
Hepatic Lipoprotein Metabolism: Current and Future In Vitro Cell-Based Systems

 , , , and
, , , and
Abstract

1. Introduction—Current Concepts in Hepatic Lipoprotein Metabolism
2. Lipoprotein Metabolism in Disease: Mechanistic Insights and Research Limitations in Model Organisms
3. Two-Dimensional In Vitro Cell and Tissue Culture Models for Lipoprotein Metabolism
3.1. Primary Human Hepatocytes and Hepatoma Cell Lines
3.2. Methodological Challenges of iPSC-Derived Hepatocytes
3.3. Applications for iPSC-Derived Hepatocytes
3.4. Current Approaches for Generating iPSC-Derived Non-Parenchymal Liver Cells
4. Human Hepatic 3D In Vitro Models and Other Hepatic Tissue Models
4.1. Precision-Cut Liver Slices (PCLSs)
4.2. Liver-on-a-Chip Technology
4.3. Three-Dimensional Spheroid Models
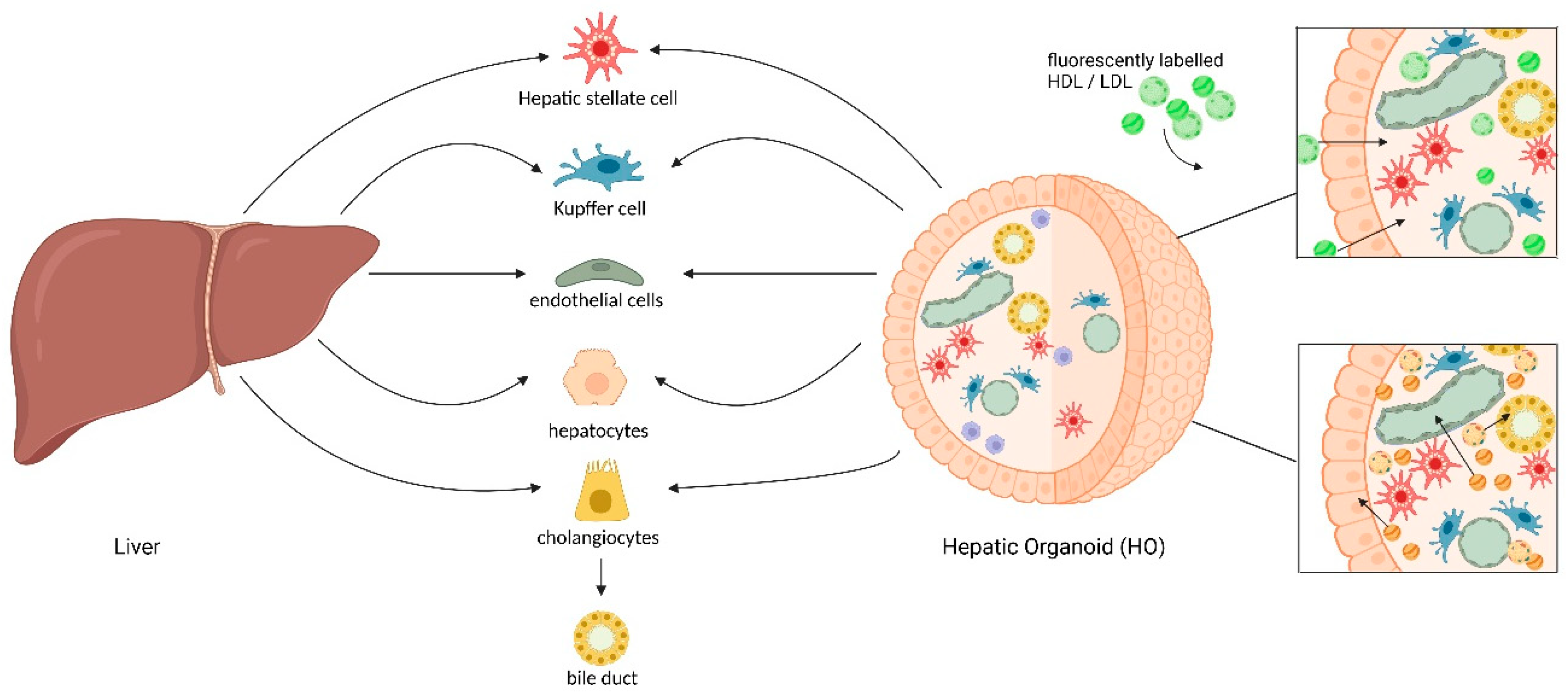
4.4. Liver Organoids: Protocols, Improvements, and Comparisons Between Tissue-Derived and iPSC-Derived Models
4.5. Exploring Lipoprotein Metabolism: Potential and Applicability of Liver Organoids
5. Future Directions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABCA1 | ATP-binding cassette subfamily A member 1 |
| ALCAM | Activated leukocyte cell adhesion molecule |
| ApoA1 | Apolipoprotein A1 |
| ApoB | Apolipoprotein B |
| ApoE | Apolipoprotein E |
| ASCVD | Atherosclerotic cardiovascular disease |
| ASO | Antisense oligonucleotides |
| CVD | Cardiovascular disease |
| CE | Cholesteryl esters |
| CETP | Cholesteryl ester transfer protein |
| CD36 | Cluster of differentiation 36 |
| CYP450 | Cytochrome P450 |
| ECM | Extracellular matrix |
| ESCs | Embryonic stem cells |
| FFAs | Free fatty acids |
| FH | Familial hypercholesterolemia |
| GalNAc | N-acetylgalactosamine |
| HCC | Hepatocellular carcinoma |
| HDL | High-density lipoprotein |
| HGF | Hepatocyte growth factor |
| HGPS | Hutchinson–Gilford progeria syndrome |
| HLCs | Human hepatocyte-like cells |
| HSCs | Hepatic stellate cells |
| iHLCs | Immature hepatocyte-like cells |
| iPSCs | Induced pluripotent stem cells |
| LXR | Liver X receptor |
| LDL | Low-density lipoprotein |
| LDL-C | LDL cholesterol |
| LDLRs | LDL receptors |
| LDLRAP1 | Low-density lipoprotein receptor adapter protein 1 |
| Lp(a) | Lipoprotein (a) |
| LSECs | Liver sinusoidal endothelial cells |
| KCs | Kupffer cells |
| MAFLD | Metabolic dysfunction-associated fatty liver disease |
| MASH | Metabolic dysfunction-associated steatohepatitis |
| MASLD | Metabolic dysfunction-associated steatotic liver disease |
| miR | MicroRNA |
| NPCs | Non-parenchymal cells |
| NRF2 | Nuclear factor erythroid-2-related factor 2 (NRF2) |
| OCA | Obeticholic acid |
| PCLSs | Precision-cut liver slices |
| PCSK9 | Proprotein convertase subtilisin/kexin type 9 |
| PHHs | Primary human hepatocytes |
| RCT | Reverse cholesterol transport |
| SR-B1 | Scavenger receptor class B type 1 |
| VLDL | Very low-density lipoprotein |
References
- Gugliucci, A. The chylomicron saga: Time to focus on postprandial metabolism. Front. Endocrinol. 2024, 14, 1322869. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R. Lipid and Lipoprotein Metabolism. Endocrinol. Metab. Clin. N. Am. 2022, 51, 437–458. [Google Scholar] [CrossRef] [PubMed]
- Dietschy, J.M.; Turley, S.D.; Spady, D.K. Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J. Lipid Res. 1993, 34, 1637–1659. [Google Scholar] [CrossRef]
- Tiwari, S.; Siddiqi, S.A. Intracellular trafficking and secretion of VLDL. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Borén, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: Pathophysiological, genetic, and therapeutic insights: A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41, 2313–2330. [Google Scholar] [CrossRef]
- Hobbs, H.H.; Brown, M.S.; Goldstein, J.L. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum. Mutat. 1992, 1, 445–466. [Google Scholar] [CrossRef]
- Nikkila, K.; Aberg, F.; Isoniemi, H. Transmission of LDLR mutation from donor through liver transplantation resulting in hypercholesterolemia in the recipient. Am. J. Transplant. 2014, 14, 2898–2902. [Google Scholar] [CrossRef]
- Lin, D.; Lu, Y.; Qiu, B.; Feng, M.; Luo, Y.; Xue, F.; Zhou, T.; Zhu, J.; Zhang, J.; Wang, L.; et al. The therapeutic effect of liver transplantation in 14 children with homozygous familial hypercholesterolemia: A prospective cohort: Liver transplant for familial hypercholesterolemia. J. Clin. Lipidol. 2024, 18, e1055–e1066. [Google Scholar] [CrossRef]
- Lambert, G.; Sjouke, B.; Choque, B.; Kastelein, J.J.; Hovingh, G.K. The PCSK9 decade. J. Lipid Res. 2012, 53, 2515–2524. [Google Scholar] [CrossRef]
- Morales, S.V.; Mahmood, A.; Pollard, J.; Mayne, J.; Figeys, D.; Wiseman, P.W. The LDL receptor is regulated by membrane cholesterol as revealed by fluorescence fluctuation analysis. Biophys. J. 2023, 122, 3783–3797. [Google Scholar] [CrossRef]
- Havel, R.J.; Hamilton, R.L. Hepatocytic lipoprotein receptors and intracellular lipoprotein catabolism. Hepatology 1988, 8, 1689–1704. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. A receptor-mediated pathway for cholesterol homeostasis. Science 1986, 232, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Krieger, M. Charting the fate of the “good cholesterol”: Identification and characterization of the high-density lipoprotein receptor SR-BI. Annu. Rev. Biochem. 1999, 68, 523–558. [Google Scholar] [CrossRef]
- Diffenderfer, M.R.; Schaefer, E.J. The composition and metabolism of large and small LDL. Curr. Opin. Lipidol. 2014, 25, 221–226. [Google Scholar] [CrossRef]
- de Beer, M.C.; van der Westhuyzen, D.; Whitaker, N.L.; Webb, N.R.; de Beer, F.C. SR-BI-mediated selective lipid uptake segregates apoA-I and apoA-II catabolism. J. Lipid Res. 2005, 46, 2143–2150. [Google Scholar] [CrossRef]
- Wang, S.; Peng, D.Q.; Yi, Y. The unsolved mystery of apoA-I recycling in adipocyte. Lipids Health Dis. 2016, 15, 35. [Google Scholar] [CrossRef]
- Yvan-Charvet, L.; Wang, N.; Tall, A.R. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Lee-Rueckert, M.; Escola-Gil, J.C.; Kovanen, P.T. HDL functionality in reverse cholesterol transport—Challenges in translating data emerging from mouse models to human disease. Biochim. Biophys. Acta 2016, 1861, 566–583. [Google Scholar] [CrossRef]
- Shrestha, S.; Wu, B.J.; Guiney, L.; Barter, P.J.; Rye, K.-A. Cholesteryl ester transfer protein and its inhibitors. J. Lipid Res. 2018, 59, 772–783. [Google Scholar] [CrossRef]
- Semmler, G.; Baumgartner, C.; Metz, M.; Gensluckner, S.; Habisch, H.; Hofer, H.; März, W.; Offner, F.; Völkerer, A.; Petrenko, O.; et al. Lipid Dysregulation in Tangier Disease: A Case Series and Metabolic Characterization. J. Clin. Endocrinol. Metab. 2025, 110, e2146–e2156. [Google Scholar] [CrossRef]
- Axmann, M.; Plochberger, B.; Mikula, M.; Weber, F.; Strobl, W.M.; Stangl, H. Plasma Membrane Lipids: An Important Binding Site for All Lipoprotein Classes. Membranes 2021, 11, 882. [Google Scholar] [CrossRef]
- Axmann, M.; Sezgin, E.; Karner, A.; Novacek, J.; Brodesser, M.D.; Rohrl, C.; Preiner, J.; Stangl, H.; Plochberger, B. Receptor-Independent Transfer of Low Density Lipoprotein Cargo to Biomembranes. Nano Lett. 2019, 19, 2562–2567. [Google Scholar] [CrossRef]
- Axmann, M.; Strobl, W.M.; Plochberger, B.; Stangl, H. Cholesterol transfer at the plasma membrane. Atherosclerosis 2019, 290, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Plochberger, B.; Axmann, M.; Rohrl, C.; Weghuber, J.; Brameshuber, M.; Rossboth, B.K.; Mayr, S.; Ros, R.; Bittman, R.; Stangl, H.; et al. Direct observation of cargo transfer from HDL particles to the plasma membrane. Atherosclerosis 2018, 277, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Plochberger, B.; Rohrl, C.; Preiner, J.; Rankl, C.; Brameshuber, M.; Madl, J.; Bittman, R.; Ros, R.; Sezgin, E.; Eggeling, C.; et al. HDL particles incorporate into lipid bilayers—A combined AFM and single molecule fluorescence microscopy study. Sci. Rep. 2017, 7, 15886. [Google Scholar] [CrossRef]
- Plochberger, B.; Sych, T.; Weber, F.; Novacek, J.; Axmann, M.; Stangl, H.; Sezgin, E. Lipoprotein Particles Interact with Membranes and Transfer Their Cargo without Receptors. Biochemistry 2020, 59, 4421–4428. [Google Scholar] [CrossRef] [PubMed]
- Lund-Katz, S.; Hammerschlag, B.; Phillips, M.C. Kinetics and mechanism of free cholesterol exchange between human serum high- and low-density lipoproteins. Biochemistry 1982, 21, 2964–2969. [Google Scholar] [CrossRef]
- Rothblat, G.H.; Mahlberg, F.H.; Johnson, W.J.; Phillips, M.C. Apolipoproteins, membrane cholesterol domains, and the regulation of cholesterol efflux. J. Lipid Res. 1992, 33, 1091–1097. [Google Scholar] [CrossRef]
- Phillips, M.C.; Johnson, W.J.; Rothblat, G.H. Mechanisms and consequences of cellular cholesterol exchange and transfer. Biochim. Biophys. Acta 1987, 906, 223–276. [Google Scholar] [CrossRef]
- Scobey, M.W.; Johnson, F.L.; Rudel, L.L. Delivery of high-density lipoprotein free and esterified cholesterol to bile by the perfused monkey liver. Am. J. Physiol. 1989, 257, G644–G652. [Google Scholar] [CrossRef]
- Schwartz, C.C.; VandenBroek, J.M.; Cooper, P.S. Lipoprotein cholesteryl ester production, transfer, and output in vivo in humans. J. Lipid Res. 2004, 45, 1594–1607. [Google Scholar] [CrossRef]
- Schwartz, C.C.; Berman, M.; Vlahcevic, Z.R.; Halloran, L.G.; Gregory, D.H.; Swell, L. Multicompartmental analysis of cholesterol metabolism in man. Characterization of the hepatic bile acid and biliary cholesterol precursor sites. J. Clin. Investig. 1978, 61, 408–423. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, C.C.; Halloran, L.G.; Vlahcevic, Z.R.; Gregory, D.H.; Swell, L. Preferential utilization of free cholesterol from high-density lipoproteins for biliary cholesterol secretion in man. Science 1978, 200, 62–64. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, C.C.; Vlahcevic, Z.R.; Berman, M.; Meadows, J.G.; Nisman, R.M.; Swell, L. Central role of high density lipoprotein in plasma free cholesterol metabolism. J. Clin. Investig. 1982, 70, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, C.C.; Zech, L.A.; VandenBroek, J.M.; Cooper, P.S. Cholesterol kinetics in subjects with bile fistula. Positive relationship between size of the bile acid precursor pool and bile acid synthetic rate. J. Clin. Investig. 1993, 91, 923–938. [Google Scholar] [CrossRef]
- Yelamanchili, D.; Gillard, B.K.; Gotto, A.M., Jr.; Achirica, M.C.; Nasir, K.; Remaley, A.T.; Rosales, C.; Pownall, H.J. HDL-free cholesterol influx into macrophages and transfer to LDL correlate with HDL-free cholesterol content. J. Lipid Res. 2025, 66, 100707. [Google Scholar] [CrossRef]
- Kutyavin, V.I.; Chawla, A. Aster: A New Star in Cholesterol Trafficking. Cell 2018, 175, 307–309. [Google Scholar] [CrossRef]
- Ferrari, A.; Tontonoz, P. Nonvesicular cholesterol transport in physiology. J. Clin. Investig. 2025, 135, e188127. [Google Scholar] [CrossRef]
- Ouimet, M.; Barrett, T.J.; Fisher, E.A. HDL and Reverse Cholesterol Transport. Circ. Res. 2019, 124, 1505–1518. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; Caparrós, E.; Fernández-Iglesias, A.; Francés, R. Role of liver sinusoidal endothelial cells in liver diseases. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 411–431. [Google Scholar] [CrossRef]
- Li, R.; Oteiza, A.; Sørensen, K.K.; McCourt, P.; Olsen, R.; Smedsrød, B.; Svistounov, D. Role of liver sinusoidal endothelial cells and stabilins in elimination of oxidized low-density lipoproteins. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G71–G81. [Google Scholar] [CrossRef]
- Van Berkel, T.J.; De Rijke, Y.B.; Kruijt, J.K. Different fate in vivo of oxidatively modified low density lipoprotein and acetylated low density lipoprotein in rats. Recognition by various scavenger receptors on Kupffer and endothelial liver cells. J. Biol. Chem. 1991, 266, 2282–2289. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Kruse, L.D.; Li, R.; Oteiza, A.; Struck, E.C.; Schürstedt, J.; Hübner, W.; Cogger, V.C.; Le Couteur, D.; Wolfson, D.L.; et al. Impact of oxidized low-density lipoprotein on rat liver sinusoidal endothelial cell morphology and function. Npj Gut Liver 2024, 1, 9. [Google Scholar] [CrossRef]
- Le Couteur, D.G.; Fraser, R.; Cogger, V.C.; McLean, A.J. Hepatic pseudocapillarisation and atherosclerosis in ageing. Lancet 2002, 359, 1612–1615. [Google Scholar] [CrossRef] [PubMed]
- Grosse, L.; Bulavin, D.V. LSEC model of aging. Aging 2020, 12, 11152–11160. [Google Scholar] [CrossRef] [PubMed]
- Grosse, L.; Wagner, N.; Emelyanov, A.; Molina, C.; Lacas-Gervais, S.; Wagner, K.D.; Bulavin, D.V. Defined p16(High) Senescent Cell Types Are Indispensable for Mouse Healthspan. Cell Metab. 2020, 32, 87–99.e86. [Google Scholar] [CrossRef]
- Mitchell, S.J.; Huizer-Pajkos, A.; Cogger, V.C.; McLachlan, A.J.; Le Couteur, D.G.; Jones, B.; de Cabo, R.; Hilmer, S.N. Age-related pseudocapillarization of the liver sinusoidal endothelium impairs the hepatic clearance of acetaminophen in rats. J. Gerontol. A Biol. Sci. Med. Sci. 2011, 66, 400–408. [Google Scholar] [CrossRef]
- Kraehling, J.R.; Chidlow, J.H.; Rajagopal, C.; Sugiyama, M.G.; Fowler, J.W.; Lee, M.Y.; Zhang, X.; Ramírez, C.M.; Park, E.J.; Tao, B.; et al. Genome-wide RNAi screen reveals ALK1 mediates LDL uptake and transcytosis in endothelial cells. Nat. Commun. 2016, 7, 13516. [Google Scholar] [CrossRef]
- Huang, L.; Chambliss, K.L.; Gao, X.; Yuhanna, I.S.; Behling-Kelly, E.; Bergaya, S.; Ahmed, M.; Michaely, P.; Luby-Phelps, K.; Darehshouri, A.; et al. SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nature 2019, 569, 565–569. [Google Scholar] [CrossRef]
- Fruhwurth, S.; Pavelka, M.; Bittman, R.; Kovacs, W.J.; Walter, K.M.; Rohrl, C.; Stangl, H. High-density lipoprotein endocytosis in endothelial cells. World J. Biol. Chem. 2013, 4, 131–140. [Google Scholar] [CrossRef]
- Jang, E.; Robert, J.; Rohrer, L.; von Eckardstein, A.; Lee, W.L. Transendothelial transport of lipoproteins. Atherosclerosis 2020, 315, 111–125. [Google Scholar] [CrossRef] [PubMed]
- von Eckardstein, A.; Robert, J. Endocytosis, Transcytosis, and Retroendocytosis of HDL: Mechanisms, Pathophysiology, and Options for Clinical Exploitation. Arterioscler. Thromb. Vasc. Biol. Online ahead of print. 2025. [Google Scholar]
- Wang, Y.; van der Tuin, S.; Tjeerdema, N.; van Dam, A.D.; Rensen, S.S.; Hendrikx, T.; Berbee, J.F.; Atanasovska, B.; Fu, J.; Hoekstra, M.; et al. Plasma cholesteryl ester transfer protein is predominantly derived from Kupffer cells. Hepatology 2015, 62, 1710–1722. [Google Scholar] [CrossRef]
- McCuskey, R.S.; McCuskey, P.A.; Urbaschek, R.; Urbaschek, B. Kupffer cell function in host defense. Rev. Infect. Dis. 1987, 9 (Suppl. 5), S616–S619. [Google Scholar] [CrossRef]
- Naito, M.; Hasegawa, G.; Ebe, Y.; Yamamoto, T. Differentiation and function of Kupffer cells. Med. Electron. Microsc. 2004, 37, 16–28. [Google Scholar] [CrossRef]
- Takeishi, T.; Hirano, K.; Kobayashi, T.; Hasegawa, G.; Hatakeyama, K.; Naito, M. The role of Kupffer cells in liver regeneration. Arch. Histol. Cytol. 1999, 62, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Bilzer, M.; Roggel, F.; Gerbes, A.L. Role of Kupffer cells in host defense and liver disease. Liver Int. 2006, 26, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Diehl, K.L.; Vorac, J.; Hofmann, K.; Meiser, P.; Unterweger, I.; Kuerschner, L.; Weighardt, H.; Forster, I.; Thiele, C. Kupffer Cells Sense Free Fatty Acids and Regulate Hepatic Lipid Metabolism in High-Fat Diet and Inflammation. Cells 2020, 9, 2258. [Google Scholar] [CrossRef]
- Kandhi, R.; Menendez, A.; Ramanathan, S.; Ilangumaran, S. Regulation of High-Fat Diet-Induced Liver Fibrosis by SOCS1 Expression in Hepatic Stellate Cells. J. Clin. Exp. Hepatol. 2024, 14, 101280. [Google Scholar] [CrossRef]
- Gangireddy, V.G.R.; Pilkerton, C.; Xiang, J.; Tinajero, R.; Ashcraft, A.M. Hepatic Fibrosis and Steatosis in Metabolic Syndrome. J. Obes. Metab. Syndr. 2022, 31, 61–69. [Google Scholar] [CrossRef]
- Chan, D.C.; Barrett, P.H.; Watts, G.F. Lipoprotein transport in the metabolic syndrome: Methodological aspects of stable isotope kinetic studies. Clin. Sci. 2004, 107, 221–232. [Google Scholar] [CrossRef]
- Chan, D.C.; Watts, G.F.; Ng, T.W.; Uchida, Y.; Sakai, N.; Yamashita, S.; Barrett, P.H. Apolipoprotein B-100 kinetics and static plasma indices of triglyceride-rich lipoprotein metabolism in overweight men. Clin. Biochem. 2005, 38, 806–812. [Google Scholar] [CrossRef]
- Chan, D.C.; Barrett, P.H.; Watts, G.F. Lipoprotein transport in the metabolic syndrome: Pathophysiological and interventional studies employing stable isotopy and modelling methods. Clin. Sci. 2004, 107, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Watts, G.F.; Redgrave, T.G.; Mori, T.A.; Barrett, P.H. Apolipoprotein B-100 kinetics in visceral obesity: Associations with plasma apolipoprotein C-III concentration. Metabolism 2002, 51, 1041–1046. [Google Scholar] [CrossRef]
- Chan, D.C.; Watts, G.F.; Gan, S.; Wong, A.T.; Ooi, E.M.; Barrett, P.H. Nonalcoholic fatty liver disease as the transducer of hepatic oversecretion of very-low-density lipoprotein-apolipoprotein B-100 in obesity. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1043–1050. [Google Scholar] [CrossRef]
- Luciani, L.; Pedrelli, M.; Parini, P. Modification of lipoprotein metabolism and function driving atherogenesis in diabetes. Atherosclerosis 2024, 394, 117545. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Gong, Y.; Zhu, N.; Shi, Y.; Zhang, C.; Qin, L. Lipids and lipid metabolism in cellular senescence: Emerging targets for age-related diseases. Ageing Res. Rev. 2024, 97, 102294. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, L.; An, Y.; Wang, Y.; Wang, G. The atherogenic index of plasma: A novel factor more closely related to non-alcoholic fatty liver disease than other lipid parameters in adults. Front. Nutr. 2022, 9, 954219. [Google Scholar] [CrossRef]
- Heeren, J.; Scheja, L. Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol. Metab. 2021, 50, 101238. [Google Scholar] [CrossRef] [PubMed]
- Björnson, E.; Adiels, M.; Taskinen, M.R.; Burgess, S.; Rawshani, A.; Borén, J.; Packard, C.J. Triglyceride-rich lipoprotein remnants, low-density lipoproteins, and risk of coronary heart disease: A UK Biobank study. Eur. Heart J. 2023, 44, 4186–4195. [Google Scholar] [CrossRef]
- Ference, B.A.; Braunwald, E.; Catapano, A.L. The LDL cumulative exposure hypothesis: Evidence and practical applications. Nat. Rev. Cardiol. 2024, 21, 701–716. [Google Scholar] [CrossRef]
- Tybjærg-Hansen, A.; Nordestgaard, B.G.; Christoffersen, M. Triglyceride-rich remnant lipoproteins are more atherogenic than LDL per particle: Is this important? Eur. Heart J. 2023, 44, 4196–4198. [Google Scholar] [CrossRef]
- Berndsen, Z.T.; Cassidy, C.K. The structure of apolipoprotein B100 from human low-density lipoprotein. Nature 2025, 638, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Reimund, M.; Dearborn, A.D.; Graziano, G.; Lei, H.; Ciancone, A.M.; Kumar, A.; Holewinski, R.; Neufeld, E.B.; O’Reilly, F.J.; Remaley, A.T.; et al. Structure of apolipoprotein B100 bound to the low-density lipoprotein receptor. Nature 2025, 638, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Won, H.H.; Peloso, G.M.; Lawson, K.S.; Bartz, T.M.; Deng, X.; van Leeuwen, E.M.; Natarajan, P.; Emdin, C.A.; Bick, A.G.; et al. Diagnostic Yield and Clinical Utility of Sequencing Familial Hypercholesterolemia Genes in Patients With Severe Hypercholesterolemia. J. Am. Coll. Cardiol. 2016, 67, 2578–2589. [Google Scholar] [CrossRef]
- Sjouke, B.; Kusters, D.M.; Kindt, I.; Besseling, J.; Defesche, J.C.; Sijbrands, E.J.; Roeters van Lennep, J.E.; Stalenhoef, A.F.; Wiegman, A.; de Graaf, J.; et al. Homozygous autosomal dominant hypercholesterolaemia in the Netherlands: Prevalence, genotype-phenotype relationship, and clinical outcome. Eur. Heart J. 2015, 36, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Soutar, A.K.; Naoumova, R.P. Mechanisms of disease: Genetic causes of familial hypercholesterolemia. Nat. Clin. Pract. Cardiovasc. Med. 2007, 4, 214–225. [Google Scholar] [CrossRef]
- Vega, G.L.; Grundy, S.M. In vivo evidence for reduced binding of low density lipoproteins to receptors as a cause of primary moderate hypercholesterolemia. J. Clin. Investig. 1986, 78, 1410–1414. [Google Scholar] [CrossRef]
- Innerarity, T.L.; Weisgraber, K.H.; Arnold, K.S.; Mahley, R.W.; Krauss, R.M.; Vega, G.L.; Grundy, S.M. Familial defective apolipoprotein B-100: Low density lipoproteins with abnormal receptor binding. Proc. Natl. Acad. Sci. USA 1987, 84, 6919–6923. [Google Scholar] [CrossRef]
- Young, S.G.; Bertics, S.J.; Curtiss, L.K.; Witztum, J.L. Characterization of an abnormal species of apolipoprotein B, apolipoprotein B-37, associated with familial hypobetalipoproteinemia. J. Clin. Investig. 1987, 79, 1831–1841. [Google Scholar] [CrossRef]
- Simons, L.A.; Reichl, D.; Myant, N.B.; Mancini, M. The metabolism of the apoprotein of plasma low density lipoprotein in familial hyperbetalipoproteinaemia in the homozygous form. Atherosclerosis 1975, 21, 283–298. [Google Scholar] [CrossRef]
- Guardamagna, O.; Abello, F.; Saracco, P.; Baracco, V.; Rolfo, E.; Pirro, M. Endothelial activation, inflammation and premature atherosclerosis in children with familial dyslipidemia. Atherosclerosis 2009, 207, 471–475. [Google Scholar] [CrossRef]
- Napoli, C.; Postiglione, A.; Triggiani, M.; Corso, G.; Palumbo, G.; Carbone, V.; Ruocco, A.; Ambrosio, G.; Montefusco, S.; Malorni, A.; et al. Oxidative structural modifications of low density lipoprotein in homozygous familial hypercholesterolemia. Atherosclerosis 1995, 118, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, J.R.; Rader, D.J.; Ikewaki, K.; Fairwell, T.; Zech, L.A.; Kindt, M.R.; Davignon, J.; Gregg, R.E.; Brewer, H.B., Jr. In vivo metabolism of apolipoprotein A-I in a patient with homozygous familial hypercholesterolemia. Arterioscler. Thromb. 1992, 12, 843–848. [Google Scholar] [CrossRef]
- Frenais, R.; Ouguerram, K.; Maugeais, C.; Marchini, J.S.; Benlian, P.; Bard, J.M.; Magot, T.; Krempf, M. Apolipoprotein A-I kinetics in heterozygous familial hypercholesterolemia: A stable isotope study. J. Lipid Res. 1999, 40, 1506–1511. [Google Scholar] [CrossRef] [PubMed]
- Bellanger, N.; Orsoni, A.; Julia, Z.; Fournier, N.; Frisdal, E.; Duchene, E.; Bruckert, E.; Carrie, A.; Bonnefont-Rousselot, D.; Pirault, J.; et al. Atheroprotective reverse cholesterol transport pathway is defective in familial hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1675–1681. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; Noureen, A.; Kronenberg, F.; Utermann, G. Structure, function, and genetics of lipoprotein (a). J. Lipid Res. 2016, 57, 1339–1359. [Google Scholar] [CrossRef] [PubMed]
- Cain, W.J.; Millar, J.S.; Himebauch, A.S.; Tietge, U.J.; Maugeais, C.; Usher, D.; Rader, D.J. Lipoprotein [a] is cleared from the plasma primarily by the liver in a process mediated by apolipoprotein [a]. J. Lipid Res. 2005, 46, 2681–2691. [Google Scholar] [CrossRef]
- Siddiqui, H.; Yevstigneyev, N.; Madani, G.; McCormick, S. Approaches to Visualising Endocytosis of LDL-Related Lipoproteins. Biomolecules 2022, 12, 158. [Google Scholar] [CrossRef]
- McCormick, S.P.A.; Schneider, W.J. Lipoprotein(a) catabolism: A case of multiple receptors. Pathology 2019, 51, 155–164. [Google Scholar] [CrossRef]
- Havekes, L.; Vermeer, B.J.; Brugman, T.; Emeis, J. Binding of LP(a) to the low density lipoprotein receptor of human fibroblasts. FEBS Lett. 1981, 132, 169–173. [Google Scholar] [CrossRef]
- Romagnuolo, R.; Scipione, C.A.; Boffa, M.B.; Marcovina, S.M.; Seidah, N.G.; Koschinsky, M.L. Lipoprotein(a) catabolism is regulated by proprotein convertase subtilisin/kexin type 9 through the low density lipoprotein receptor. J. Biol. Chem. 2015, 290, 11649–11662. [Google Scholar] [CrossRef] [PubMed]
- Romagnuolo, R.; Scipione, C.A.; Marcovina, S.M.; Gemin, M.; Seidah, N.G.; Boffa, M.B.; Koschinsky, M.L. Roles of the low density lipoprotein receptor and related receptors in inhibition of lipoprotein(a) internalization by proprotein convertase subtilisin/kexin type 9. PLoS ONE 2017, 12, e0180869. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.L.; Eaton, D.L.; Brown, M.S.; McConathy, W.J.; Goldstein, J.L.; Hammer, R.E. Overexpression of human low density lipoprotein receptors leads to accelerated catabolism of Lp(a) lipoprotein in transgenic mice. J. Clin. Investig. 1990, 85, 1542–1547. [Google Scholar] [CrossRef] [PubMed]
- Lingenhel, A.; Kraft, H.G.; Kotze, M.; Peeters, A.V.; Kronenberg, F.; Kruse, R.; Utermann, G. Concentrations of the atherogenic Lp(a) are elevated in FH. Eur. J. Hum. Genet. 1998, 6, 50–60. [Google Scholar] [CrossRef]
- Carmena, R.; Lussier-Cacan, S.; Roy, M.; Minnich, A.; Lingenhel, A.; Kronenberg, F.; Davignon, J. Lp(a) levels and atherosclerotic vascular disease in a sample of patients with familial hypercholesterolemia sharing the same gene defect. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 129–136. [Google Scholar] [CrossRef]
- Yang, X.-P.; Amar, M.J.; Vaisman, B.; Bocharov, A.V.; Vishnyakova, T.G.; Freeman, L.A.; Kurlander, R.J.; Patterson, A.P.; Becker, L.C.; Remaley, A.T. Scavenger receptor-BI is a receptor for lipoprotein(a). J. Lipid Res. 2013, 54, 2450–2457. [Google Scholar] [CrossRef]
- Sharma, M.; Von Zychlinski-Kleffmann, A.; Porteous, C.M.; Jones, G.T.; Williams, M.J.; McCormick, S.P. Lipoprotein (a) upregulates ABCA1 in liver cells via scavenger receptor-B1 through its oxidized phospholipids. J. Lipid Res. 2015, 56, 1318–1328. [Google Scholar] [CrossRef]
- Borén, J.; Packard, C.J.; Binder, C.J. Apolipoprotein B-containing lipoproteins in atherogenesis. Nat. Rev. Cardiol. 2025, 22, 399–413. [Google Scholar] [CrossRef]
- van der Valk, F.M.; Bekkering, S.; Kroon, J.; Yeang, C.; Van den Bossche, J.; van Buul, J.D.; Ravandi, A.; Nederveen, A.J.; Verberne, H.J.; Scipione, C.; et al. Oxidized Phospholipids on Lipoprotein(a) Elicit Arterial Wall Inflammation and an Inflammatory Monocyte Response in Humans. Circulation 2016, 134, 611–624. [Google Scholar] [CrossRef]
- Kronenberg, F.; Mora, S.; Stroes, E.S.G.; Ference, B.A.; Arsenault, B.J.; Berglund, L.; Dweck, M.R.; Koschinsky, M.; Lambert, G.; Mach, F.; et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: A European Atherosclerosis Society consensus statement. Eur. Heart J. 2022, 43, 3925–3946. [Google Scholar] [CrossRef]
- Björnson, E.; Adiels, M.; Taskinen, M.R.; Burgess, S.; Chapman, M.J.; Packard, C.J.; Borén, J. Lipoprotein(a) Is Markedly More Atherogenic Than LDL: An Apolipoprotein B-Based Genetic Analysis. J. Am. Coll. Cardiol. 2024, 83, 385–395. [Google Scholar] [CrossRef]
- Roudaut, M.; Caillaud, A.; Souguir, Z.; Bray, L.; Girardeau, A.; Rimbert, A.; Croyal, M.; Lambert, G.; Patitucci, M.; Delpouve, G.; et al. Human induced pluripotent stem cells-derived liver organoids grown on a Biomimesys® hyaluronic acid-based hydroscaffold as a new model for studying human lipoprotein metabolism. Bioeng. Transl. Med. 2024, 9, e10659. [Google Scholar] [CrossRef]
- Boren, J.; Taskinen, M.R.; Adiels, M. Kinetic studies to investigate lipoprotein metabolism. J. Intern. Med. 2012, 271, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Adiels, M.; Taskinen, M.R.; Packard, C.; Caslake, M.J.; Soro-Paavonen, A.; Westerbacka, J.; Vehkavaara, S.; Hakkinen, A.; Olofsson, S.O.; Yki-Jarvinen, H.; et al. Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia 2006, 49, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Getz, G.S.; Reardon, C.A. Animal models of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1104–1115. [Google Scholar] [CrossRef]
- Gistera, A.; Ketelhuth, D.F.J.; Malin, S.G.; Hansson, G.K. Animal Models of Atherosclerosis-Supportive Notes and Tricks of the Trade. Circ. Res. 2022, 130, 1869–1887. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, S.; Brown, M.S.; Goldstein, J.L.; Gerard, R.D.; Hammer, R.E.; Herz, J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J. Clin. Investig. 1993, 92, 883–893. [Google Scholar] [CrossRef]
- Zhang, S.H.; Reddick, R.L.; Piedrahita, J.A.; Maeda, N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 1992, 258, 468–471. [Google Scholar] [CrossRef]
- Plump, A.S.; Smith, J.D.; Hayek, T.; Aalto-Setala, K.; Walsh, A.; Verstuyft, J.G.; Rubin, E.M.; Breslow, J.L. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell 1992, 71, 343–353. [Google Scholar] [CrossRef]
- Dillard, A.; Matthan, N.R.; Lichtenstein, A.H. Use of hamster as a model to study diet-induced atherosclerosis. Nutr. Metab. 2010, 7, 89. [Google Scholar] [CrossRef]
- Fernandez, M.L.; Volek, J.S. Guinea pigs: A suitable animal model to study lipoprotein metabolism, atherosclerosis and inflammation. Nutr. Metab. 2006, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Clay, M.A.; Hopkins, G.J.; Ehnholm, C.P.; Barter, P.J. The rabbit as an animal model of hepatic lipase deficiency. Biochim. Et Biophys. Acta 1989, 1002, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Rudel, L.L.; Bond, M.G.; Bullock, B.C. LDL heterogeneity and atherosclerosis in nonhuman primates. Ann. N. Y Acad. Sci. 1985, 454, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Kingsley, D.M.; Krieger, M. Receptor-mediated endocytosis of low density lipoprotein: Somatic cell mutants define multiple genes required for expression of surface-receptor activity. Proc. Natl. Acad. Sci. USA 1984, 81, 5454–5458. [Google Scholar] [CrossRef]
- Chang, T.Y.; Hasan, M.T.; Chin, J.; Chang, C.C.; Spillane, D.M.; Chen, J. Chinese hamster ovary cell mutants affecting cholesterol metabolism. Curr. Opin. Lipidol. 1997, 8, 65–71. [Google Scholar] [CrossRef]
- Harder, C.J.; Vassiliou, G.; McBride, H.M.; McPherson, R. Hepatic SR-BI-mediated cholesteryl ester selective uptake occurs with unaltered efficiency in the absence of cellular energy. J. Lipid Res. 2006, 47, 492–503. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Dana, S.E.; Brown, M.S. Esterification of low density lipoprotein cholesterol in human fibroblasts and its absence in homozygous familial hypercholesterolemia. Proc. Natl. Acad. Sci. USA 1974, 71, 4288–4292. [Google Scholar] [CrossRef]
- Hoeg, J.M.; Demosky, S.J., Jr.; Lackner, K.J.; Osborne, J.C., Jr.; Oliver, C.; Brewer, H.B., Jr. The expressed human hepatic receptor for low-density lipoproteins differs from the fibroblast low-density lipoprotein receptor. Biochim. Biophys. Acta 1986, 876, 13–21. [Google Scholar] [CrossRef]
- Sparrow, C.P.; Pittman, R.C. Cholesterol esters selectively taken up from high-density lipoproteins are hydrolyzed extralysosomally. Biochim. Biophys. Acta 1990, 1043, 203–210. [Google Scholar] [CrossRef]
- Rinninger, F.; Pittman, R.C. The selective uptake of HDL cholesteryl esters and its role in reverse cholesterol transport. Agents Actions Suppl. 1988, 26, 181–187. [Google Scholar]
- Goldstein, J.L.; Brown, M.S. Familial hypercholesterolemia: Identification of a defect in the regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity associated with overproduction of cholesterol. Proc. Natl. Acad. Sci. USA 1973, 70, 2804–2808. [Google Scholar] [CrossRef] [PubMed]
- Hoeg, J.M.; Edge, S.B.; Demosky, S.J., Jr.; Starzl, T.E.; Triche, T.; Gregg, R.E.; Brewer, H.B., Jr. Metabolism of low-density lipoproteins by cultured hepatocytes from normal and homozygous familial hypercholesterolemic subjects. Biochim. Et Biophys. Acta 1986, 876, 646–657. [Google Scholar] [CrossRef]
- Rinninger, F.; Pittman, R.C. Regulation of the selective uptake of high density lipoprotein-associated cholesteryl esters by human fibroblasts and Hep G2 hepatoma cells. J. Lipid Res. 1988, 29, 1179–1194. [Google Scholar] [CrossRef] [PubMed]
- Heeren, J.; Beisiegel, U.; Grewal, T. Apolipoprotein E recycling: Implications for dyslipidemia and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 442–448. [Google Scholar] [CrossRef]
- Pittman, R.C.; Knecht, T.P.; Rosenbaum, M.S.; Taylor, C.A., Jr. A nonendocytotic mechanism for the selective uptake of high density lipoprotein-associated cholesterol esters. J. Biol. Chem. 1987, 262, 2443–2450. [Google Scholar] [CrossRef] [PubMed]
- Rohrl, C.; Stangl, H. HDL endocytosis and resecretion. Biochim. Biophys. Acta 2013, 1831, 1626–1633. [Google Scholar] [CrossRef]
- Cruz, P.M.; Mo, H.; McConathy, W.J.; Sabnis, N.; Lacko, A.G. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: A review of scientific findings, relevant to future cancer therapeutics. Front. Pharmacol. 2013, 4, 119. [Google Scholar] [CrossRef]
- Blidisel, A.; Marcovici, I.; Coricovac, D.; Hut, F.; Dehelean, C.A.; Cretu, O.M. Experimental Models of Hepatocellular Carcinoma-A Preclinical Perspective. Cancers 2021, 13, 3651. [Google Scholar] [CrossRef]
- Heeren, J.; Grewal, T.; Laatsch, A.; Rottke, D.; Rinninger, F.; Enrich, C.; Beisiegel, U. Recycling of apoprotein E is associated with cholesterol efflux and high density lipoprotein internalization. J. Biol. Chem. 2003, 278, 14370–14378. [Google Scholar] [CrossRef]
- Meex, S.J.; Andreo, U.; Sparks, J.D.; Fisher, E.A. Huh-7 or HepG2 cells: Which is the better model for studying human apolipoprotein-B100 assembly and secretion? J. Lipid Res. 2011, 52, 152–158. [Google Scholar] [CrossRef]
- Lalanne, F.; Lambert, G.; Amar, M.J.; Chetiveaux, M.; Zair, Y.; Jarnoux, A.L.; Ouguerram, K.; Friburg, J.; Seidah, N.G.; Brewer, H.B., Jr.; et al. Wild-type PCSK9 inhibits LDL clearance but does not affect apoB-containing lipoprotein production in mouse and cultured cells. J. Lipid Res. 2005, 46, 1312–1319. [Google Scholar] [CrossRef] [PubMed]
- Rhainds, D.; Brodeur, M.; Lapointe, J.; Charpentier, D.; Falstrault, L.; Brissette, L. The role of human and mouse hepatic scavenger receptor class B type I (SR-BI) in the selective uptake of low-density lipoprotein-cholesteryl esters. Biochemistry 2003, 42, 7527–7538. [Google Scholar] [CrossRef]
- Rohrl, C.; Meisslitzer-Ruppitsch, C.; Bittman, R.; Li, Z.; Pabst, G.; Prassl, R.; Strobl, W.; Neumuller, J.; Ellinger, A.; Pavelka, M.; et al. Combined light and electron microscopy using diaminobenzidine photooxidation to monitor trafficking of lipids derived from lipoprotein particles. Curr. Pharm. Biotechnol. 2012, 13, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Rohrl, C.; Pagler, T.A.; Strobl, W.; Ellinger, A.; Neumuller, J.; Pavelka, M.; Stangl, H.; Meisslitzer-Ruppitsch, C. Characterization of endocytic compartments after holo-high density lipoprotein particle uptake in HepG2 cells. Histochem. Cell Biol. 2010, 133, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Rohrl, C.; Eigner, K.; Fruhwurth, S.; Stangl, H. Bile acids reduce endocytosis of high-density lipoprotein (HDL) in HepG2 cells. PLoS ONE 2014, 9, e102026. [Google Scholar] [CrossRef]
- Gerets, H.H.J.; Tilmant, K.; Gerin, B.; Chanteux, H.; Depelchin, B.O.; Dhalluin, S.; Atienzar, F.A. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biol. Toxicol. 2012, 28, 69–87. [Google Scholar] [CrossRef]
- Zhou, B.; Ho, S.S.; Greer, S.U.; Spies, N.; Bell, J.M.; Zhang, X.; Zhu, X.; Arthur, J.G.; Byeon, S.; Pattni, R.; et al. Haplotype-resolved and integrated genome analysis of the cancer cell line HepG2. Nucleic Acids Res. 2019, 47, 3846–3861. [Google Scholar] [CrossRef]
- Thrift, R.N.; Forte, T.M.; Cahoon, B.E.; Shore, V.G. Characterization of lipoproteins produced by the human liver cell line, Hep G2, under defined conditions. J. Lipid Res. 1986, 27, 236–250. [Google Scholar] [CrossRef]
- Olsavsky, K.M.; Page, J.L.; Johnson, M.C.; Zarbl, H.; Strom, S.C.; Omiecinski, C.J. Gene expression profiling and differentiation assessment in primary human hepatocyte cultures, established hepatoma cell lines, and human liver tissues. Toxicol. Appl. Pharmacol. 2007, 222, 42–56. [Google Scholar] [CrossRef]
- Guo, L.; Dial, S.; Shi, L.; Branham, W.; Liu, J.; Fang, J.L.; Green, B.; Deng, H.; Kaput, J.; Ning, B. Similarities and differences in the expression of drug-metabolizing enzymes between human hepatic cell lines and primary human hepatocytes. Drug Metab. Dispos. 2011, 39, 528–538. [Google Scholar] [CrossRef]
- Kvist, A.J.; Kanebratt, K.P.; Walentinsson, A.; Palmgren, H.; O’Hara, M.; Björkbom, A.; Andersson, L.C.; Ahlqvist, M.; Andersson, T.B. Critical differences in drug metabolic properties of human hepatic cellular models, including primary human hepatocytes, stem cell derived hepatocytes, and hepatoma cell lines. Biochem. Pharmacol. 2018, 155, 124–140. [Google Scholar] [CrossRef] [PubMed]
- Graf, J.; Gautam, A.; Boyer, J.L. Isolated rat hepatocyte couplets: A primary secretory unit for electrophysiologic studies of bile secretory function. Proc. Natl. Acad. Sci. USA 1984, 81, 6516–6520. [Google Scholar] [CrossRef] [PubMed]
- Gautam, A.; Ng, O.C.; Boyer, J.L. Isolated rat hepatocyte couplets in short-term culture: Structural characteristics and plasma membrane reorganization. Hepatology 1987, 7, 216–223. [Google Scholar] [CrossRef]
- Rowe, C.; Gerrard, D.T.; Jenkins, R.; Berry, A.; Durkin, K.; Sundstrom, L.; Goldring, C.E.; Park, B.K.; Kitteringham, N.R.; Hanley, K.P.; et al. Proteome-wide analyses of human hepatocytes during differentiation and dedifferentiation. Hepatology 2013, 58, 799–809. [Google Scholar] [CrossRef]
- Ling, J.; Lewis, J.; Douglas, D.; Kneteman, N.M.; Vance, D.E. Characterization of lipid and lipoprotein metabolism in primary human hepatocytes. Biochim. Biophys. Acta 2013, 1831, 387–397. [Google Scholar] [CrossRef]
- Huggett, Z.J.; Smith, A.; De Vivo, N.; Gomez, D.; Jethwa, P.; Brameld, J.M.; Bennett, A.; Salter, A.M. A Comparison of Primary Human Hepatocytes and Hepatoma Cell Lines to Model the Effects of Fatty Acids, Fructose and Glucose on Liver Cell Lipid Accumulation. Nutrients 2023, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Gissen, P.; Arias, I.M. Structural and functional hepatocyte polarity and liver disease. J. Hepatol. 2015, 63, 1023–1037. [Google Scholar] [CrossRef]
- Geraud, C.; Schledzewski, K.; Demory, A.; Klein, D.; Kaus, M.; Peyre, F.; Sticht, C.; Evdokimov, K.; Lu, S.; Schmieder, A.; et al. Liver sinusoidal endothelium: A microenvironment-dependent differentiation program in rat including the novel junctional protein liver endothelial differentiation-associated protein-1. Hepatology 2010, 52, 313–326. [Google Scholar] [CrossRef]
- Si-Tayeb, K.; Idriss, S.; Champon, B.; Caillaud, A.; Pichelin, M.; Arnaud, L.; Lemarchand, P.; Le May, C.; Zibara, K.; Cariou, B. Urine-sample-derived human induced pluripotent stem cells as a model to study PCSK9-mediated autosomal dominant hypercholesterolemia. Dis. Model. Mech. 2016, 9, 81–90. [Google Scholar] [CrossRef]
- Cayo, M.A.; Cai, J.; DeLaForest, A.; Noto, F.K.; Nagaoka, M.; Clark, B.S.; Collery, R.F.; Si-Tayeb, K.; Duncan, S.A. JD induced pluripotent stem cell-derived hepatocytes faithfully recapitulate the pathophysiology of familial hypercholesterolemia. Hepatology 2012, 56, 2163–2171. [Google Scholar] [CrossRef]
- Caron, J.; Pene, V.; Tolosa, L.; Villaret, M.; Luce, E.; Fourrier, A.; Heslan, J.M.; Saheb, S.; Bruckert, E.; Gomez-Lechon, M.J.; et al. Low-density lipoprotein receptor-deficient hepatocytes differentiated from induced pluripotent stem cells allow familial hypercholesterolemia modeling, CRISPR/Cas-mediated genetic correction, and productive hepatitis C virus infection. Stem Cell Res. Ther. 2019, 10, 221. [Google Scholar] [CrossRef]
- Si-Tayeb, K.; Lemaigre, F.P.; Duncan, S.A. Organogenesis and development of the liver. Dev. Cell 2010, 18, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Koui, Y.; Kido, T.; Ito, T.; Oyama, H.; Chen, S.W.; Katou, Y.; Shirahige, K.; Miyajima, A. An In Vitro Human Liver Model by iPSC-Derived Parenchymal and Non-Parenchymal Cells. Stem Cell Rep. 2017, 9, 490–498. [Google Scholar] [CrossRef]
- Takayama, K.; Inamura, M.; Kawabata, K.; Sugawara, M.; Kikuchi, K.; Higuchi, M.; Nagamoto, Y.; Watanabe, H.; Tashiro, K.; Sakurai, F.; et al. Generation of metabolically functioning hepatocytes from human pluripotent stem cells by FOXA2 and HNF1alpha transduction. J. Hepatol. 2012, 57, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Feng, Y.; Qiu, D.; Xu, Y.; Pang, M.; Cai, N.; Xiang, A.P.; Zhang, Q. Highly efficient and expedited hepatic differentiation from human pluripotent stem cells by pure small-molecule cocktails. Stem Cell Res. Ther. 2018, 9, 58. [Google Scholar] [CrossRef] [PubMed]
- Nagamoto, Y.; Tashiro, K.; Takayama, K.; Ohashi, K.; Kawabata, K.; Sakurai, F.; Tachibana, M.; Hayakawa, T.; Furue, M.K.; Mizuguchi, H. The promotion of hepatic maturation of human pluripotent stem cells in 3D co-culture using type I collagen and Swiss 3T3 cell sheets. Biomaterials 2012, 33, 4526–4534. [Google Scholar] [CrossRef]
- Takayama, K.; Kawabata, K.; Nagamoto, Y.; Kishimoto, K.; Tashiro, K.; Sakurai, F.; Tachibana, M.; Kanda, K.; Hayakawa, T.; Furue, M.K.; et al. 3D spheroid culture of hESC/hiPSC-derived hepatocyte-like cells for drug toxicity testing. Biomaterials 2013, 34, 1781–1789. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Malinen, M.M.; Lauren, P.; Lou, Y.-R.; Kuisma, S.W.; Kanninen, L.; Lille, M.; Corlu, A.; GuGuen-Guillouzo, C.; Ikkala, O.; et al. Nanofibrillar cellulose hydrogel promotes three-dimensional liver cell culture. J. Control. Release 2012, 164, 291–298. [Google Scholar] [CrossRef]
- Jalil, S.; Keskinen, T.; Maldonado, R.; Sokka, J.; Trokovic, R.; Otonkoski, T.; Wartiovaara, K. Simultaneous high-efficiency base editing and reprogramming of patient fibroblasts. Stem Cell Rep. 2021, 16, 3064–3075. [Google Scholar] [CrossRef]
- Okada, H.; Nakanishi, C.; Yoshida, S.; Shimojima, M.; Yokawa, J.; Mori, M.; Tada, H.; Yoshimuta, T.; Hayashi, K.; Yamano, T.; et al. Function and Immunogenicity of Gene-corrected iPSC-derived Hepatocyte-Like Cells in Restoring Low Density Lipoprotein Uptake in Homozygous Familial Hypercholesterolemia. Sci. Rep. 2019, 9, 4695. [Google Scholar] [CrossRef]
- Smiriglia, A.; Lorito, N.; Bacci, M.; Subbiani, A.; Bonechi, F.; Comito, G.; Kowalik, M.A.; Perra, A.; Morandi, A. Estrogen-dependent activation of TRX2 reverses oxidative stress and metabolic dysfunction associated with steatotic disease. Cell Death Dis. 2025, 16, 57. [Google Scholar] [CrossRef] [PubMed]
- Tavian, D.; Missaglia, S.; Castagnetta, M.; Degiorgio, D.; Pennisi, E.M.; Coleman, R.A.; Dell’Era, P.; Mora, C.; Angelini, C.; Coviello, D.A. Generation of induced Pluripotent Stem Cells as disease modelling of NLSDM. Mol. Genet. Metab. 2017, 121, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Doueiry, C.; Kappler, C.S.; Martinez-Morant, C.; Duncan, S.A. A PNPLA3-Deficient iPSC-Derived Hepatocyte Screen Identifies Pathways to Potentially Reduce Steatosis in Metabolic Dysfunction-Associated Fatty Liver Disease. Int. J. Mol. Sci. 2024, 25, 7277. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.T.; Doueiry, C.; Jiang, Y.L.; Blaszkiewicz, J.; Lamprecht, M.P.; Heslop, J.A.; Peterson, Y.K.; Carten, J.D.; Traktman, P.; Yuan, Y.; et al. A human iPSC-derived hepatocyte screen identifies compounds that inhibit production of Apolipoprotein B. Commun. Biol. 2023, 6, 452. [Google Scholar] [CrossRef]
- Cayo, M.A.; Mallanna, S.K.; Di Furio, F.; Jing, R.; Tolliver, L.B.; Bures, M.; Urick, A.; Noto, F.K.; Pashos, E.E.; Greseth, M.D.; et al. A Drug Screen using Human iPSC-Derived Hepatocyte-like Cells Reveals Cardiac Glycosides as a Potential Treatment for Hypercholesterolemia. Cell Stem Cell 2017, 20, 478–489.e475. [Google Scholar] [CrossRef]
- Blaszkiewicz, J.; Duncan, S.A. Use of stem cell-derived hepatocytes to model liver disease. J. Hepatol. 2024, 80, 826–828. [Google Scholar] [CrossRef]
- Deguchi, S.; Takayama, K.; Mizuguchi, H. Generation of Human Induced Pluripotent Stem Cell-Derived Hepatocyte-Like Cells for Cellular Medicine. Biol. Pharm. Bull. 2020, 43, 608–615. [Google Scholar] [CrossRef]
- Si-Tayeb, K.; Noto, F.K.; Nagaoka, M.; Li, J.; Battle, M.A.; Duris, C.; North, P.E.; Dalton, S.; Duncan, S.A. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology 2010, 51, 297–305. [Google Scholar] [CrossRef]
- Si-Tayeb, K.; Noto, F.K.; Sepac, A.; Sedlic, F.; Bosnjak, Z.J.; Lough, J.W.; Duncan, S.A. Generation of human induced pluripotent stem cells by simple transient transfection of plasmid DNA encoding reprogramming factors. BMC Dev. Biol. 2010, 10, 81. [Google Scholar] [CrossRef]
- Zanoni, P.; Khetarpal, S.A.; Larach, D.B.; Hancock-Cerutti, W.F.; Millar, J.S.; Cuchel, M.; DerOhannessian, S.; Kontush, A.; Surendran, P.; Saleheen, D.; et al. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science 2016, 351, 1166–1171. [Google Scholar] [CrossRef]
- Ramakrishnan, V.M.; Yang, J.Y.; Tien, K.T.; McKinley, T.R.; Bocard, B.R.; Maijub, J.G.; Burchell, P.O.; Williams, S.K.; Morris, M.E.; Hoying, J.B.; et al. Restoration of Physiologically Responsive Low-Density Lipoprotein Receptor-Mediated Endocytosis in Genetically Deficient Induced Pluripotent Stem Cells. Sci. Rep. 2015, 5, 13231. [Google Scholar] [CrossRef] [PubMed]
- Kerr, A.G.; Tam, L.C.; Hale, A.B.; Cioroch, M.; Douglas, G.; Channon, K.M.; Wade-Martins, R. Episomal Nonviral Gene Therapy Vectors Slow Progression of Atherosclerosis in a Model of Familial Hypercholesterolemia. Mol. Ther. Nucleic Acids 2016, 5, e383. [Google Scholar] [CrossRef] [PubMed]
- Koui, Y.; Kido, T. Using human induced pluripotent stem cell-derived liver cells to investigate the mechanisms of liver fibrosis in vitro. Biochem. Soc. Trans. 2023, 51, 1271–1277. [Google Scholar] [CrossRef]
- Tian, S.P.; Ge, J.Y.; Song, Y.M.; Yu, X.Q.; Chen, W.H.; Chen, Y.Y.; Ye, D.; Zheng, Y.W. A novel efficient strategy to generate liver sinusoidal endothelial cells from human pluripotent stem cells. Sci. Rep. 2024, 14, 13831. [Google Scholar] [CrossRef]
- Tasnim, F.; Xing, J.; Huang, X.; Mo, S.; Wei, X.; Tan, M.H.; Yu, H. Generation of mature kupffer cells from human induced pluripotent stem cells. Biomaterials 2019, 192, 377–391. [Google Scholar] [CrossRef]
- Poulis, N.; Martin, M.; Hoerstrup, S.P.; Emmert, M.Y.; Fioretta, E.S. Development of an iPSC-derived tissue-resident macrophage-based platform for the in vitro immunocompatibility assessment of human tissue engineered matrices. Sci. Rep. 2024, 14, 12171. [Google Scholar] [CrossRef] [PubMed]
- Gage, B.K.; Liu, J.C.; Innes, B.T.; MacParland, S.A.; McGilvray, I.D.; Bader, G.D.; Keller, G.M. Generation of Functional Liver Sinusoidal Endothelial Cells from Human Pluripotent Stem-Cell-Derived Venous Angioblasts. Cell Stem Cell 2020, 27, 254–269. [Google Scholar] [CrossRef]
- Edge, S.B.; Hoeg, J.M.; Triche, T.; Schneider, P.D.; Brewer, H.B., Jr. Cultured human hepatocytes. Evidence for metabolism of low density lipoproteins by a pathway independent of the classical low density lipoprotein receptor. J. Biol. Chem. 1986, 261, 3800–3806. [Google Scholar] [CrossRef]
- Ose, L.; Ose, T.; Norum, K.R.; Berg, T. Uptake and degradation of 125I-labelled high density lipoproteins in rat liver cells in vivo and in vitro. Biochim. Biophys. Acta Lipids Lipid Metab. 1979, 574, 521–536. [Google Scholar] [CrossRef]
- Silver, D.L.; Wang, N.; Tall, A.R. Defective HDL particle uptake in ob/ob hepatocytes causes decreased recycling, degradation, and selective lipid uptake. J. Clin. Investig. 2000, 105, 151–159. [Google Scholar] [CrossRef]
- Mee, C.J.; Harris, H.J.; Farquhar, M.J.; Wilson, G.; Reynolds, G.; Davis, C.; van IJzendoorn, I.S.C.; Balfe, P.; McKeating, J.A. Polarization restricts hepatitis C virus entry into HepG2 hepatoma cells. J. Virol. 2009, 83, 6211–6221. [Google Scholar] [CrossRef]
- Ramos, M.J.; Bandiera, L.; Menolascina, F.; Fallowfield, J.A. In vitro models for non-alcoholic fatty liver disease: Emerging platforms and their applications. iScience 2022, 25, 103549. [Google Scholar] [CrossRef] [PubMed]
- Borén, J.; Wettesten, M.; Sjöberg, A.; Thorlin, T.; Bondjers, G.; Wiklund, O.; Olofsson, S.O. The assembly and secretion of apoB 100 containing lipoproteins in Hep G2 cells. Evidence for different sites for protein synthesis and lipoprotein assembly. J. Biol. Chem. 1990, 265, 10556–10564. [Google Scholar] [CrossRef]
- Ellsworth, J.L.; Erickson, S.K.; Cooper, A.D. Very low and low density lipoprotein synthesis and secretion by the human hepatoma cell line Hep-G2: Effects of free fatty acid. J. Lipid Res. 1986, 27, 858–874. [Google Scholar] [CrossRef]
- Dashti, N.; Wolfbauer, G. Secretion of lipids, apolipoproteins, and lipoproteins by human hepatoma cell line, HepG2: Effects of oleic acid and insulin. J. Lipid Res. 1987, 28, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, G.F.; Khurana, R.; Odwell, A.; Seelaender, M.C. Lipid balance in HepG2 cells: Active synthesis and impaired mobilization. J. Lipid Res. 1994, 35, 1801–1808. [Google Scholar] [CrossRef] [PubMed]
- Hugo-Wissemann, D.; Anundi, I.; Lauchart, W.; Viebahn, R.; de Groot, H. Differences in glycolytic capacity and hypoxia tolerance between hepatoma cells and hepatocytes. Hepatology 1991, 13, 297–303. [Google Scholar] [CrossRef]
- Javitt, N.B. Hep G2 cells as a resource for metabolic studies: Lipoprotein, cholesterol, and bile acids. Faseb J 1990, 4, 161–168. [Google Scholar] [CrossRef]
- Mavri-Damelin, D.; Damelin, L.H.; Eaton, S.; Rees, M.; Selden, C.; Hodgson, H.J.F. Cells for bioartificial liver devices: The human hepatoma-derived cell line C3A produces urea but does not detoxify ammonia. Biotechnol. Bioeng. 2008, 99, 644–651. [Google Scholar] [CrossRef]
- Iyer, V.V.; Yang, H.; Ierapetritou, M.G.; Roth, C.M. Effects of glucose and insulin on HepG2-C3A cell metabolism. Biotechnol. Bioeng. 2010, 107, 347–356. [Google Scholar] [CrossRef]
- Larsen, L.E.; Smith, M.A.; Abbey, D.; Korn, A.; Reeskamp, L.F.; Hand, N.J.; Holleboom, A.G. Hepatocyte-like cells derived from induced pluripotent stem cells: A versatile tool to understand lipid disorders. Atherosclerosis 2020, 303, 8–14. [Google Scholar] [CrossRef]
- Robinson, J.F.; Hamilton, E.G.; Lam, J.; Chen, H.; Woodruff, T.J. Differences in cytochrome p450 enzyme expression and activity in fetal and adult tissues. Placenta 2020, 100, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Kido, T.; Koui, Y.; Suzuki, K.; Kobayashi, A.; Miura, Y.; Chern, E.Y.; Tanaka, M.; Miyajima, A. CPM Is a Useful Cell Surface Marker to Isolate Expandable Bi-Potential Liver Progenitor Cells Derived from Human iPS Cells. Stem Cell Rep. 2015, 5, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Zhang, W.; Safarikia, S.; Isidan, A.; Chen, A.M.; Li, P.; Francis, H.; Kennedy, L.; Baiocchi, L.; Alvaro, D.; et al. Organoids and Spheroids as Models for Studying Cholestatic Liver Injury and Cholangiocarcinoma. Hepatology 2021, 74, 491–502. [Google Scholar] [CrossRef]
- Guan, Y.; Xu, D.; Garfin, P.M.; Ehmer, U.; Hurwitz, M.; Enns, G.; Michie, S.; Wu, M.; Zheng, M.; Nishimura, T.; et al. Human hepatic organoids for the analysis of human genetic diseases. JCI Insight 2017, 2, e94954. [Google Scholar] [CrossRef]
- de Graaf, I.A.; Olinga, P.; de Jager, M.H.; Merema, M.T.; de Kanter, R.; van de Kerkhof, E.G.; Groothuis, G.M. Preparation and incubation of precision-cut liver and intestinal slices for application in drug metabolism and toxicity studies. Nat. Protoc. 2010, 5, 1540–1551. [Google Scholar] [CrossRef] [PubMed]
- van Swelm, R.P.; Hadi, M.; Laarakkers, C.M.; Masereeuw, R.; Groothuis, G.M.; Russel, F.G. Proteomic profiling in incubation medium of mouse, rat and human precision-cut liver slices for biomarker detection regarding acute drug-induced liver injury. J. Appl. Toxicol. 2014, 34, 993–1001. [Google Scholar] [CrossRef]
- Konsue, N.; Ioannides, C. Modulation of carcinogen-metabolising cytochromes P450 in human liver by the chemopreventive phytochemical phenethyl isothiocyanate, a constituent of cruciferous vegetables. Toxicology 2010, 268, 184–190. [Google Scholar] [CrossRef]
- van de Bovenkamp, M.; Groothuis, G.M.; Meijer, D.K.; Olinga, P. Liver slices as a model to study fibrogenesis and test the effects of anti-fibrotic drugs on fibrogenic cells in human liver. Toxicol Vitr. 2008, 22, 771–778. [Google Scholar] [CrossRef]
- Wang, Y.; Leaker, B.; Qiao, G.; Sojoodi, M.; Eissa, I.R.; Epstein, E.T.; Eddy, J.; Dimowo, O.; Lauer, G.M.; Qadan, M.; et al. Precision-cut liver slices as an ex vivo model to evaluate antifibrotic therapies for liver fibrosis and cirrhosis. Hepatol. Commun. 2024, 8, e0558. [Google Scholar] [CrossRef]
- Palma, E.; Doornebal, E.J.; Chokshi, S. Precision-cut liver slices: A versatile tool to advance liver research. Hepatol. Int. 2019, 13, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Larsen, F.T.; van den Heuvel, M.C.; Gier, K.; Gorter, A.R.; Oosterhuis, D.; Bijzet, J.; de Meijer, V.E.; Ravnskjaer, K.; Nagelkerke, A.; et al. Metabolic Dysfunction-Associated Steatotic Liver Disease in a Dish: Human Precision-Cut Liver Slices as a Platform for Drug Screening and Interventions. Nutrients 2024, 16, 626. [Google Scholar] [CrossRef]
- Hammoutene, A.; Laouirem, S.; Albuquerque, M.; Colnot, N.; Brzustowski, A.; Valla, D.; Provost, N.; Delerive, P.; Paradis, V. A new NRF2 activator for the treatment of human metabolic dysfunction-associated fatty liver disease. JHEP Rep. 2023, 5, 100845. [Google Scholar] [CrossRef]
- van Midwoud, P.M.; Merema, M.T.; Verpoorte, E.; Groothuis, G.M. Microfluidics enables small-scale tissue-based drug metabolism studies with scarce human tissue. J. Lab. Autom. 2011, 16, 468–476. [Google Scholar] [CrossRef]
- Vickers, A.E.; Saulnier, M.; Cruz, E.; Merema, M.T.; Rose, K.; Bentley, P.; Olinga, P. Organ slice viability extended for pathway characterization: An in vitro model to investigate fibrosis. Toxicol. Sci. 2004, 82, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Vickers, A.E.; Fisher, R.; Olinga, P.; Dial, S. Repair pathways evident in human liver organ slices. Toxicol. Vitr. 2011, 25, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- Ijssennagger, N.; Janssen, A.W.F.; Milona, A.; Ramos Pittol, J.M.; Hollman, D.A.A.; Mokry, M.; Betzel, B.; Berends, F.J.; Janssen, I.M.; van Mil, S.W.C.; et al. Gene expression profiling in human precision cut liver slices in response to the FXR agonist obeticholic acid. J. Hepatol. 2016, 64, 1158–1166. [Google Scholar] [CrossRef]
- Mirzababaei, S.; Navaei-Nigjeh, M.; Abdollahi, M.; Shamloo, A. Chapter 7—Liver-on-a-chip. In Principles of Human Organs-on-Chips; Mozafari, M., Ed.; Woodhead Publishing: Sawston, UK, 2023; pp. 195–249. [Google Scholar]
- Suurmond, C.-A.E.; Lasli, S.; van den Dolder, F.W.; Ung, A.; Kim, H.-J.; Bandaru, P.; Lee, K.; Cho, H.-J.; Ahadian, S.; Ashammakhi, N.; et al. In Vitro Human Liver Model of Nonalcoholic Steatohepatitis by Coculturing Hepatocytes, Endothelial Cells, and Kupffer Cells. Adv. Healthc. Mater. 2019, 8, 1901379. [Google Scholar] [CrossRef]
- Lasli, S.; Kim, H.J.; Lee, K.; Suurmond, C.E.; Goudie, M.; Bandaru, P.; Sun, W.; Zhang, S.; Zhang, N.; Ahadian, S.; et al. A Human Liver-on-a-Chip Platform for Modeling Nonalcoholic Fatty Liver Disease. Adv. Biosyst. 2019, 3, e1900104. [Google Scholar] [CrossRef]
- Gori, M.; Simonelli, M.C.; Giannitelli, S.M.; Businaro, L.; Trombetta, M.; Rainer, A. Investigating Nonalcoholic Fatty Liver Disease in a Liver-on-a-Chip Microfluidic Device. PLoS ONE 2016, 11, e0159729. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Deng, R.; Hao Tong, W.; Huan, L.; Chan Way, N.; IslamBadhan, A.; Iliescu, C.; Yu, H. A perfusion incubator liver chip for 3D cell culture with application on chronic hepatotoxicity testing. Sci. Rep. 2017, 7, 14528. [Google Scholar] [CrossRef] [PubMed]
- Majer, J.; Alex, A.; Shi, J.; Chaney, E.J.; Mukherjee, P.; Spillman, D.R., Jr.; Marjanovic, M.; Newman, C.F.; Groseclose, R.M.; Watson, P.D.; et al. Multimodal imaging of a liver-on-a-chip model using labelled and label-free optical microscopy techniques. Lab. Chip 2024, 24, 4594–4608. [Google Scholar] [CrossRef]
- Sun, M.; Han, K.; Hu, R.; Liu, D.; Fu, W.; Liu, W. Advances in Micro/Nanoporous Membranes for Biomedical Engineering. Adv. Healthc. Mater. 2021, 10, 2001545. [Google Scholar] [CrossRef]
- Li, L.; Tan, D.; Liu, S.; Jiao, R.; Yang, X.; Li, F.; Wu, H.; Huang, W. Optimization of Factor Combinations for Stem Cell Differentiations on a Design-of-Experiment Microfluidic Chip. Anal. Chem. 2020, 92, 14228–14235. [Google Scholar] [CrossRef]
- Qiu, L.; Kong, B.; Kong, T.; Wang, H. Recent advances in liver-on-chips: Design, fabrication, and applications. Smart Med. 2023, 2, e20220010. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.C.; Hendriks, D.F.; Moro, S.M.; Ellis, E.; Walsh, J.; Renblom, A.; Fredriksson Puigvert, L.; Dankers, A.C.; Jacobs, F.; Snoeys, J.; et al. Characterization of primary human hepatocyte spheroids as a model system for drug-induced liver injury, liver function and disease. Sci. Rep. 2016, 6, 25187. [Google Scholar] [CrossRef]
- Kanebratt, K.P.; Janefeldt, A.; Vilen, L.; Vildhede, A.; Samuelsson, K.; Milton, L.; Bjorkbom, A.; Persson, M.; Leandersson, C.; Andersson, T.B.; et al. Primary Human Hepatocyte Spheroid Model as a 3D In Vitro Platform for Metabolism Studies. J. Pharm. Sci. 2021, 110, 422–431. [Google Scholar] [CrossRef]
- Tao, F.; Hanada, S.; Matsushima, K.; Arakawa, H.; Ishida, N.; Kato, Y.; Okimura, S.; Watanabe, T.; Kojima, N. Enhancement and maintenance of hepatic metabolic functions by controlling 3D aggregation of cryopreserved human iPS cell-derived hepatocyte-like cells. J. Biosci. Bioeng. 2023, 135, 134–142. [Google Scholar] [CrossRef]
- Prill, S.; Caddeo, A.; Baselli, G.; Jamialahmadi, O.; Dongiovanni, P.; Rametta, R.; Kanebratt, K.P.; Pujia, A.; Pingitore, P.; Mancina, R.M.; et al. The TM6SF2 E167K genetic variant induces lipid biosynthesis and reduces apolipoprotein B secretion in human hepatic 3D spheroids. Sci. Rep. 2019, 9, 11585. [Google Scholar] [CrossRef]
- Holmgren, G.; Ulfenborg, B.; Asplund, A.; Toet, K.; Andersson, C.X.; Hammarstedt, A.; Hanemaaijer, R.; Küppers-Munther, B.; Synnergren, J. Characterization of Human Induced Pluripotent Stem Cell-Derived Hepatocytes with Mature Features and Potential for Modeling Metabolic Diseases. Int. J. Mol. Sci. 2020, 21, 469. [Google Scholar] [CrossRef]
- Tao, F.; Mihara, H.; Kojima, N. Generation of Hepatic Tissue Structures Using Multicellular Spheroid Culture. In Hepatic Stem Cells: Methods and Protocols; Tanimizu, N., Ed.; Springer New York: New York, NY, USA, 2019; pp. 157–165. [Google Scholar]
- Pingitore, P.; Sasidharan, K.; Ekstrand, M.; Prill, S.; Lindén, D.; Romeo, S. Human Multilineage 3D Spheroids as a Model of Liver Steatosis and Fibrosis. Int. J. Mol. Sci. 2019, 20, 1629. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, Y.; Hu, P.; Zhang, Y.; Liu, Y.; Yang, Q.; Xu, L.; Gong, Z.; Yang, J.; Sun, W.; et al. Impact of enniatins and beauvericin on lipid metabolism: Insights from a 3D HepaRG spheroid model. Environ. Int. 2024, 191, 108969. [Google Scholar] [CrossRef] [PubMed]
- Kurano, M.; Iso, O.N.; Hara, M.; Ishizaka, N.; Moriya, K.; Koike, K.; Tsukamoto, K. LXR agonist increases apoE secretion from HepG2 spheroid, together with an increased production of VLDL and apoE-rich large HDL. Lipids Health Dis. 2011, 10, 134. [Google Scholar] [CrossRef]
- Tao, F.; Sayo, K.; Sugimoto, K.; Aoki, S.; Kojima, N. Development of a tunable method to generate various three-dimensional microstructures by replenishing macromolecules such as extracellular matrix components and polysaccharides. Sci. Rep. 2020, 10, 6567. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.J.; Bhandari, R.; Barrett, D.A.; Bennett, A.J.; Fry, J.R.; Powe, D.; Thomson, B.J.; Shakesheff, K.M. The effect of three-dimensional co-culture of hepatocytes and hepatic stellate cells on key hepatocyte functions in vitro. Cells Tissues Organs 2005, 181, 67–79. [Google Scholar] [CrossRef]
- Kozyra, M.; Johansson, I.; Nordling, A.; Ullah, S.; Lauschke, V.M.; Ingelman-Sundberg, M. Human hepatic 3D spheroids as a model for steatosis and insulin resistance. Sci. Rep. 2018, 8, 14297. [Google Scholar] [CrossRef]
- Fujisawa, K.; Takami, T.; Sasai, N.; Matsumoto, T.; Yamamoto, N.; Sakaida, I. Metabolic Alterations in Spheroid-Cultured Hepatic Stellate Cells. Int. J. Mol. Sci. 2020, 21, 3451. [Google Scholar] [CrossRef]
- Hurrell, T.; Kastrinou-Lampou, V.; Fardellas, A.; Hendriks, D.F.G.; Nordling, A.; Johansson, I.; Baze, A.; Parmentier, C.; Richert, L.; Ingelman-Sundberg, M. Human Liver Spheroids as a Model to Study Aetiology and Treatment of Hepatic Fibrosis. Cells 2020, 9, 964. [Google Scholar] [CrossRef]
- Pennisi, G.; Maurotti, S.; Ciociola, E.; Jamialahmadi, O.; Bertolazzi, G.; Mirarchi, A.; Bergh, P.O.; Scionti, F.; Mancina, R.M.; Spagnuolo, R.; et al. ANGPTL3 Downregulation Increases Intracellular Lipids by Reducing Energy Utilization. Arterioscler. Thromb. Vasc. Biol. 2024, 44, 1086–1097. [Google Scholar] [CrossRef]
- Harrison, S.P.; Siller, R.; Tanaka, Y.; Chollet, M.E.; de la Morena-Barrio, M.E.; Xiang, Y.; Patterson, B.; Andersen, E.; Bravo-Perez, C.; Kempf, H.; et al. Scalable production of tissue-like vascularized liver organoids from human PSCs. Exp. Mol. Med. 2023, 55, 2005–2024. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Wu, D.; Ren, Y.; Huang, Y.; Feng, B.; Zhao, N.; Zhang, T.; Chen, X.; Chen, S.; Xu, A. Generation of hepatobiliary organoids from human induced pluripotent stem cells. J. Hepatol. 2019, 70, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Takebe, T.; Sekine, K.; Kimura, M.; Yoshizawa, E.; Ayano, S.; Koido, M.; Funayama, S.; Nakanishi, N.; Hisai, T.; Kobayashi, T.; et al. Massive and Reproducible Production of Liver Buds Entirely from Human Pluripotent Stem Cells. Cell Rep. 2017, 21, 2661–2670. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, X.; Dowbaj, A.M.; Sljukic, A.; Bratlie, K.; Lin, L.; Fong, E.L.S.; Balachander, G.M.; Chen, Z.; Soragni, A.; et al. Organoids. Nat. Rev. Methods Primers 2022, 2, 94. [Google Scholar] [CrossRef] [PubMed]
- Kammerer, S. Three-Dimensional Liver Culture Systems to Maintain Primary Hepatic Properties for Toxicological Analysis In Vitro. Int. J. Mol. Sci. 2021, 22, 10214. [Google Scholar] [CrossRef]
- Huch, M.; Gehart, H.; van Boxtel, R.; Hamer, K.; Blokzijl, F.; Verstegen, M.M.A.; Ellis, E.; van Wenum, M.; Fuchs, S.A.; de Ligt, J.; et al. Long-Term Culture of Genome-Stable Bipotent Stem Cells from Adult Human Liver. Cell 2015, 160, 299–312. [Google Scholar] [CrossRef]
- De Crignis, E.; Hossain, T.; Romal, S.; Carofiglio, F.; Moulos, P.; Khalid, M.M.; Rao, S.; Bazrafshan, A.; Verstegen, M.M.; Pourfarzad, F.; et al. Application of human liver organoids as a patient-derived primary model for HBV infection and related hepatocellular carcinoma. Elife 2021, 10, e60747. [Google Scholar] [CrossRef]
- Hendriks, D.; Brouwers, J.F.; Hamer, K.; Geurts, M.H.; Luciana, L.; Massalini, S.; Lopez-Iglesias, C.; Peters, P.J.; Rodriguez-Colman, M.J.; Chuva de Sousa Lopes, S.; et al. Engineered human hepatocyte organoids enable CRISPR-based target discovery and drug screening for steatosis. Nat. Biotechnol. 2023, 41, 1567–1581. [Google Scholar] [CrossRef]
- Cho, H.J.; Kim, H.J.; Lee, K.; Lasli, S.; Ung, A.; Hoffman, T.; Nasiri, R.; Bandaru, P.; Ahadian, S.; Dokmeci, M.R.; et al. Bioengineered Multicellular Liver Microtissues for Modeling Advanced Hepatic Fibrosis Driven Through Non-Alcoholic Fatty Liver Disease. Small 2021, 17, e2007425. [Google Scholar] [CrossRef]
- Matsumoto, K.; Yoshitomi, H.; Rossant, J.; Zaret, K.S. Liver organogenesis promoted by endothelial cells prior to vascular function. Science 2001, 294, 559–563. [Google Scholar] [CrossRef]
- Schmidt, C.; Bladt, F.; Goedecke, S.; Brinkmann, V.; Zschiesche, W.; Sharpe, M.; Gherardi, E.; Birchmeier, C. Scatter factor/hepatocyte growth factor is essential for liver development. Nature 1995, 373, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Wang, N.; Zhang, J.; Yang, F.; Chen, Y.; Zhuang, Y.; Xu, Y.; Fang, J.; You, K.; Lin, X.; et al. Efficiently generate functional hepatic cells from human pluripotent stem cells by complete small-molecule strategy. Stem Cell Res. Ther. 2022, 13, 159. [Google Scholar] [CrossRef] [PubMed]
- Nahmias, Y.; Casali, M.; Barbe, L.; Berthiaume, F.; Yarmush, M.L. Liver endothelial cells promote LDL-R expression and the uptake of HCV-like particles in primary rat and human hepatocytes. Hepatology 2006, 43, 257–265. [Google Scholar] [CrossRef]
- Sych, T.; Schlegel, J.; Barriga, H.M.G.; Ojansivu, M.; Hanke, L.; Weber, F.; Beklem Bostancioglu, R.; Ezzat, K.; Stangl, H.; Plochberger, B.; et al. High-throughput measurement of the content and properties of nano-sized bioparticles with single-particle profiler. Nat. Biotechnol. 2024, 42, 587–590. [Google Scholar] [CrossRef]
- Wijers, M.; Zanoni, P.; Liv, N.; Vos, D.Y.; Jackstein, M.Y.; Smit, M.; Wilbrink, S.; Wolters, J.C.; van der Veen, Y.T.; Huijkman, N.; et al. The hepatic WASH complex is required for efficient plasma LDL and HDL cholesterol clearance. JCI Insight 2019, 4, e126462. [Google Scholar] [CrossRef]
- Xu, Y.; Gao, J.; Gong, Y.; Chen, M.; Chen, J.; Zhao, W.; Tan, S. Hsa-miR-140-5p down-regulates LDL receptor and attenuates LDL-C uptake in human hepatocytes. Atherosclerosis 2020, 297, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Gerold, G.; Moeller, R.; Pietschmann, T. Hepatitis C Virus Entry: Protein Interactions and Fusion Determinants Governing Productive Hepatocyte Invasion. Cold Spring Harb. Perspect. Med. 2020, 10, a036830. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Vallee, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Ouchi, R.; Togo, S.; Kimura, M.; Shinozawa, T.; Koido, M.; Koike, H.; Thompson, W.; Karns, R.A.; Mayhew, C.N.; McGrath, P.S.; et al. Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids. Cell Metab. 2019, 30, 374–384.e6. [Google Scholar] [CrossRef]
- Ramli, M.N.B.; Lim, Y.S.; Koe, C.T.; Demircioglu, D.; Tng, W.; Gonzales, K.A.U.; Tan, C.P.; Szczerbinska, I.; Liang, H.; Soe, E.L.; et al. Human Pluripotent Stem Cell-Derived Organoids as Models of Liver Disease. Gastroenterology 2020, 159, 1471–1486e.12. [Google Scholar] [CrossRef]
- Mun, S.J.; Ryu, J.S.; Lee, M.O.; Son, Y.S.; Oh, S.J.; Cho, H.S.; Son, M.Y.; Kim, D.S.; Kim, S.J.; Yoo, H.J.; et al. Generation of expandable human pluripotent stem cell-derived hepatocyte-like liver organoids. J. Hepatol. 2019, 71, 970–985. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
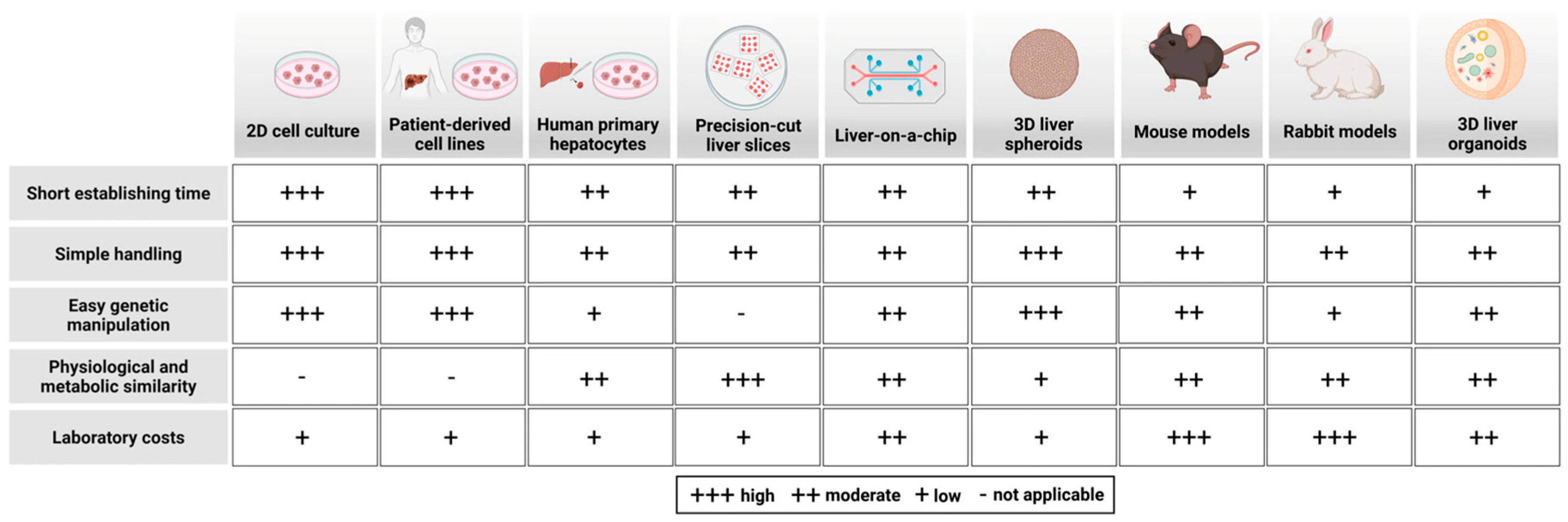
| Source | Achievements/Use | Advantages | Drawbacks | References |
|---|---|---|---|---|
| CHO cells | Analysis of cholesterol metabolism and LDL catabolism | Easy to handle | Not a valid model for liver metabolism | [86,115,116,117] |
| Fibroblasts/patient-derived fibroblasts | Detailed LDL uptake studies in normal and FH-derived fibroblasts | Reflect disease, easy to cultivate | Do not reflect all facets of hepatic metabolism | [12,79,118,119,120,121,122,123,124] |
| Hepatic cell lines/hepatoma cell lines (HepG2, HUH6, HUH7, HeparRG, Hep-3B) | Detailed studies on lipoprotein metabolism, e.g., ApoE recycling, ApoB assembly and secretion, LDLR-PCSK9 axis, SR-B1 function in selective cholesterol ester uptake, differences between LDL and HDL uptake | Non-limited proliferation, easy genetic manipulations, partial hepatocyte polarization, to some extent similar gene expression profile, can be used for a liver-on-a-chip model | Non-physiological, cancer origin, aneuploidy, low VLDL secretion, unphysiologically high SR-B1 levels, altered ApoB lipidation, low CYP450, preferential anaerobic glycolysis, altered bile acid and urea synthesis | [124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142] |
| Primary hepatocytes/patient-derived hepatocytes | Linear non-saturable LDL uptake (FH patients), alternative uptake routes (FH patients) | Fresh primary cultures, reflect lipoprotein disease and physiological models, can be used for a liver-on-a-chip model | Limited human donors, diseased donor tissue poorly reflects physiological lipid metabolism, loss of polarization upon culture, less efficient genetic manipulations | [117,119,123,143,144,145,146,147,148] |
| Hepatic non-parenchymal cells | Delineation of their role in the maintenance of hepatic homeostasis | Analysis of their role in lipoprotein metabolism, detailed analysis possible in combination with a chip and by co-culturing with PC cells | Polarization generally lost after isolation, lack of interaction with hepatocytes and lack of 3D microenvironment | [40,41,42,43,53,54,55,56,57,58,59,149] |
| iPSC-derived hepatocytes | Proof-of-principle studies, e.g., FH disease model, deciphering PCSK9 mutations and function | Circumvent liver donor shortage problem, very good liver disease models, generation directly from patients possible, recapitulate to high extent human hepatocyte metabolism, genetic manipulations possible, can be co-cultured with NPCs, can used for liver-on-a-chip | Varying degrees of hepatocyte-like cell phenotype (dependent on the method of generation), absence of 3D microenvironment, possible contamination with undifferentiated iPSC derivates | [150,151,152,153,154,155,156,157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173] |
| iPSC-derived non-parenchymal cells | Establishment of functional human liver model in vitro from LPCs, LSECs, and HSCs derived from hiPSCs | Analysis of cell-specific functions like vitamin A storage, CETP production, or fibrosis development possible; can be used in combination with other cell types and a chip to create a 3D microenvironment | Absent 3D microenvironment, possible contamination with undifferentiated iPSC derivates | [154,174,175,176,177,178] |
| Source | Achievements/Use | Advantages | Drawbacks | References |
|---|---|---|---|---|
| Precision-cut liver slices | Used to study liver injuries and hepatic drug metabolism, suitable for the identification of novel therapeutics | Intact intercellular and cell-matrix interactions, resembles the in vivo pathology, preserves structural and cellular composition of native liver | Limited access to freshly resected human tissue, slices are usually from diseased tissues, short lifespan, repair and regenerative response after slice preparation | [196,197,198,199,200,201,202,203,204,205,206,207,208] |
| Liver-on-a-chip | Valid model for the analysis of hepatic lipoprotein metabolism, used to investigate liver injuries and diseases, suitable for drug screenings | Recapitulates key structure and function of native liver, a variety of chips with different microstructures and-channels are available, a 3D environment can be created, easy administration of nutrients and drugs due to a microfluidic system | Very expensive, time-consuming, lacks applicable standard methods | [209,210,211,212,213,214,215,216,217] |
| Spheroids (PHHs; HLCs; immortalized cancer cell lines, e.g., HepG2 and NPCs) | Good models to study drug metabolism and metabolic changes | Improved liver functions due to 3D environment, easy generation, co-culturing possible | Ethical constraints, limited number of donors, genetic variability, cancer origin, not all liver cell types included | [195,218,219,220,221,222,223,224,225,226,227,228,229,230,231,232] |
| PSC-derived liver organoids (hESCs, hiPSCs) | Good models to study hepatic metabolism and liver diseases | Consist of more or all liver cell types, exhibit key liver functions (e.g., albumin secretion) and structure (e.g., bile ducts and vascular network), accessible to genetic manipulation, can be used for regenerative medicine, efficient, reproducible, no genetic variability | Long generation time (e.g., 20 days) | [103,233,234,235] |
| Tissue-derived hepatic organoids | Good models to study liver diseases and drug metabolism | Reflect disease, consist of (all) liver cell types | Ethical constraints, usually diseased tissues, limited human sample availability, genetic variability | [236,237,238,239,240] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiss, I.; Neuwert, N.; Oberle, R.; Hengstschläger, M.; Osmanagic-Myers, S.; Stangl, H. Hepatic Lipoprotein Metabolism: Current and Future In Vitro Cell-Based Systems. Biomolecules 2025, 15, 956. https://doi.org/10.3390/biom15070956
Kiss I, Neuwert N, Oberle R, Hengstschläger M, Osmanagic-Myers S, Stangl H. Hepatic Lipoprotein Metabolism: Current and Future In Vitro Cell-Based Systems. Biomolecules. 2025; 15(7):956. https://doi.org/10.3390/biom15070956
Chicago/Turabian StyleKiss, Izabella, Nicole Neuwert, Raimund Oberle, Markus Hengstschläger, Selma Osmanagic-Myers, and Herbert Stangl. 2025. "Hepatic Lipoprotein Metabolism: Current and Future In Vitro Cell-Based Systems" Biomolecules 15, no. 7: 956. https://doi.org/10.3390/biom15070956
APA StyleKiss, I., Neuwert, N., Oberle, R., Hengstschläger, M., Osmanagic-Myers, S., & Stangl, H. (2025). Hepatic Lipoprotein Metabolism: Current and Future In Vitro Cell-Based Systems. Biomolecules, 15(7), 956. https://doi.org/10.3390/biom15070956