
Uric Acid, the End-Product of Purine Metabolism, Mitigates Tau-Related Abnormalities: Comparison with DOT, a Non-Antibiotic Oxytetracycline Derivative
 , , , ,
, , , ,  , , ,
, , ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
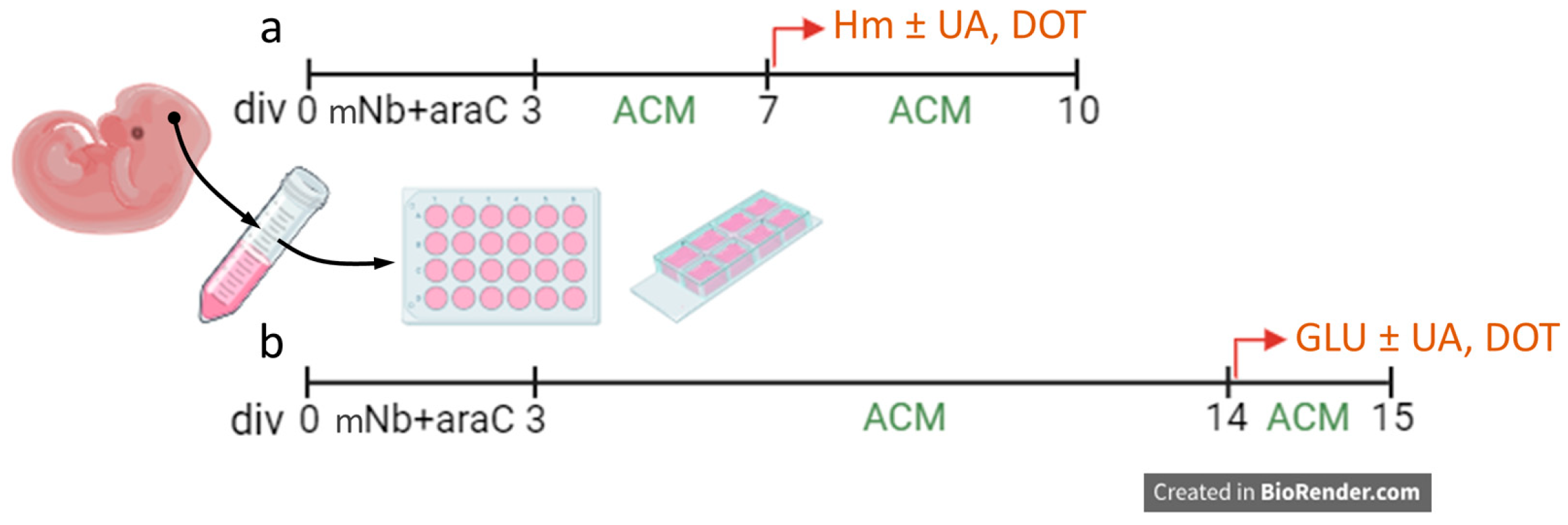
2.1. Use of Animals
2.2. Cortical Neuronal Cultures
2.3. The Non-Antibiotic TC Compound, DOT
2.3.1. Synthesis of DOT
2.3.2. Capacity of DOT to Penetrate the Brain
2.4. Uric Acid and Related Purine Derivatives
2.5. Other Pharmacological Reagents
2.6. Immunocytochemical Procedures
2.7. Cell Counting Procedures
2.8. Confocal Imaging
2.9. Assessment of ROS Emission and Changes in Mitochondrial Membrane Potential
2.10. Tau Aggregation Assay
2.11. Statistical Analysis
3. Results
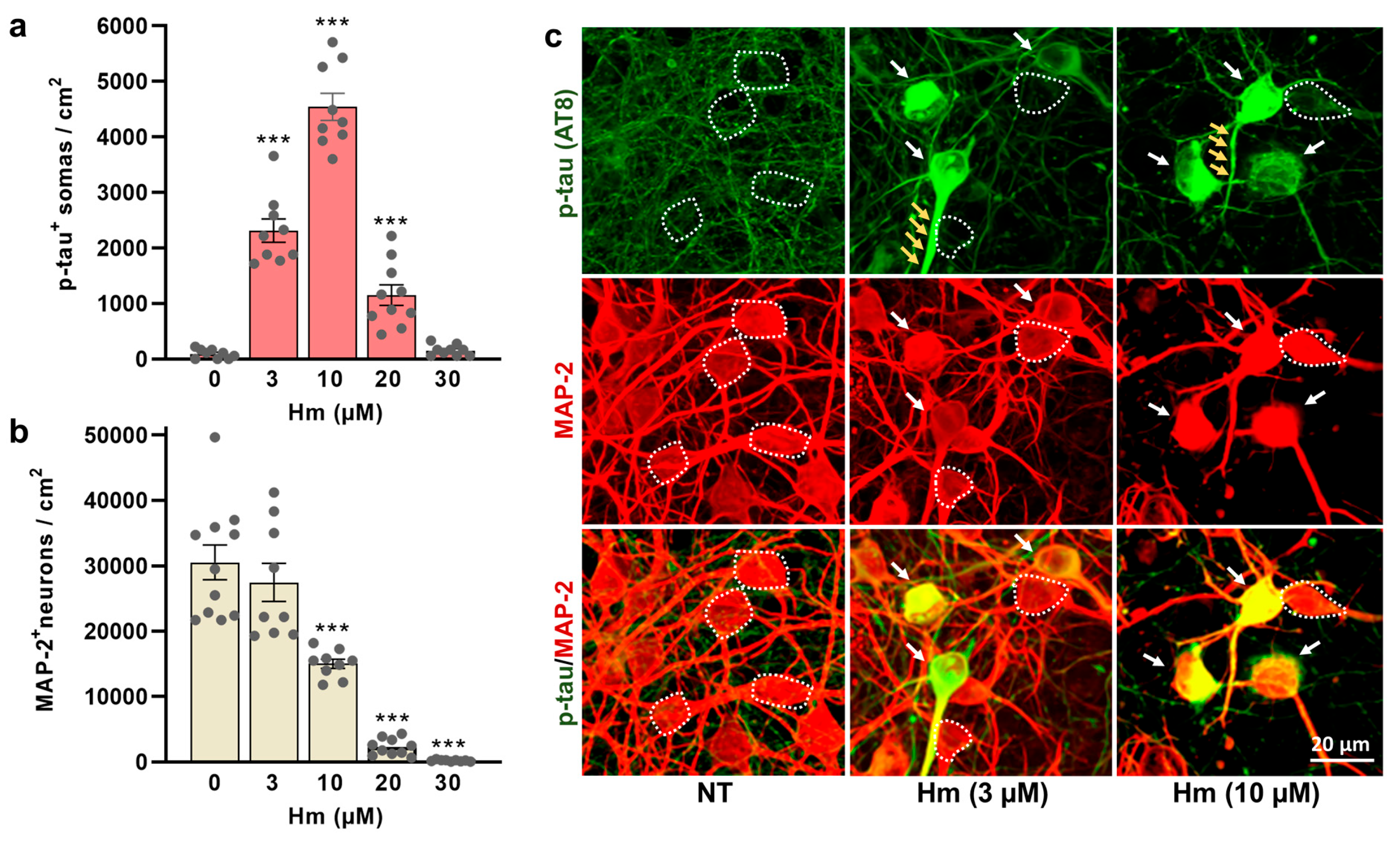
3.1. Induction of P-Tau Abnormalities by Hemin
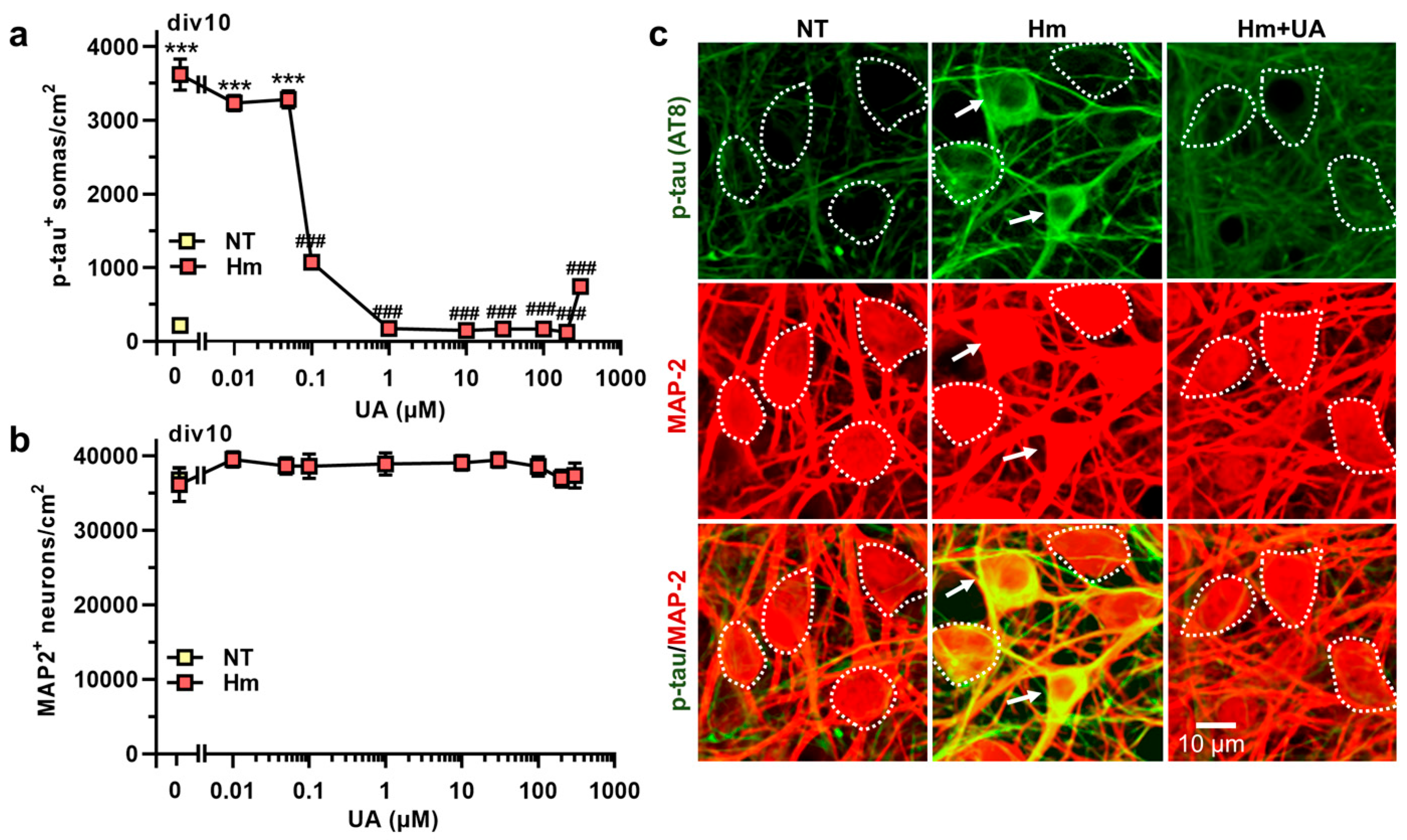
3.2. Hm-Induced P-Tau Abnormalities Are Curtailed by the Purine Metabolic End-Product UA
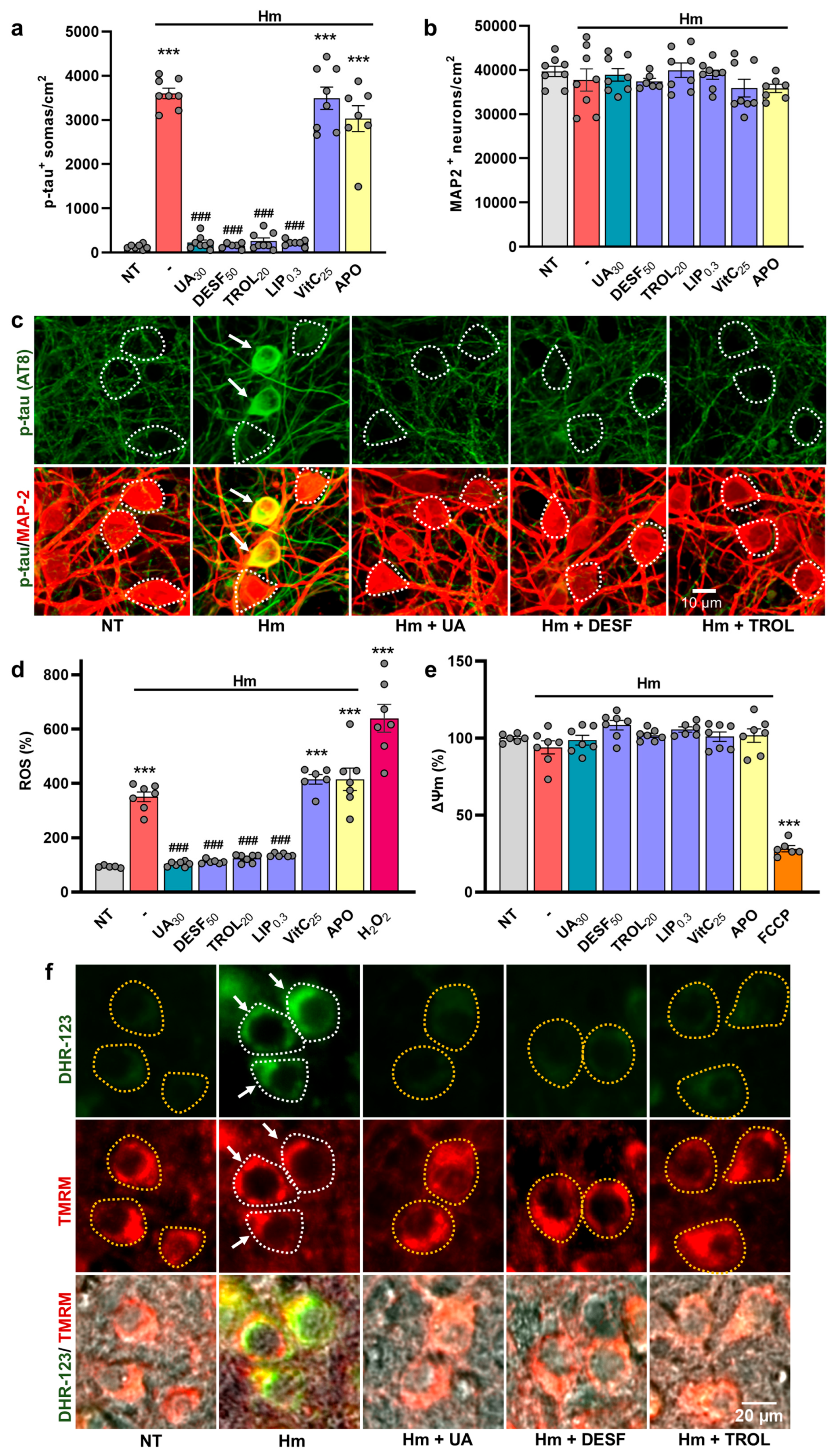
3.3. UA Exerts Anti-Tau Effects by Curtailing Hm-Mediated Oxidative Insults
3.4. The Suppression of P-Tau Build-Up by UA Is Reproduced by Compounds Inhibiting Iron-Mediated Lipid Peroxidation
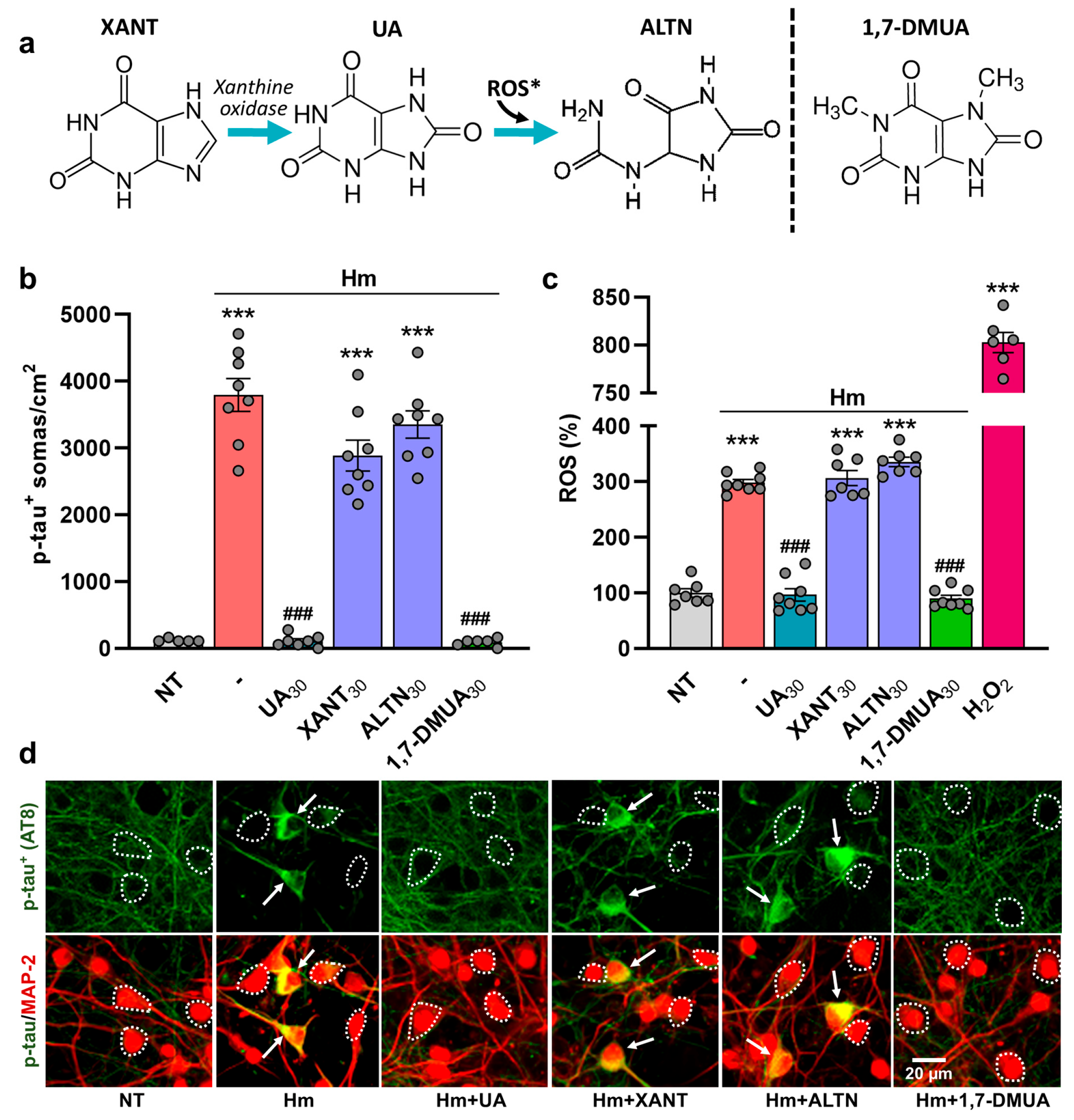
3.5. The Anti-Tau Effect of UA Is Not Reproduced Either by Its Immediate Precursor Xanthine or Its Oxidized Metabolite, Allantoin
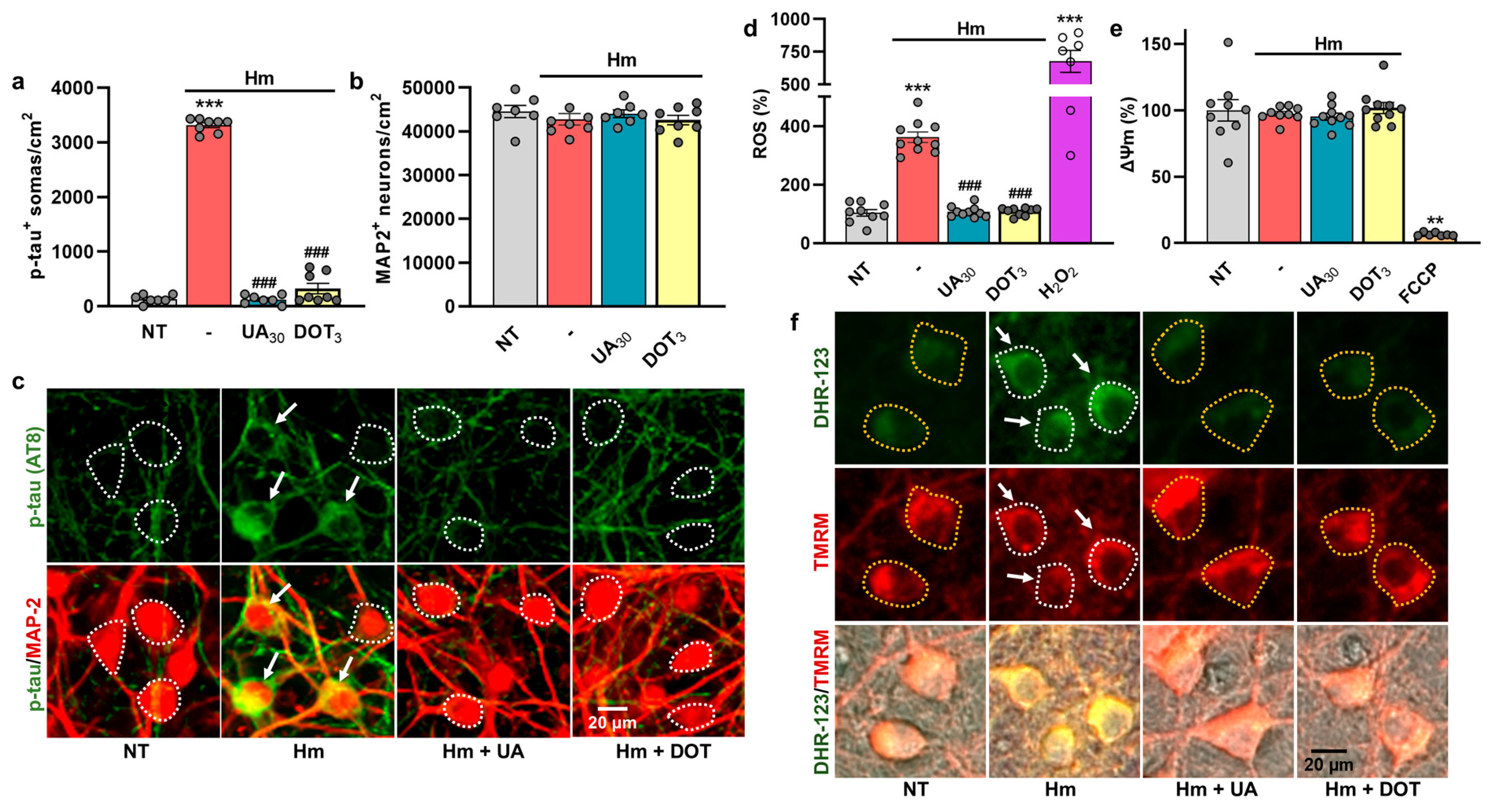
3.6. The Repressive Action of Uric Acid Against Hm-Induced P-Tau Accumulation Is Mimicked by DOT, a Non-Antibiotic Oxytetracycline Derivative
3.7. Glutamate-Mediated Tau Neurodegenerative Events Are Similarly Preventable by UA and DOT
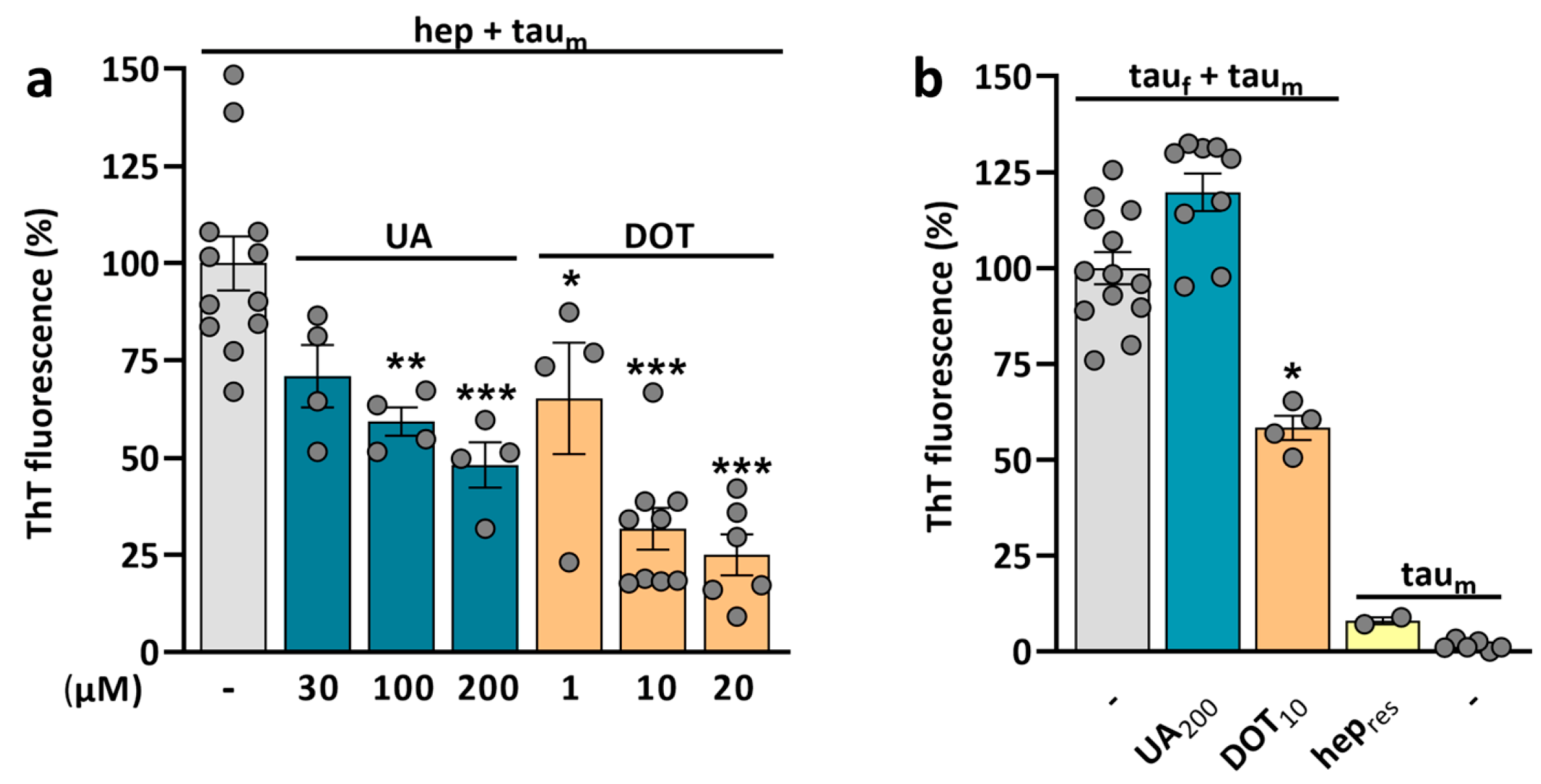
3.8. Potential of UA and DOT to Prevent Tau Amyloid Aggregation
4. Discussion
4.1. Hemin Promotes Pathological P-Tau Accumulation in Cultured Cortical Neurons
4.2. UA, the End Metabolic Product of Purines in Humans, Prevents Hm-Induced P-Tau Build-Up
4.3. UA Exerts Anti-Tau Effects by Curtailing Hm-Mediated Oxidative Stress
4.4. UA Exerts Anti-Tau Effects by Preventing Hm-Mediated Lipid Peroxidation
4.5. The Anti-Tau Effect of UA Is Not Reproduced by Either Its Immediate Precursor XANT or Its Oxidative Product ALTN
4.6. DOT, a Non-Antibiotic Oxytetracycline Derivative, Mimics the Anti-Tau Effects of UA
4.7. P-Tau Build-Up Induced by Glutamate Is Preventable by UA and DOT
4.8. UA and DOT Have the Potential to Restrain Tau Aggregation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ACM | astrocyte-conditioned medium |
| ALTN | allantoin |
| cNb | complete neurobasal medium |
| ΔΨm | mitochondrial membrane potential |
| DESF | desferioxamine |
| DHR-123 | dihydrorhodamine-123 |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| div | day(s) in vitro |
| DMUA | 1,7-dimethyluric acid |
| DOT | 12a-deoxy-dedimethylamino-oxytetracycline |
| FCCP | carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone |
| Hb | hemoglobin |
| Hm | hemin |
| LIP | Liproxstatin-1 |
| NFTs | neurofibrillary tangles |
| PBS | Dulbecco’s phosphate-buffered saline |
| Phaco | phase contrast |
| ROS | reactive oxygen species |
| tauf | tau fibrils |
| taum | tau monomers |
| TC | tetracycline |
| TMRM | tetramethylrhodamine methyl ester |
| TROL | trolox |
| UA | uric acid |
| XANT | xanthine |
References
- Braak, H.; Braak, E. Neuropathological Stageing of Alzheimer-Related Changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Goedert, M. Tau Pathology and Neurodegeneration. Lancet. Neurol. 2013, 12, 609–622. [Google Scholar] [CrossRef]
- Ganguly, J.; Jog, M. Tauopathy and Movement Disorders-Unveiling the Chameleons and Mimics. Front. Neurol. 2020, 11, 599384. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Gonzalez, M.I.; Pritchard, M.C.; May, P.C.; Toledo-Sherman, L.M.; Harris, G.A. The Therapeutic Landscape of Tauopathies: Challenges and Prospects. Alzheimer’s Res. Ther. 2023, 15, 168. [Google Scholar] [CrossRef]
- Lee, G.; Leugers, C.J. Tau and Tauopathies. Prog. Mol. Biol. Transl. Sci. 2012, 107, 263–293. [Google Scholar] [CrossRef]
- Chung, D.C.; Roemer, S.; Petrucelli, L.; Dickson, D.W. Cellular and Pathological Heterogeneity of Primary Tauopathies. Mol. Neurodegener. 2021, 16, 57. [Google Scholar] [CrossRef]
- Vaquer-Alicea, J.; Diamond, M.I.; Joachimiak, L.A. Tau Strains Shape Disease. Acta Neuropathol. 2021, 142, 57–71. [Google Scholar] [CrossRef]
- Tagge, C.A.; Fisher, A.M.; Minaeva, O.V.; Gaudreau-Balderrama, J.A.; Zhang, X.L.; Mark, W.; Wojnarowicz, N.C.; Lu, H.; Kokiko-Cochran, O.N.; Saman, S.; et al. Concussion, Microvascular Injury, and Early Tauopathy in Young Athletes after Impact Head Injury and an Impact Concussion Mouse Model. Brain 2018, 141, 422–458. [Google Scholar] [CrossRef] [PubMed]
- Cleret de Langavant, L.; Roze, E.; Petit, A.; Tressières, B.; Gharbi-Meliani, A.; Chaumont, H.; Michel, P.P.; Bachoud-Lévi, A.C.; Remy, P.; Edragas, R.; et al. Annonaceae Consumption Worsens Disease Severity and Cognitive Deficits in Degenerative Parkinsonism. Movement Disorders: Official Journal of the Movement Disorder Society. Mov. Disord. 2022, 37, 2355–2366. [Google Scholar] [CrossRef]
- González-Lizárraga, F.; Boluda, S.; Ruiz Hidalgo, J.; Avila, C.L.; Dos Santos, C.; Socias, B.; Medina, L.; Chaumont, H.; Akbar, D.; Roze, E.; et al. Protein coaggregation in Caribbean atypical parkinsonism: The contribution of annonacin. Neuropathol. Appl. Neurobiol. 2024, in press.
- Götz, J.; Probst, A.; Spillantini, M.G.; Schäfer, T.; Jakes, R.; Bürki, K.; Goedert, M. Somatodendritic Localization and Hyperphosphorylation of Tau Protein in Transgenic Mice Expressing the Longest Human Brain Tau Isoform. EMBO J. 1995, 14, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Yilmazer-Hanke, D.M. Pathogenesis of Alzheimer-Related Neuritic Plaques: AT8 Immunoreactive Dystrophic Neurites Precede Argyrophilic Neurites in Plaques of the Entorhinal Region, Hippocampal Formation, and Amygdala. Clin. Neuropathol. 1998, 17, 194–198. [Google Scholar] [PubMed]
- Uchihara, T.; Nakamura, A.; Yamazaki, M.; Mori, O. Evolution from Pretangle Neurons to Neurofibrillary Tangles Monitored by Thiazin Red Combined with Gallyas Method and Double Immunofluorescence. Acta Neuropathol. 2001, 101, 535–539. [Google Scholar] [CrossRef]
- Despres, C.; Byrne, C.; Qi, H.; Cantrelle, F.X.; Huvent, I.; Chambraud, B.; Baulieu, E.E.; Jacquot, Y.; Landrieu, I.; Lippens, G.; et al. Identification of the Tau Phosphorylation Pattern That Drives Its Aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 9080–9085. [Google Scholar] [CrossRef]
- Xia, Y.; Prokop, S.; Gorion, K.M.; Kim, J.D.; Sorrentino, Z.A.; Bell, B.M.; Manaois, A.N.; Chakrabarty, P.; Davies, P.; Giasson, B.I. Tau Ser208 Phosphorylation Promotes Aggregation and Reveals Neuropathologic Diversity in Alzheimer’s Disease and Other Tauopathies. Acta Neuropathol. Commun. 2020, 8, 88. [Google Scholar] [CrossRef] [PubMed]
- Malia, T.J.; Teplyakov, A.; Ernst, R.; Wu, S.J.; Lacy, E.R.; Liu, X.; Vandermeeren, M.; Mercken, M.; Luo, J.; Sweet, R.W.; et al. Epitope Mapping and Structural Basis for the Recognition of Phosphorylated Tau by the Anti-Tau Antibody AT8. Proteins 2016, 84, 427–434. [Google Scholar] [CrossRef]
- Goldstein, L.; Teng, Z.P.; Zeserson, E.; Patel, M.; Regan, R.F. Hemin Induces an Iron-Dependent, Oxidative Injury to Human Neuron-like Cells. J. Neurosci. Res. 2003, 73, 113–121. [Google Scholar] [CrossRef]
- Zille, M.; Oses-Prieto, J.A.; Savage, S.R.; Karuppagounder, S.S.; Chen, Y.; Kumar, A.; Morris, J.H.; Scheidt, K.A.; Burlingame, A.L.; Ratan, R.R. Hemin-Induced Death Models Hemorrhagic Stroke and Is a Variant of Classical Neuronal Ferroptosis. J. Neurosci. Off. J. Soc. Neurosci. 2022, 42, 2065–2079. [Google Scholar] [CrossRef] [PubMed]
- Schubert, D.; Piasecki, D. Oxidative Glutamate Toxicity Can Be a Component of the Excitotoxicity Cascade. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 7455–7462. [Google Scholar] [CrossRef]
- Spotorno, N.; Acosta-Cabronero, J.; Stomrud, E.; Lampinen, B.; Strandberg, O.T.; van Westen, D.; Hansson, O. Relationship between cortical iron and tau aggregation in Alzheimer’s disease. Brain A J. Neurol. 2020, 143, 1341–1349. [Google Scholar] [CrossRef]
- Yamamoto, A.; Shin, R.W.; Hasegawa, K.; Naiki, H.; Sato, H.; Yoshimasu, F.; Kitamoto, T. Iron (III) induces aggregation of hyperphosphorylated tau and its reduction to iron (II) reverses the aggregation: Implications in the formation of neurofibrillary tangles of Alzheimer’s disease. J. Neurochem. 2002, 82, 1137–1147. [Google Scholar] [CrossRef]
- Kazim, S.F.; Seo, J.H.; Bianchi, R.; Larson, C.S.; Sharma, A.; Wong, R.K.S.; Gorbachev, K.Y.; Pereira, A.C. Neuronal Network Excitability in Alzheimer’s Disease: The Puzzle of Similar versus Divergent Roles of Amyloid β and Tau. eNeuro 2021, 8, ENEURO.0418-20.2020. [Google Scholar] [CrossRef]
- Targa Dias Anastacio, H.; Matosin, N.; Ooi, L. Neuronal Hyperexcitability in Alzheimer’s Disease: What Are the Drivers behind This Aberrant Phenotype? Transl. Psychiatry 2022, 12, 257. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R.; Vanmechelen, E. Monoclonal Antibody AT8 Recognises Tau Protein Phosphorylated at Both Serine 202 and Threonine 205. Neurosci. Lett. 1995, 189, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.L.; Tsai, M.Y.; Aloe, S.; Bechberger, K.; König, S.; Morfini, G.; Brady, S.T. Defined Tau Phosphospecies Differentially Inhibit Fast Axonal Transport Through Activation of Two Independent Signaling Pathways. Front. Mol. Neurosci. 2021, 13, 610037. [Google Scholar] [CrossRef]
- Schirinzi, T.; Di Lazzaro, G.; Colona, V.L.; Imbriani, P.; Alwardat, M.; Sancesario, G.M.; Martorana, A.; Pisani, A. Assessment of Serum Uric Acid as Risk Factor for Tauopathies. J. Neural Transm. 2017, 124, 1105–1108. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhong, S.; Liang, Y.; Zhang, X.; Zhang, R.; Kang, K.; Qu, H.; Xu, Y.; Zhao, C.; Zhao, M. Serum Uric Acid and the Risk of Dementia: A Systematic Review and Meta-Analysis. Front. Aging Neurosci. 2021, 13, 625690. [Google Scholar] [CrossRef] [PubMed]
- Alves Fernandes, T.A.; Tourville, A.; Ciss, I.; Ribeiro Silva, R.; Andretto de Mattos, B.; Dos Santos Pereira, M.; Oblaza, M.; Brunel, J.M.; Ferrié, L.; Raisman-Vozari, R.; et al. Oxytetracycline and Its Non-Antibiotic Derivative DOT Protect Midbrain Dopamine Neurons from Iron-Driven Oxidative Damage. Neurotox. Res. 2025, 43, 16. [Google Scholar] [CrossRef]
- Medina, L.; González-Lizárraga, F.; Dominguez-Meijide, A.; Ploper, D.; Parrales, V.; Sequeira, S.; Cima-Omori, M.S.; Zweckstetter, M.; Del Bel, E.; Michel, P.P.; et al. Doxycycline Interferes with Tau Aggregation and Reduces Its Neuronal Toxicity. Front. Aging Neurosci. 2021, 13, 635760. [Google Scholar] [CrossRef]
- Montgomery, K.M.; Carroll, E.C.; Thwin, A.C.; Quddus, A.Y.; Hodges, P.; Southworth, D.R.; Gestwicki, J.E. Chemical Features of Polyanions Modulate Tau Aggregation and Conformational States. J. Am. Chem. Soc. 2023, 145, 3926–3936. [Google Scholar] [CrossRef]
- Tourville, A.; Akbar, D.; Corti, O.; Prehn, J.H.M.; Melki, R.; Hunot, S.; Michel, P.P. Modelling α-Synuclein Aggregation and Neurodegeneration with Fibril Seeds in Primary Cultures of Mouse Dopaminergic Neurons. Cells 2022, 11, 1640. [Google Scholar] [CrossRef] [PubMed]
- Ferreira Junior, N.C.; dos Santos Pereira, M.; Francis, N.; Ramirez, P.; Martorell, P.; González-Lizarraga, F.; Figadère, B.; Chehin, R.; Del Bel, E.; Raisman-Vozari, R.; et al. The Chemically-Modified Tetracycline COL-3 and Its Parent Compound Doxycycline Prevent Microglial Inflammatory Responses by Reducing Glucose-Mediated Oxidative Stress. Cells 2021, 10, 2163. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos Pereira, M.; Dias de Abreu, G.H.; Vanderlei, L.C.A.; Raisman-Vozari, R.; Guimarães, F.S.; Lu, H.C.; Michel, P.P.; Del Bel, E. 4′-Fluorocannabidiol Associated with Capsazepine Restrains L-DOPA-Induced Dyskinesia in Hemiparkinsonian Mice: Contribution of Anti-Inflammatory and Anti-Glutamatergic Mechanisms. Neuropharmacology 2024, 251, 109926. [Google Scholar] [CrossRef] [PubMed]
- Michel, P.P.; Ruberg, M.; Agid, Y. Rescue of Mesencephalic Dopamine Neurons by Anticancer Drug Cytosine Arabinoside. J. Neurochem. 1997, 69, 1499–1507. [Google Scholar] [CrossRef]
- Tourville, A.; Viguier, S.; González-Lizárraga, F.; Tomas-Grau, R.H.; Ramirez, P.; Brunel, J.M.; Dos Santos Pereira, M.; Del-Bel, E.; Chehin, R.; Ferrié, L.; et al. Rescue of Dopamine Neurons from Iron-Dependent Ferroptosis by Doxycycline and Demeclocycline and Their Non-Antibiotic Derivatives. Antioxidants 2023, 12, 575. [Google Scholar] [CrossRef]
- dos Santos Pereira, M.; Abreu, G.H.D.; Rocca, J.; Hamadat, S.; Raisman-Vozari, R.; Michel, P.P.; Del Bel, E. Contributive Role of TNF-α to L-DOPA-Induced Dyskinesia in a Unilateral 6-OHDA Lesion Model of Parkinson’s Disease. Front. Pharmacol. 2021, 11, 617085. [Google Scholar] [CrossRef]
- Golub, L.M.; Ramamurthy, N.S.; McNamara, T.F.; Greenwald, R.A.; Rifkin, B.R. Tetracyclines Inhibit Connective Tissue Breakdown: New Therapeutic Implications for an Old Family of Drugs. Crit. Rev. Oral Biol. Med. 1991, 2, 297–321. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Joshi, D.C.; Bakowska, J.C. Determination of Mitochondrial Membrane Potential and Reactive Oxygen Species in Live Rat Cortical Neurons. J. Vis. Exp. JoVE 2011, 2704. [Google Scholar] [CrossRef]
- Parrales-Macias, V.; Michel, P.P.; Tourville, A.; Raisman-Vozari, R.; Haïk, S.; Hunot, S.; Bizat, N.; Lannuzel, A. The Pesticide Chlordecone Promotes Parkinsonism-like Neurodegeneration with Tau Lesions in Midbrain Cultures and C. elegans Worms. Cells 2023, 12, 1336. [Google Scholar] [CrossRef]
- Barghorn, S.; Biernat, J.; Mandelkow, E. Purification of Recombinant Tau Protein and Preparation of Alzheimer-Paired Helical Filaments in Vitro. Methods Mol. Biol. 2005, 299, 35–51. [Google Scholar] [CrossRef] [PubMed]
- LeVine, H., 3rd. Quantification of Beta-Sheet Amyloid Fibril Structures with Thioflavin, T. Methods Enzymol. 1999, 309, 274–284. [Google Scholar] [CrossRef]
- Upchurch, C.M.; Yeudall, S.; Pavelec, C.M.; Merk, D.; Greulich, J.; Manjegowda, M.; Raghavan, S.S.; Bochkis, I.M.; Scott, M.M.; Perez-Reyes, E.; et al. Targeting Oxidized Phospholipids by AAV-Based Gene Therapy in Mice with Established Hepatic Steatosis Prevents Progression to Fibrosis. Sci. Adv. 2022, 8, Eabn0050. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Pandey, C.M.; Singh, U.; Gupta, A.; Sahu, C.; Keshri, A. Descriptive Statistics and Normality Tests for Statistical Data. Ann. Card. Anaesth. 2019, 22, 67–72. [Google Scholar] [CrossRef]
- Ames, B.N.; Cathcart, R.; Schwiers, E.; Hochstein, P. Uric Acid Provides an Antioxidant Defense in Humans against Oxidant- and Radical-Caused Aging and Cancer: A Hypothesis. Proc. Natl. Acad. Sci. USA 1981, 78, 6858–6862. [Google Scholar] [CrossRef]
- Guerreiro, S.; Ponceau, A.; Toulorge, D.; Martin, E.; Alvarez-Fischer, D.; Hirsch, E.C.; Michel, P.P. Protection of Midbrain Dopaminergic Neurons by the End-Product of Purine Metabolism Uric Acid: Potentiation by Low-Level Depolarization. J. Neurochem. 2009, 109, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Michel, P.P.; Vyas, S.; Agid, Y. Toxic Effects of Iron for Cultured Mesencephalic Dopaminergic Neurons Derived from Rat Embryonic Brains. J. Neurochem. 1992, 59, 118–127. [Google Scholar] [CrossRef]
- Kobayashi, S.; Tanaka, T.; Soeda, Y.; Almeida, O.F.X.; Takashima, A. Local Somatodendritic Translation and Hyperphosphorylation of Tau Protein Triggered by AMPA and NMDA Receptor Stimulation. EBioMedicine 2017, 20, 120–126. [Google Scholar] [CrossRef]
- Huynh, M.B.; Rebergue, N.; Merrick, H.; Gomez-Henao, W.; Jospin, E.; Biard, D.S.F.; Papy-Garcia, D. HS3ST2 Expression Induces the Cell Autonomous Aggregation of Tau. Sci. Rep. 2022, 12, 10850. [Google Scholar] [CrossRef]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the Pathologic Process in Alzheimer Disease: Age Categories from 1 to 100 Years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef]
- Attems, J.; Thal, D.R.; Jellinger, K.A. The Relationship between Subcortical Tau Pathology and Alzheimer’s Disease. Biochem. Soc. Trans. 2012, 40, 711–715. [Google Scholar] [CrossRef]
- Connor, J.R.; Snyder, B.S.; Beard, J.L.; Fine, R.E.; Mufson, E.J. Regional Distribution of Iron and Iron-Regulatory Proteins in the Brain in Aging and Alzheimer’s Disease. J. Neurosci. Res. 1992, 31, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Chen, K.M.; Ling, H.W.; Sun, F.; Li, X.; Wan, T.; Chai, W.M.; Zhang, H.; Zhan, Y.; Guan, Y.J. Correlation of Iron in the Hippocampus with MMSE in Patients with Alzheimer’s Disease. J. Magn. Reson. Imaging JMRI 2009, 29, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Wang, Y.; Rogers, J.T.; Wang, F. Perturbed Iron Distribution in Alzheimer’s Disease Serum, Cerebrospinal Fluid, and Selected Brain Regions: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. JAD 2014, 42, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Alosco, M.L.; Su, Y.; Stein, T.D.; Protas, H.; Cherry, J.D.; Adler, C.H.; Balcer, L.J.; Bernick, C.; Pulukuri, S.V.; Abdolmohammadi, B.; et al. Associations between near End-of-Life Flortaucipir PET and Postmortem CTE-Related Tau Neuropathology in Six Former American Football Players. Eur. J. Nucl. Med. Mol. Imaging 2023, 50, 435–452. [Google Scholar] [CrossRef]
- Paterno, G.; Bell, B.M.; Gorion, K.M.; Prokop, S.; Giasson, B.I. Reassessment of Neuronal Tau Distribution in Adult Human Brain and Implications for Tau Pathobiology. Acta Neuropathol. Commun. 2022, 10, 94. [Google Scholar] [CrossRef]
- Combs, B.; Mueller, R.L.; Morfini, G.; Brady, S.T.; Kanaan, N.M. Tau and Axonal Transport Misregulation in Tauopathies. Adv. Exp. Med. Biol. 2019, 1184, 81–95. [Google Scholar] [CrossRef]
- Wan, W.; Cao, L.; Kalionis, B.; Murthi, P.; Xia, S.; Guan, Y. Iron Deposition Leads to Hyperphosphorylation of Tau and Disruption of Insulin Signaling. Front. Neurol. 2019, 10, 607. [Google Scholar] [CrossRef]
- Fu, H.; Possenti, A.; Freer, R.; Nakano, Y.; Hernandez Villegas, N.C.; Tang, M.; Cauhy, P.V.; Lassus, B.A.; Chen, S.; Fowler, S.L.; et al. A Tau Homeostasis Signature Is Linked with the Cellular and Regional Vulnerability of Excitatory Neurons to Tau Pathology. Nat. Neurosci. 2019, 22, 47–56. [Google Scholar] [CrossRef]
- Carroll, T.; Guha, S.; Nehrke, K.; Johnson, G.V.W. Tau Post-Translational Modifications: Potentiators of Selective Vulnerability in Sporadic Alzheimer’s Disease. Biology 2021, 10, 1047. [Google Scholar] [CrossRef]
- Otero-Garcia, M.; Mahajani, S.U.; Wakhloo, D.; Tang, W.; Xue, Y.Q.; Morabito, S.; Pan, J.; Oberhauser, J.; Madira, A.E.; Shakouri, T.; et al. Molecular Signatures Underlying Neurofibrillary Tangle Susceptibility in Alzheimer’s Disease. Neuron 2022, 110, 2929–2948.e8. [Google Scholar] [CrossRef]
- Chiasseu, M.; Alarcon-Martinez, L.; Belforte, N.; Quintero, H.; Dotigny, F.; Destroismaisons, L.; Vande Velde, C.; Panayi, F.; Louis, C.; Di Polo, A. Tau Accumulation in the Retina Promotes Early Neuronal Dysfunction and Precedes Brain Pathology in a Mouse Model of Alzheimer’s Disease. Mol. Neurodegener. 2017, 12, 58. [Google Scholar] [CrossRef]
- Buée, L.; Troquier, L.; Burnouf, S.; Belarbi, K.; Van Der Jeugd, A.; Ahmed, T.; Fernandez-Gomez, F.; Caillierez, R.; Grosjean, M.E.; Begard, S.; et al. From Tau Phosphorylation to Tau Aggregation: What about Neuronal Death? Biochem. Soc. Trans. 2010, 38, 967–972. [Google Scholar] [CrossRef]
- Hediger, M.A.; Johnson, R.J.; Miyazaki, H.; Endou, H. Molecular Physiology of Urate Transport. Physiology 2005, 20, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.S.; Lee, W.W.; Ham, J.H.; Lee, J.J.; Lee, P.H.; Sohn, Y.H. Alzheimer’s Disease Neuroimaging Initiative Does Serum Uric Acid Act as a Modulator of Cerebrospinal Fluid Alzheimer’s Disease Biomarker Related Cognitive Decline? Eur. J. Neurol. 2016, 23, 948–957. [Google Scholar] [CrossRef]
- Cao, B.; Li, Q.; Xiong, L.; Ruan, H.; Lu, Y.; Peng, F.; Li, H.; Huang, Y.; Luo, M.; Shu, Y. Cerebrospinal Fluid Uric Acid Levels Associated with Disease Severity in Patients with Anti-N-Methyl-d-Aspartate Receptor Encephalitis. J. Neuroimmunol. 2023, 384, 578221. [Google Scholar] [CrossRef] [PubMed]
- Hasíková, L.; Závada, J.; Serranová, T.; Kozlík, P.; Kalíková, K.; Kotačková, L.; Trnka, J.; Zogala, D.; Šonka, K.; Růžička, E.; et al. Serum but Not Cerebrospinal Fluid Levels of Allantoin Are Increased in de Novo Parkinson’s Disease. NPJ Park. Dis. 2023, 9, 60. [Google Scholar] [CrossRef]
- Crawley, W.T.; Jungels, C.G.; Stenmark, K.R.; Fini, M.A. U-Shaped Association of Uric Acid to Overall-Cause Mortality and Its Impact on Clinical Management of Hyperuricemia. Redox Biol. 2022, 51, 102271. [Google Scholar] [CrossRef]
- Su, B.; Wang, X.; Lee, H.G.; Tabaton, M.; Perry, G.; Smith, M.A.; Zhu, X. Chronic Oxidative Stress Causes Increased Tau Phosphorylation in M17 Neuroblastoma Cells. Neurosci. Lett. 2010, 468, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Shao, C.; Zhang, L.; Siedlak, S.L.; Meabon, J.S.; Peskind, E.R.; Lu, Y.; Wang, W.; Perry, G.; Cook, D.G.; et al. Oxidative Stress Signaling in Blast TBI-Induced Tau Phosphorylation. Antioxidants 2021, 10, 955. [Google Scholar] [CrossRef]
- Zambrano, C.A.; Egaña, J.T.; Núñez, M.T.; Maccioni, R.B.; González-Billault, C. Oxidative Stress Promotes Tau Dephosphorylation in Neuronal Cells: The Roles of Cdk5 and PP1. Free Radic. Biol. Med. 2004, 36, 1393–1402. [Google Scholar] [CrossRef]
- Gulcin, I.; Oktay, M.; Koksal, E.; Serbetci, H.; Beydemir, S.; Kufrevioglu, O.I. Antioxidant and Radical Scavenging Activities of Uric Acid. Asian J. Chem. 2008, 20, 2079–2090. [Google Scholar]
- Bakshi, R.; Zhang, H.; Logan, R.; Joshi, I.; Xu, Y.; Chen, X.; Schwarzschild, M.A. Neuroprotective effects of urate are mediated by augmenting astrocytic glutathione synthesis and release. Neurobiol. Dis. 2015, 82, 574–579. [Google Scholar] [CrossRef]
- Escobar-Khondiker, M.; Höllerhage, M.; Muriel, M.P.; Champy, P.; Bach, A.; Depienne, C.; Respondek, G.; Yamada, E.S.; Lannuzel, A.; Yagi, T.; et al. Annonacin, a natural mitochondrial complex I inhibitor, causes tau pathology in cultured neurons. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 7827–7837. [Google Scholar] [CrossRef]
- Eckert, A.; Nisbet, R.; Grimm, A.; Götz, J. March Separate, Strike Together--Role of Phosphorylated TAU in Mitochondrial Dysfunction in Alzheimer’s Disease. Biochim. Et Biophys. Acta 2014, 1842, 1258–1266. [Google Scholar] [CrossRef]
- Samluk, L.; Ostapczuk, P.; Dziembowska, M. Long-Term Mitochondrial Stress Induces Early Steps of Tau Aggregation by Increasing Reactive Oxygen Species Levels and Affecting Cellular Proteostasis. Mol. Biol. Cell 2022, 33, Ar67. [Google Scholar] [CrossRef]
- Hershko, C.; Abrahamov, A.; Konijn, A.M.; Breuer, W.; Cabantchik, I.Z.; Pootrakul, P.; Link, G. Objectives and Methods of Iron Chelation Therapy. Bioinorg. Chem. Appl. 2003, 1, 151–168. [Google Scholar] [CrossRef]
- Zilka, O.; Shah, R.; Li, B.; Friedmann Angeli, J.P.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, Present and Future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Von Krusenstiern, A.N.; Robson, R.N.; Qian, N.; Qiu, B.; Hu, F.; Reznik, E.; Smith, N.; Zandkarimi, F.; Estes, V.M.; Dupont, M.; et al. Identification of Essential Sites of Lipid Peroxidation in Ferroptosis. Nat. Chem. Biol. 2023, 19, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, T.H.; Frezzatti, W.A., Jr.; Schreier, S. Hemin-Induced Lipid Membrane Disorder and Increased Permeability: A Molecular Model for the Mechanism of Cell Lysis. Arch. Biochem. Biophys. 1993, 307, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Balla, G.; Jacob, H.S.; Eaton, J.W.; Belcher, J.D.; Vercellotti, G.M. Hemin: A Possible Physiological Mediator of Low Density Lipoprotein Oxidation and Endothelial Injury. Arterioscler. Thromb. A J. Vasc. Biol. 1991, 11, 1700–1711. [Google Scholar] [CrossRef]
- Muraoka, S.; Miura, T. Inhibition by Uric Acid of Free Radicals That Damage Biological Molecules. Pharmacol. Toxicol. 2003, 93, 284–289. [Google Scholar] [CrossRef]
- Howell, R.R.; Wyngaarden, J.B. On the Mechanism of Peroxidation of Uric Acids by Hemoproteins. J. Biol. Chem. 1960, 235, 3544–3550. [Google Scholar] [CrossRef] [PubMed]
- Regoli, F.; Winston, G.W. Quantification of Total Oxidant Scavenging Capacity of Antioxidants for Peroxynitrite, Peroxyl Radicals, and Hydroxyl Radicals. Toxicol. Appl. Pharmacol. 1999, 156, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J.; Sevanian, A.; Muakkassah-Kelly, S.F.; Hochstein, P. Uric acid-iron ion complexes. A new aspect of the antioxidant functions of uric acid. Biochem. J. 1986, 235, 747–754. [Google Scholar] [CrossRef]
- Sarkar, A.R.; Pal, S.; Sarkar, A.K.; Jana, N.R. Hemin-Based Cell Therapy via Nanoparticle-Assisted Uptake, Intracellular Reactive Oxygen Species Generation and Autophagy Induction. New J. Chem. 2022, 46, 21746–21755. [Google Scholar] [CrossRef]
- Cahill, C.M.; Sarang, S.S.; Bakshi, R.; Xia, N.; Lahiri, D.K.; Rogers, J.T. Neuroprotective Strategies and Cell-Based Biomarkers for Manganese-Induced Toxicity in Human Neuroblastoma (SH-SY5Y) Cells. Biomolecules 2024, 14, 647. [Google Scholar] [CrossRef]
- Pandithavidana, D.R.; Jayawardana, S.B. Comparative Study of Antioxidant Potential of Selected Dietary Vitamins; Computational Insights. Molecules 2019, 24, 1646. [Google Scholar] [CrossRef]
- Pongratz, M.; Schluga, P.; Jakupec, M.A.; Arion, V.B.; Hartinger, C.G.; Allmaier, G.; Keppler, B.K. Transferrin Binding and Transferrin-Mediated Cellular Uptake of the Ruthenium Coordination Compound KP1019, Studied by Means of AAS, ESI-MS and CD Spectroscopy. J. Anal. At. Spectrom. 2004, 19, 46–51. [Google Scholar] [CrossRef]
- Maiuolo, J.; Oppedisano, F.; Gratteri, S.; Muscoli, C.; Mollace, V. Regulation of Uric Acid Metabolism and Excretion. Int. J. Cardiol. 2016, 213, 8–14. [Google Scholar] [CrossRef]
- Smith, R.C.; Reeves, J.C.; Dage, R.C.; Schnettler, R.A. Antioxidant Properties of 2-Imidazolones and 2-Imidazolthiones. Biochem. Pharmacol. 1987, 36, 1457–1460. [Google Scholar] [CrossRef]
- Tomas-Grau, R.; González-Lizárraga, F.; Ploper, D.; Avila, C.L.; Socías, S.B.; Besnault, P.; Tourville, A.; Mella, R.M.; Villacé, P.; Salado, C.; et al. Neuroprotective Effects of a Novel Demeclocycline Derivative Lacking Antibiotic Activity: From a Hit to a Promising Lead Compound. Cells 2022, 11, 2759. [Google Scholar] [CrossRef]
- Rose, C.; Tomas-Grau, R.H.; Zabala, B.; Michel, P.P.; Brunel, J.M.; Chehín, R.; Raisman-Vozari, R.; Ferrié, L.; Figadère, B. C9-Functionalized Doxycycline Analogs as Drug Candidates to Prevent Pathological α-Synuclein Aggregation and Neuroinflammation in Parkinson’s Disease Degeneration. ChemMedChem 2024, 19, E202300597. [Google Scholar] [CrossRef]
- Hwang, K.; Vaknalli, R.N.; Addo-Osafo, K.; Vicente, M.; Vossel, K. Tauopathy and Epilepsy Comorbidities and Underlying Mechanisms. Front. Aging Neurosci. 2022, 14, 903973. [Google Scholar] [CrossRef]
- Couratier, P.; Lesort, M.; Sindou, P.; Esclaire, F.; Yardin, C.; Hugon, J. Modifications of Neuronal Phosphorylated Tau Immunoreactivity Induced by NMDA Toxicity. Mol. Chem. Neuropathol. 1996, 27, 259–273. [Google Scholar] [CrossRef]
- Yu, Z.F.; Bruce-Keller, A.J.; Goodman, Y.; Mattson, M.P. Uric Acid Protects Neurons against Excitotoxic and Metabolic Insults in Cell Culture, and against Focal Ischemic Brain Injury in Vivo. J. Neurosci. Res. 1998, 53, 613–625. [Google Scholar] [CrossRef]
- Du, Y.; Chen, C.P.; Tseng, C.Y.; Eisenberg, Y.; Firestein, B.L. Astroglia-Mediated Effects of Uric Acid to Protect Spinal Cord Neurons from Glutamate Toxicity. Glia 2007, 55, 463–472. [Google Scholar] [CrossRef]
- Lu, Y.; Yang, Y.; Chen, W.; Du, N.; Du, Y.; Gu, H.; Liu, Q. Minocycline, but Not Doxycycline Attenuates NMDA-Induced [Ca2+]i and Excitotoxicity. NeuroReport 2021, 32, 38–43. [Google Scholar] [CrossRef]
- Brennan, A.M.; Suh, S.W.; Won, S.J.; Narasimhan, P.; Kauppinen, T.M.; Lee, H.; Edling, Y.; Chan, P.H.; Swanson, R.A. NADPH Oxidase Is the Primary Source of Superoxide Induced by NMDA Receptor Activation. Nat. Neurosci. 2009, 12, 857–863. [Google Scholar] [CrossRef]
- Lam, T.I.; Brennan-Minnella, A.M.; Won, S.J.; Shen, Y.; Hefner, C.; Shi, Y.; Sun, D.; Swanson, R.A. Intracellular pH Reduction Prevents Excitotoxic and Ischemic Neuronal Death by Inhibiting NADPH Oxidase. Proc. Natl. Acad. Sci. USA 2013, 110, E4362–E4368. [Google Scholar] [CrossRef] [PubMed]
- Lavaur, J.; Le Nogue, D.; Lemaire, M.; Pype, J.; Farjot, G.; Hirsch, E.C.; Michel, P.P. The Noble Gas Xenon Provides Protection and Trophic Stimulation to Midbrain Dopamine Neurons. J. Neurochem. 2017, 142, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Kampers, T.; Pangalos, M.; Geerts, H.; Wiech, H.; Mandelkow, E. Assembly of Paired Helical Filaments from Mouse Tau: Implications for the Neurofibrillary Pathology in Transgenic Mouse Models for Alzheimer’s Disease. FEBS Lett. 1999, 451, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R.; Spillantini, M.G.; Hasegawa, M.; Smith, M.J.; Crowther, R.A. Assembly of Microtubule-Associated Protein Tau into Alzheimer-like Filaments Induced by Sulphated Glycosaminoglycans. Nature 1996, 383, 550–553. [Google Scholar] [CrossRef]
- Finlayson, B.; Du Bois, L. Adsorption of Heparin on Sodium Acid Urate. Clin. Chim. Acta Int. J. Clin. Chem. 1978, 84, 203–206. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andretto de Mattos, B.; Tomas-Grau, R.H.; Alves Fernandes, T.A.; González-Lizárraga, F.; Tourville, A.; Ciss, I.; Brunel, J.-M.; Chehin, R.; Lannuzel, A.; Ferrié, L.; et al. Uric Acid, the End-Product of Purine Metabolism, Mitigates Tau-Related Abnormalities: Comparison with DOT, a Non-Antibiotic Oxytetracycline Derivative. Biomolecules 2025, 15, 941. https://doi.org/10.3390/biom15070941
Andretto de Mattos B, Tomas-Grau RH, Alves Fernandes TA, González-Lizárraga F, Tourville A, Ciss I, Brunel J-M, Chehin R, Lannuzel A, Ferrié L, et al. Uric Acid, the End-Product of Purine Metabolism, Mitigates Tau-Related Abnormalities: Comparison with DOT, a Non-Antibiotic Oxytetracycline Derivative. Biomolecules. 2025; 15(7):941. https://doi.org/10.3390/biom15070941
Chicago/Turabian StyleAndretto de Mattos, Bianca, Rodrigo Hernán Tomas-Grau, Thaís Antonia Alves Fernandes, Florencia González-Lizárraga, Aurore Tourville, Ismaila Ciss, Jean-Michel Brunel, Rosana Chehin, Annie Lannuzel, Laurent Ferrié, and et al. 2025. "Uric Acid, the End-Product of Purine Metabolism, Mitigates Tau-Related Abnormalities: Comparison with DOT, a Non-Antibiotic Oxytetracycline Derivative" Biomolecules 15, no. 7: 941. https://doi.org/10.3390/biom15070941
APA StyleAndretto de Mattos, B., Tomas-Grau, R. H., Alves Fernandes, T. A., González-Lizárraga, F., Tourville, A., Ciss, I., Brunel, J.-M., Chehin, R., Lannuzel, A., Ferrié, L., Raisman-Vozari, R., Figadère, B., Del Bel, E., & Michel, P. P. (2025). Uric Acid, the End-Product of Purine Metabolism, Mitigates Tau-Related Abnormalities: Comparison with DOT, a Non-Antibiotic Oxytetracycline Derivative. Biomolecules, 15(7), 941. https://doi.org/10.3390/biom15070941