNA-Derived Fragment TRF-17-79MP9PP Attenuates Cell Invasion and Migration via THBS1/TGF-Β1/Smad3 Axis in Breast Cancer. Front. Oncol. 2021, 11, 656078. [Google Scholar] [CrossRef]
- Zhu, L.; Li, Z.; Yu, X.; Ruan, Y.; Shen, Y.; Shao, Y.; Zhang, X.; Ye, G.; Guo, J. The TRNA-Derived Fragment 5026a Inhibits the Proliferation of Gastric Cancer Cells by Regulating the PTEN/PI3K/AKT Signaling Pathway. Stem Cell Res. Ther. 2021, 12, 418. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zheng, J.; Wang, X.; Zhou, B.; Chen, H.; Li, G.; Yan, F. TRF-Val-CAC-016 Modulates the Transduction of CACNA1d-Mediated MAPK Signaling Pathways to Suppress the Proliferation of Gastric Carcinoma. Cell Commun. Signal. 2022, 20, 68. [Google Scholar] [CrossRef]
- Han, Y.; Peng, Y.; Liu, S.; Wang, X.; Cai, C.; Guo, C.; Chen, Y.; Gao, L.; Huang, Q.; He, M.; et al. TRF3008A Suppresses the Progression and Metastasis of Colorectal Cancer by Destabilizing FOXK1 in an AGO-Dependent Manner. J. Exp. Clin. Cancer Res. 2022, 41, 32. [Google Scholar] [CrossRef]
- Wang, Y.; Xia, W.; Shen, F.; Zhou, J.; Gu, Y.; Chen, Y. TRNA-derived Fragment TRF-Glu49 Inhibits Cell Proliferation, Migration and Invasion in Cervical Cancer by Targeting FGL1. Oncol. Lett. 2022, 24, 334. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Z.; Weng, Q.; Zheng, Y.; Lin, Y.; Guo, J.; Ye, G. Clinical Diagnostic Values of Transfer RNA-Derived Fragment TRF-41-YDLBRY73W0K5KKOVD and Its Effects on the Growth of Gastric Cancer Cells. DNA Cell Biol. 2023, 42, 176–187. [Google Scholar] [CrossRef]
- Zhang, S.; Gu, Y.; Ge, J.; Xie, Y.; Yu, X.; Wu, X.; Sun, D.; Zhang, X.; Guo, J.; Guo, J. TRF-33-P4R8YP9LON4VDP Inhibits Gastric Cancer Progression via Modulating STAT3 Signaling Pathway in an AGO2-Dependent Manner. Oncogene 2024, 43, 2160–2171. [Google Scholar] [CrossRef]
- Su, J.; Cheng, J.; Hu, Y.; Yu, Q.; Li, Z.; Li, J.; Zheng, N.; Zhang, Z.; Yang, J.; Li, X.; et al. Transfer RNA-Derived Small RNAs and Their Potential Roles in the Therapeutic Heterogeneity of Sacubitril/Valsartan in Heart Failure Patients after Acute Myocardial Infarction. Front. Cardiovasc. Med. 2022, 9, 961700. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Qian, B.; Wang, F.; Huang, Y.; Yan, X.; Li, P.; Zhang, Q.; Li, Y.; Sun, K. Global Profile of TRNA-Derived Small RNAs in Pathological Cardiac Hypertrophy Plasma and Identification of TRF-21-NB8PLML3E as a New Hypertrophy Marker. Diagnostics 2023, 13, 2065. [Google Scholar] [CrossRef]
- Sun, B.; Chen, Z.; Chi, Q.; Zhang, Y.; Gao, B. Endogenous TRNA-derived Small RNA (TRF3-Thr-AGT) Inhibits ZBP1/NLRP3 Pathway-mediated Cell Pyroptosis to Attenuate Acute Pancreatitis (AP). J. Cell. Mol. Med. 2021, 25, 10441–10453. [Google Scholar] [CrossRef]
- Green, J.A.; Ansari, M.Y.; Ball, H.C.; Haqqi, T.M. TRNA-Derived Fragments (TRFs) Regulate Post-Transcriptional Gene Expression via AGO-Dependent Mechanism in IL-1β Stimulated Chondrocytes. Osteoarthr. Cartil. 2020, 28, 1102–1110. [Google Scholar] [CrossRef]
- Du, L.; Chen, W.; Zhang, D.; Cui, Y.; He, Z. The Functions and Mechanisms of PiRNAs in Mediating Mammalian Spermatogenesis and Their Applications in Reproductive Medicine. Cell. Mol. Life Sci. 2024, 81, 379. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Borja, E.; Siegl, F.; Mateu, R.; Slaby, O.; Sedo, A.; Busek, P.; Sana, J. Critical Appraisal of the PiRNA-PIWI Axis in Cancer and Cancer Stem Cells. Biomark. Res. 2024, 12, 15. [Google Scholar] [CrossRef]
- Gomes Fernandes, M.; He, N.; Wang, F.; Van Iperen, L.; Eguizabal, C.; Matorras, R.; Roelen, B.A.J.; Chuva De Sousa Lopes, S.M. Human-Specific Subcellular Compartmentalization of P-Element Induced Wimpy Testis-like (PIWIL) Granules during Germ Cell Development and Spermatogenesis. Hum. Reprod. 2018, 33, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Shiohama, A.; Minoshima, S.; Shimizu, N. Identification of Eight Members of the Argonaute Family in the Human Genome☆. Genomics 2003, 82, 323–330. [Google Scholar] [CrossRef]
- Li, Z.; Xu, Q.; Zhong, J.; Zhang, Y.; Zhang, T.; Ying, X.; Lu, X.; Li, X.; Wan, L.; Xue, J.; et al. Structural Insights into RNA Cleavage by PIWI Argonaute. Nature 2025, 639, 250–259. [Google Scholar] [CrossRef]
- Vourekas, A.; Alexiou, P.; Vrettos, N.; Maragkakis, M.; Mourelatos, Z. Sequence-Dependent but Not Sequence-Specific PiRNA Adhesion Traps MRNAs to the Germ Plasm. Nature 2016, 531, 390–394. [Google Scholar] [CrossRef]
- Gainetdinov, I.; Vega-Badillo, J.; Cecchini, K.; Bagci, A.; Colpan, C.; De, D.; Bailey, S.; Arif, A.; Wu, P.-H.; MacRae, I.J.; et al. Relaxed Targeting Rules Help PIWI Proteins Silence Transposons. Nature 2023, 619, 394–402. [Google Scholar] [CrossRef]
- Roovers, E.F.; Rosenkranz, D.; Mahdipour, M.; Han, C.-T.; He, N.; Chuva de Sousa Lopes, S.M.; van der Westerlaken, L.A.J.; Zischler, H.; Butter, F.; Roelen, B.A.J.; et al. Piwi Proteins and PiRNAs in Mammalian Oocytes and Early Embryos. Cell Rep. 2015, 10, 2069–2082. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Xing, S.; Shen, B.-Y.; Chen, H.-T.; Sun, B.; Wang, Z.-T.; Wang, J.-W.; Lu, X.-X. PIWIL1 Interacting RNA PiR-017061 Inhibits Pancreatic Cancer Growth via Regulating EFNA5. Hum. Cell 2021, 34, 550–563. [Google Scholar] [CrossRef]
- Ding, L.; Wang, R.; Xu, W.; Shen, D.; Cheng, S.; Wang, H.; Lu, Z.; Zheng, Q.; Wang, L.; Xia, L.; et al. PIWI-Interacting RNA 57125 Restrains Clear Cell Renal Cell Carcinoma Metastasis by Downregulating CCL3 Expression. Cell Death Discov. 2021, 7, 333. [Google Scholar] [CrossRef]
- Wang, X.; Huang, P.; Lei, M.; Ma, Y.; Chen, H.; Sun, J.; Hu, Y.; Shi, J. Global Expression and Functional Analysis of Human PiRNAs during HSV-1 Infection. Virus Res. 2023, 328, 199087. [Google Scholar] [CrossRef] [PubMed]
- Qi, T.; Cao, H.; Sun, H.; Feng, H.; Li, N.; Wang, C.; Wang, L. PiR-19166 Inhibits Migration and Metastasis through CTTN/MMPs Pathway in Prostate Carcinoma. Aging 2020, 12, 18209–18220. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Chen, Q.; Zhou, Z.; Tian, Z.; Zheng, X.; Wang, K. PiRNA-18 Inhibition Cell Proliferation, Migration and Invasion in Colorectal Cancer. Biochem. Genet. 2023, 61, 1881–1897. [Google Scholar] [CrossRef]
- Johnson, K.C.; Kilikevicius, A.; Hofman, C.; Hu, J.; Liu, Y.; Aguilar, S.; Graswich, J.; Han, Y.; Wang, T.; Westcott, J.M.; et al. Nuclear Localization of Argonaute 2 Is Affected by Cell Density and May Relieve Repression by MicroRNAs. Nucleic Acids Res. 2024, 52, 1930–1952. [Google Scholar] [CrossRef]
- Patutina, O.A.; Bichenkova, E.V.; Miroshnichenko, S.K.; Mironova, N.L.; Trivoluzzi, L.T.; Burusco, K.K.; Bryce, R.A.; Vlassov, V.V.; Zenkova, M.A. MiRNases: Novel Peptide-Oligonucleotide Bioconjugates That Silence MiR-21 in Lymphosarcoma Cells. Biomaterials 2017, 122, 163–178. [Google Scholar] [CrossRef]
- Chiglintseva, D.; Clarke, D.J.; Sen’kova, A.; Heyman, T.; Miroshnichenko, S.; Shan, F.; Vlassov, V.; Zenkova, M.; Patutina, O.; Bichenkova, E. Engineering Supramolecular Dynamics of Self-Assembly and Turnover of Oncogenic MicroRNAs to Drive Their Synergistic Destruction in Tumor Models. Biomaterials 2024, 309, 122604. [Google Scholar] [CrossRef]
- Patutina, O.; Chiglintseva, D.; Amirloo, B.; Clarke, D.; Gaponova, S.; Vlassov, V.; Bichenkova, E.; Zenkova, M. Bulge-Forming MiRNases Cleave Oncogenic MiRNAs at the Central Loop Region in a Sequence-Specific Manner. Int. J. Mol. Sci. 2022, 23, 6562. [Google Scholar] [CrossRef] [PubMed]
- Zellmann, F.; Thomas, L.; Scheffer, U.; Hartmann, R.K.; Göbel, M.W. Site-Specific Cleavage of RNAs Derived from the PIM1 3'-UTR by a Metal-Free Artificial Ribonuclease. Molecules 2019, 24, 807. [Google Scholar] [CrossRef] [PubMed]
- Luige, O.; Karalė, K.; Bose, P.P.; Bollmark, M.; Tedebark, U.; Murtola, M.; Strömberg, R. Influence of Sequence Variation on the RNA Cleavage Activity of Zn2+-Dimethyl-Dppz-PNA-Based Artificial Enzymes. RSC Adv. 2022, 12, 5398–5406. [Google Scholar] [CrossRef]
- Danneberg, F.; Ghidini, A.; Dogandzhiyski, P.; Kalden, E.; Strömberg, R.; Göbel, M.W. Sequence-Specific RNA Cleavage by PNA Conjugates of the Metal-Free Artificial Ribonuclease Tris(2-Aminobenzimidazole). Beilstein J. Org. Chem. 2015, 11, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Weinrich, T.; Scheffer, U.; Kalden, E.; Göbel, M.W. Click Conjugates of Artificial Ribonucleases: Sequence Specific Cleavage with Multiple Turnover. Chem.—A Eur. J. 2025, 31, e202500451. [Google Scholar] [CrossRef]
- Patutina, O.A.; Miroshnichenko, S.K.; Mironova, N.L.; Sen’kova, A.V.; Bichenkova, E.V.; Clarke, D.J.; Vlassov, V.V.; Zenkova, M.A. Catalytic Knockdown of MiR-21 by Artificial Ribonuclease: Biological Performance in Tumor Model. Front. Pharmacol. 2019, 10, 879. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiglintseva, D.A.; Patutina, O.A.; Zenkova, M.A. Endogenous Ribonucleases: Therapeutic Targeting of the Transcriptome Through Oligonucleotide-Triggered RNA Inactivation. Biomolecules 2025, 15, 965. https://doi.org/10.3390/biom15070965
Chiglintseva DA, Patutina OA, Zenkova MA. Endogenous Ribonucleases: Therapeutic Targeting of the Transcriptome Through Oligonucleotide-Triggered RNA Inactivation. Biomolecules. 2025; 15(7):965. https://doi.org/10.3390/biom15070965
Chicago/Turabian StyleChiglintseva, Daria A., Olga A. Patutina, and Marina A. Zenkova. 2025. "Endogenous Ribonucleases: Therapeutic Targeting of the Transcriptome Through Oligonucleotide-Triggered RNA Inactivation" Biomolecules 15, no. 7: 965. https://doi.org/10.3390/biom15070965
APA StyleChiglintseva, D. A., Patutina, O. A., & Zenkova, M. A. (2025). Endogenous Ribonucleases: Therapeutic Targeting of the Transcriptome Through Oligonucleotide-Triggered RNA Inactivation. Biomolecules, 15(7), 965. https://doi.org/10.3390/biom15070965