
Overcoming Immune Barriers in Allogeneic CAR-NK Therapy: From Multiplex Gene Editing to AI-Driven Precision Design


Abstract
1. Introduction
2. CAR Design Considerations in NK Cell Therapy
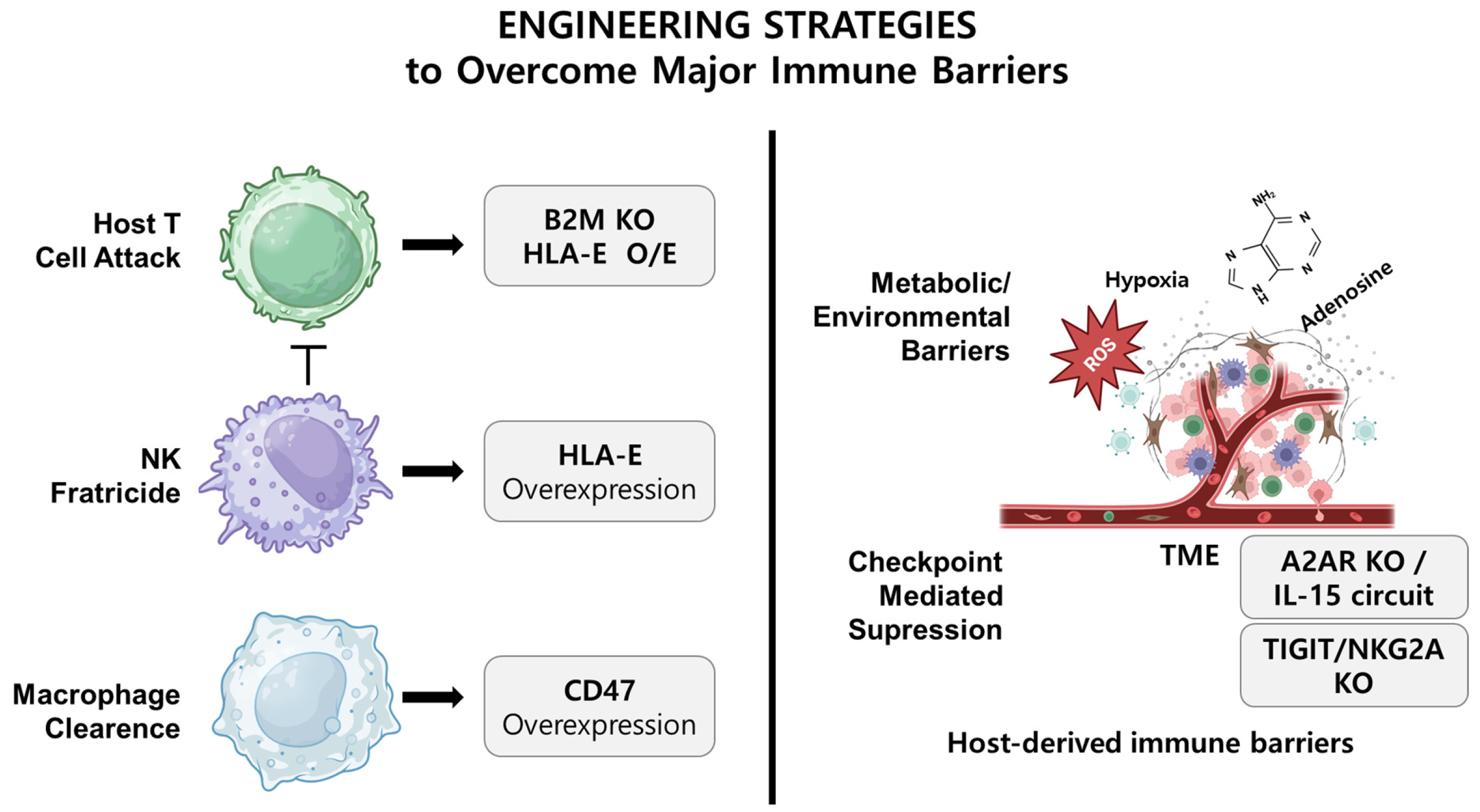
3. Immunological Barriers in Allogeneic NK Cell Therapy
3.1. CAR Engineering and Combinatorial Strategies to Overcome TME-Induced Suppression
3.2. Host T-Cell-Mediated Rejection and the Challenges of HLA Modulation
3.3. NK-Cell-Mediated Clearance: The Complexity of the “Missing Self”
3.4. Phagocyte-Mediated Clearance and the Duality of CD47
3.5. Suppression Within the Tumor Microenvironment
4. Gene Editing Strategies to Enable Immune Evasion
4.1. Modulating HLA Class I Expression: From Deletion to Precision Retention
4.2. Overexpression of CD47 to Evade Phagocytosis
4.3. Engineering HLA-E to Suppress Host NK Activation
4.4. KIR Editing: Balancing Autonomy and Tolerance
5. Functional Enhancement via Intrinsic Regulator Editing
5.1. Targeting CISH to Unlock IL-15 Responsiveness
5.2. Disrupting Transcriptional Exhaustion via FOXO1
5.3. Enhancing Infiltration and Response via IL-1R8 Knockout
5.4. Beyond Checkpoints: Toward Durable NK Cell States
6. Emerging Integrative Strategies and Platforms
6.1. Multiplex Gene Editing: Opportunities and Trade-Offs
6.2. Engineering Against the Tumor Microenvironment
6.3. iPSC-Based Platforms for Programmable NK Cell Design
6.4. From Universal Cells to Contextual Precision
6.5. AI-Guided Rational Design and Predictive Optimization in CAR-NK Therapy
6.6. Practical and Regulatory Challenges
7. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef]
- Brudno, J.N.; Kochenderfer, J.N. Toxicities of chimeric antigen receptor T cells: Recognition and management. Blood 2016, 127, 3321–3330. [Google Scholar] [CrossRef]
- Rezvani, K.; Rouce, R.H. The Application of Natural Killer Cell Immunotherapy for the Treatment of Cancer. Front. Immunol. 2015, 6, 578. [Google Scholar] [CrossRef]
- Kennedy, P.R.; Felices, M.; Miller, J.S. Challenges to the broad application of allogeneic natural killer cell immunotherapy of cancer. Stem Cell Res. Ther. 2022, 13, 165. [Google Scholar] [CrossRef] [PubMed]
- Afolabi, L.O.; Adeshakin, A.O.; Sani, M.M.; Bi, J.; Wan, X. Genetic reprogramming for NK cell cancer immunotherapy with CRISPR/Cas9. Immunology 2019, 158, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, K.; Rouce, R.; Liu, E.; Shpall, E. Engineering Natural Killer Cells for Cancer Immunotherapy. Mol. Ther. 2017, 25, 1769–1781. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Dwivedi, A.; Karulkar, A.; Ghosh, S.; Rafiq, A.; Purwar, R. Lymphocytes in Cellular Therapy: Functional Regulation of CAR T Cells. Front. Immunol. 2018, 9, 3180. [Google Scholar] [CrossRef]
- Parayath, N.N.; Stephan, S.B.; Koehne, A.L.; Nelson, P.S.; Stephan, M.T. In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo. Nat. Commun. 2020, 11, 6080. [Google Scholar] [CrossRef]
- Rath, J.A.; Arber, C. Engineering Strategies to Enhance TCR-Based Adoptive T Cell Therapy. Cells 2020, 9, 1485. [Google Scholar] [CrossRef]
- Wei, F.; Cheng, X.X.; Xue, J.Z.; Xue, S.A. Emerging Strategies in TCR-Engineered T Cells. Front. Immunol. 2022, 13, 850358. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Kent, A.; Davila, E. Chimeric non-antigen receptors in T cell-based cancer therapy. J. Immunother. Cancer 2021, 9, e002628. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Gu, J.; Xu, H. Prospects for chimeric antigen receptor-modified T cell therapy for solid tumors. Mol. Cancer 2018, 17, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Husman, T.; Cen, X.; Tsao, T.; Brown, J.; Bajpai, A.; Li, M.; Zhou, K.; Yang, L. Interleukin 15 in Cell-Based Cancer Immunotherapy. Int. J. Mol. Sci. 2022, 23, 7311. [Google Scholar] [CrossRef]
- Andrea, A.E.; Chiron, A.; Sarrabayrouse, G.; Bessoles, S.; Hacein-Bey-Abina, S. A structural, genetic and clinical comparison of CAR-T cells and CAR-NK cells: Companions or competitors? Front. Immunol. 2024, 15, 1459818. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.Y.; Du, Z.; Zhang, X.; Chng, W.J.; Wang, S. CXCR4 and anti-BCMA CAR co-modified natural killer cells suppress multiple myeloma progression in a xenograft mouse model. Cancer Gene Ther. 2022, 29, 475–483. [Google Scholar] [CrossRef]
- Guo, Y.; Xu, B.; Wu, Z.; Bo, J.; Tong, C.; Chen, D.; Wang, J.; Wang, H.; Wang, Y.; Han, W. Mutant B2M-HLA-E and B2M-HLA-G fusion proteins protects universal chimeric antigen receptor-modified T cells from allogeneic NK cell-mediated lysis. Eur. J. Immunol. 2021, 51, 2513–2521. [Google Scholar] [CrossRef]
- Sim, M.J.; Malaker, S.A.; Khan, A.; Stowell, J.M.; Shabanowitz, J.; Peterson, M.E.; Rajagopalan, S.; Hunt, D.F.; Altmann, D.M.; Long, E.O.; et al. Canonical and Cross-reactive Binding of NK Cell Inhibitory Receptors to HLA-C Allotypes Is Dictated by Peptides Bound to HLA-C. Front. Immunol. 2017, 8, 193. [Google Scholar] [CrossRef]
- Hammer, Q.; Ruckert, T.; Romagnani, C. Natural killer cell specificity for viral infections. Nat. Immunol. 2018, 19, 800–808. [Google Scholar] [CrossRef]
- Weiskopf, K.; Jahchan, N.S.; Schnorr, P.J.; Cristea, S.; Ring, A.M.; Maute, R.L.; Volkmer, A.K.; Volkmer, J.P.; Liu, J.; Lim, J.S.; et al. CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J. Clin. Investig. 2016, 126, 2610–2620. [Google Scholar] [CrossRef]
- Sun, H.; Sun, C. The Rise of NK Cell Checkpoints as Promising Therapeutic Targets in Cancer Immunotherapy. Front. Immunol. 2019, 10, 2354. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef]
- Kumar, A.; Lee, S.J.; Liu, Q.; Chan, A.K.N.; Pokharel, S.P.; Yu, J.; Chen, C.W.; Swaminathan, S. Generation and validation of CRISPR-engineered human natural killer cell lines for research and therapeutic applications. STAR Protoc. 2021, 2, 100874. [Google Scholar] [CrossRef] [PubMed]
- Deuse, T.; Hu, X.; Agbor-Enoh, S.; Jang, M.K.; Alawi, M.; Saygi, C.; Gravina, A.; Tediashvili, G.; Nguyen, V.Q.; Liu, Y.; et al. The SIRPalpha-CD47 immune checkpoint in NK cells. J. Exp. Med. 2021, 218, e20200839. [Google Scholar] [CrossRef]
- Andrechak, J.C.; Dooling, L.J.; Discher, D.E. The macrophage checkpoint CD47: SIRPalpha for recognition of ‘self’ cells: From clinical trials of blocking antibodies to mechanobiological fundamentals. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20180217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Q.; Thomas, F.; Fang, J.; Austgen, K.; Cowan, C.; Welstead, G.G. Universal protection of allogeneic T-cell therapies from natural killer cells via CD300a agonism. Blood Adv. 2025, 9, 254–264. [Google Scholar] [CrossRef]
- Huang, R.S.; Lai, M.C.; Shih, H.A.; Lin, S. A robust platform for expansion and genome editing of primary human natural killer cells. J. Exp. Med. 2021, 218, e20201529. [Google Scholar] [CrossRef]
- Wang, X.; Xiong, H.; Ning, Z. Implications of NKG2A in immunity and immune-mediated diseases. Front. Immunol. 2022, 13, 960852. [Google Scholar] [CrossRef]
- Li, Y.; Li, Z.; Tang, Y.; Zhuang, X.; Feng, W.; Boor, P.P.C.; Buschow, S.; Sprengers, D.; Zhou, G. Unlocking the therapeutic potential of the NKG2A-HLA-E immune checkpoint pathway in T cells and NK cells for cancer immunotherapy. J. Immunother. Cancer 2024, 12, e009934. [Google Scholar] [CrossRef]
- Isitman, G.; Tremblay-McLean, A.; Lisovsky, I.; Bruneau, J.; Lebouche, B.; Routy, J.P.; Bernard, N.F. NK Cells Expressing the Inhibitory Killer Immunoglobulin-Like Receptors (iKIR) KIR2DL1, KIR2DL3 and KIR3DL1 Are Less Likely to Be CD16+ than Their iKIR Negative Counterparts. PLoS ONE 2016, 11, e0164517. [Google Scholar] [CrossRef]
- Leijonhufvud, C.; Sanz-Ortega, L.; Schlums, H.; Gaballa, A.; Andersson, A.; Eriksson, C.; Segerberg, F.; Uhlin, M.; Bryceson, Y.T.; Carlsten, M. KIR2DS1 and KIR2DL1-C245 Dominantly Repress NK Cell Degranulation Triggered by Monoclonal or Bispecific Antibodies, whereas Education by Uptuning Inhibitory Killer Ig-related Receptors Exerts No Advantage in Ab-dependent Cellular Cytotoxicity. J. Immunol. 2024, 212, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.S.; Rezvani, K. Can we make a better match or mismatch with KIR genotyping? Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 106–118. [Google Scholar] [CrossRef]
- Marcenaro, E.; Carlomagno, S.; Pesce, S.; Della Chiesa, M.; Moretta, A.; Sivori, S. Role of alloreactive KIR2DS1+ NK cells in haploidentical hematopoietic stem cell transplantation. J. Leukoc. Biol. 2011, 90, 661–667. [Google Scholar] [CrossRef]
- Le Luduec, J.B.; Boudreau, J.E.; Freiberg, J.C.; Hsu, K.C. Novel Approach to Cell Surface Discrimination Between KIR2DL1 Subtypes and KIR2DS1 Identifies Hierarchies in NK Repertoire, Education, and Tolerance. Front. Immunol. 2019, 10, 734. [Google Scholar] [CrossRef]
- Edris, B.; Weiskopf, K.; Volkmer, A.K.; Volkmer, J.P.; Willingham, S.B.; Contreras-Trujillo, H.; Liu, J.; Majeti, R.; West, R.B.; Fletcher, J.A.; et al. Antibody therapy targeting the CD47 protein is effective in a model of aggressive metastatic leiomyosarcoma. Proc. Natl. Acad. Sci. USA 2012, 109, 6656–6661. [Google Scholar] [CrossRef] [PubMed]
- van Hall, T.; Andre, P.; Horowitz, A.; Ruan, D.F.; Borst, L.; Zerbib, R.; Narni-Mancinelli, E.; van der Burg, S.H.; Vivier, E. Monalizumab: Inhibiting the novel immune checkpoint NKG2A. J. Immunother. Cancer 2019, 7, 263. [Google Scholar] [CrossRef]
- Lambert, M.; Leijonhufvud, C.; Segerberg, F.; Melenhorst, J.J.; Carlsten, M. CRISPR/Cas9-Based Gene Engineering of Human Natural Killer Cells: Protocols for Knockout and Readouts to Evaluate Their Efficacy. Methods Mol. Biol. 2020, 2121, 213–239. [Google Scholar] [CrossRef]
- Delconte, R.B.; Kolesnik, T.B.; Dagley, L.F.; Rautela, J.; Shi, W.; Putz, E.M.; Stannard, K.; Zhang, J.G.; Teh, C.; Firth, M.; et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat. Immunol. 2016, 17, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Hills, L.B.; Abdullah, L.; Lust, H.E.; Degefu, H.; Huang, Y.H. Foxo1 Serine 209 Is a Critical Regulatory Site of CD8 T Cell Differentiation and Survival. J. Immunol. 2021, 206, 89–100. [Google Scholar] [CrossRef]
- Molgora, M.; Bonavita, E.; Ponzetta, A.; Riva, F.; Barbagallo, M.; Jaillon, S.; Popovic, B.; Bernardini, G.; Magrini, E.; Gianni, F.; et al. IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity. Nature 2017, 551, 110–114. [Google Scholar] [CrossRef]
- Bernard, P.L.; Delconte, R.; Pastor, S.; Laletin, V.; Costa Da Silva, C.; Goubard, A.; Josselin, E.; Castellano, R.; Krug, A.; Vernerey, J.; et al. Targeting CISH enhances natural cytotoxicity receptor signaling and reduces NK cell exhaustion to improve solid tumor immunity. J. Immunother. Cancer 2022, 10, e004244. [Google Scholar] [CrossRef] [PubMed]
- Daher, M.; Basar, R.; Gokdemir, E.; Baran, N.; Uprety, N.; Nunez Cortes, A.K.; Mendt, M.; Kerbauy, L.N.; Banerjee, P.P.; Shanley, M.; et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood 2021, 137, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Krueger, J.B.; Gilkey, A.K.; Stelljes, E.M.; Kluesner, M.G.; Pomeroy, E.J.; Skeate, J.G.; Slipek, N.J.; Lahr, W.S.; Vazquez, P.N.C.; et al. Precision enhancement of CAR-NK cells through non-viral engineering and highly multiplexed base editing. J. Immunother. Cancer 2025, 13, e009560. [Google Scholar] [CrossRef]
- Yang, C.; Siebert, J.R.; Burns, R.; Zheng, Y.; Mei, A.; Bonacci, B.; Wang, D.; Urrutia, R.A.; Riese, M.J.; Rao, S.; et al. Single-cell transcriptome reveals the novel role of T-bet in suppressing the immature NK gene signature. eLife 2020, 9, e51339. [Google Scholar] [CrossRef]
- Simonetta, F.; Pradier, A.; Roosnek, E. T-bet and Eomesodermin in NK Cell Development, Maturation, and Function. Front. Immunol. 2016, 7, 241. [Google Scholar] [CrossRef]
- Huang, P.; Wang, F.; Yang, Y.; Lai, W.; Meng, M.; Wu, S.; Peng, H.; Wang, L.; Zhan, R.; Imani, S.; et al. Hematopoietic-Specific Deletion of Foxo1 Promotes NK Cell Specification and Proliferation. Front. Immunol. 2019, 10, 1016. [Google Scholar] [CrossRef]
- Miao, L.; Lu, C.; Zhang, B.; Li, H.; Zhao, X.; Chen, H.; Liu, Y.; Cui, X. Advances in metabolic reprogramming of NK cells in the tumor microenvironment on the impact of NK therapy. J. Transl. Med. 2024, 22, 229. [Google Scholar] [CrossRef] [PubMed]
- Marchal, I. FOXO1 enhances CAR T cell fitness and function. Nat. Biotechnol. 2024, 42, 699. [Google Scholar] [CrossRef]
- Landolina, N.; Mariotti, F.R.; Ingegnere, T.; Alicata, C.; Ricci, B.; Pelosi, A.; Veneziani, I.; Azzarone, B.G.; Garlanda, C.; Mantovani, A.; et al. IL-1R8 silencing improves the anti-tumor function of freshly isolated human NK cells. J. Immunother. Cancer 2022, 10, e003858. [Google Scholar] [CrossRef]
- Ghaedrahmati, F.; Akbari, V.; Seyedhosseini-Ghaheh, H.; Esmaeil, N. Strong capacity of differentiated PD-L1 CAR-modified UCB-CD34+ cells and PD-L1 CAR-modified UCB-CD34+-derived NK cells in killing target cells and restoration of the anti-tumor function of PD-1-high exhausted T Cells. Stem Cell Res. Ther. 2024, 15, 257. [Google Scholar] [CrossRef]
- Zhou, A.Y.; Marin, N.D.; Afrin, S.; Wong, P.; Tran, J.; Jacobs, M.T.; Becker-Hapak, M.; Marsala, L.; Foster, M.; Foltz, J.A.; et al. Memory-like NK cell differentiation, inhibitory NKG2A blockade, and improved recognition via antibody or CAR engineering combine to enhance NK cell attack against multiple myeloma. J. Immunol. 2025, 214, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, T.J.; Rubinsteyn, A.; Laserson, U. MHCflurry 2.0: Improved Pan-Allele Prediction of MHC Class I-Presented Peptides by Incorporating Antigen Processing. Cell Syst. 2020, 11, 42–48.e7. [Google Scholar] [CrossRef]
- Chauvin, J.M.; Ka, M.; Pagliano, O.; Menna, C.; Ding, Q.; DeBlasio, R.; Sanders, C.; Hou, J.; Li, X.Y.; Ferrone, S.; et al. IL15 Stimulation with TIGIT Blockade Reverses CD155-mediated NK-Cell Dysfunction in Melanoma. Clin. Cancer Res. 2020, 26, 5520–5533. [Google Scholar] [CrossRef]
- Villalona-Calero, M.A.; Tian, L.; Li, X.; Palmer, J.M.; Aceves, C.; Meisen, H.; Cortez, C.; Synold, T.W.; Egelston, C.; VanDeusen, J.; et al. Interim report on engineered NK cell trial in lung cancer refractory to immune checkpoint inhibitors. JCI Insight 2025, 10, e186890. [Google Scholar] [CrossRef]
- Cichocki, F.; van der Stegen, S.J.C.; Miller, J.S. Engineered and banked iPSCs for advanced NK- and T-cell immunotherapies. Blood 2023, 141, 846–855. [Google Scholar] [CrossRef]
- Hoerster, K.; Uhrberg, M.; Wiek, C.; Horn, P.A.; Hanenberg, H.; Heinrichs, S. HLA Class I Knockout Converts Allogeneic Primary NK Cells Into Suitable Effectors for “Off-the-Shelf” Immunotherapy. Front. Immunol. 2020, 11, 586168. [Google Scholar] [CrossRef]
- He, P.; Wei, X.; Xu, Y.; Huang, J.; Tang, N.; Yan, T.; Yang, C.; Lu, K. Analysis of complex chromosomal rearrangements using a combination of current molecular cytogenetic techniques. Mol. Cytogenet. 2022, 15, 20. [Google Scholar] [CrossRef] [PubMed]
- Taki, T.; Morimoto, K.; Mizuno, S.; Kuno, A. KOnezumi-AID: Automation Software for Efficient Multiplex Gene Knockout Using Target-AID. Int. J. Mol. Sci. 2024, 25, 13500. [Google Scholar] [CrossRef]
- Petazzi, P.; Menendez, P.; Sevilla, A. CRISPR/Cas9-Mediated Gene Knockout and Knockin Human iPSCs. Methods Mol. Biol. 2022, 2454, 559–574. [Google Scholar] [CrossRef]
- Li, C.; Zong, Y.; Jin, S.; Zhu, H.; Lin, D.; Li, S.; Qiu, J.L.; Wang, Y.; Gao, C. SWISS: Multiplexed orthogonal genome editing in plants with a Cas9 nickase and engineered CRISPR RNA scaffolds. Genome Biol. 2020, 21, 141. [Google Scholar] [CrossRef] [PubMed]
- Ghobadi, A.; Bachanova, V.; Patel, K.; Park, J.H.; Flinn, I.; Riedell, P.A.; Bachier, C.; Diefenbach, C.S.; Wong, C.; Bickers, C.; et al. Induced pluripotent stem-cell-derived CD19-directed chimeric antigen receptor natural killer cells in B-cell lymphoma: A phase 1, first-in-human trial. Lancet 2025, 405, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Lai, W.; Tian, B.; Xu, X.; Xie, S.; Zhong, W.; Kang, H.; Chen, X.; Li, H.; Xu, J.; et al. Application and prospects of genetic engineering in CAR-NK cell therapy. Front. Immunol. 2025, 16, 1600411. [Google Scholar] [CrossRef]
- Ji, S.; Jin, C.; Cui, X. Enhancing the physiological characteristics of chimeric antigen receptor natural killer cells by synthetic biology. Front. Immunol. 2025, 16, 1592121. [Google Scholar] [CrossRef]
- Zhao, T.; You, J.; Wang, C.; Li, B.; Liu, Y.; Shao, M.; Zhao, W.; Zhou, C. Cell-based immunotherapies for solid tumors: Advances, challenges, and future directions. Front. Oncol. 2025, 15, 1551583. [Google Scholar] [CrossRef]
- White, L.G.; Goy, H.E.; Rose, A.J.; McLellan, A.D. Controlling Cell Trafficking: Addressing Failures in CAR T and NK Cell Therapy of Solid Tumours. Cancers 2022, 14, 978. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, Y.; He, Z.; Li, L.; Liu, S.; Jiang, M.; Zhao, B.; Deng, M.; Wang, W.; Mi, X.; et al. Breakthrough of solid tumor treatment: CAR-NK immunotherapy. Cell Death Discov. 2024, 10, 40. [Google Scholar] [CrossRef]
- Chu, X.; Tian, W.; Wang, Z.; Zhang, J.; Zhou, R. Correction: Co-inhibition of TIGIT and PD-1/PD-L1 in Cancer Immunotherapy: Mechanisms and Clinical Trials. Mol. Cancer 2023, 22, 101. [Google Scholar] [CrossRef]
- Sun, J.; Tian, Y.; Yang, C. Target therapy of TIGIT; a novel approach of immunotherapy for the treatment of colorectal cancer. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2025, 398, 231–241. [Google Scholar] [CrossRef]
- Xie, Q.; Liu, X.; Liu, R.; Pan, J.; Liang, J. Cellular mechanisms of combining innate immunity activation with PD-1/PD-L1 blockade in treatment of colorectal cancer. Mol. Cancer 2024, 23, 252. [Google Scholar] [CrossRef] [PubMed]
- Chuai, G.; Ma, H.; Yan, J.; Chen, M.; Hong, N.; Xue, D.; Zhou, C.; Zhu, C.; Chen, K.; Duan, B.; et al. DeepCRISPR: Optimized CRISPR guide RNA design by deep learning. Genome Biol. 2018, 19, 80. [Google Scholar] [CrossRef]
- Zhang, G.; Luo, Y.; Dai, X.; Dai, Z. Benchmarking deep learning methods for predicting CRISPR/Cas9 sgRNA on- and off-target activities. Brief. Bioinform. 2023, 24, bbad333. [Google Scholar] [CrossRef]
- Derraz, B.; Breda, G.; Kaempf, C.; Baenke, F.; Cotte, F.; Reiche, K.; Kohl, U.; Kather, J.N.; Eskenazy, D.; Gilbert, S. New regulatory thinking is needed for AI-based personalised drug and cell therapies in precision oncology. Npj Precis. Oncol. 2024, 8, 23. [Google Scholar] [CrossRef]
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Iyer, B.; Prasath, V.B.S.; Ni, Y.; Salomonis, N. DeepImmuno: Deep learning-empowered prediction and generation of immunogenic peptides for T-cell immunity. Brief. Bioinform. 2021, 22, bbab160. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Q.; Zhang, X.; Huang, H.; Tang, S.; Chai, Y.; Xu, Z.; Li, M.; Chen, X.; Liu, J.; et al. Recent advances in exosome-mediated nucleic acid delivery for cancer therapy. J. Nanobiotechnol. 2022, 20, 279. [Google Scholar] [CrossRef]
- Soroudi, S.; Jaafari, M.R.; Arabi, L. Lipid nanoparticle (LNP) mediated mRNA delivery in cardiovascular diseases: Advances in genome editing and CAR T cell therapy. J. Control. Release 2024, 372, 113–140. [Google Scholar] [CrossRef] [PubMed]
- Esmaeilzadeh, A.; Hadiloo, K.; Yaghoubi, S.; Makoui, M.H.; Mostanadi, P. State of the art in CAR-based therapy: In vivo CAR production as a revolution in cell-based cancer treatment. Cell Oncol. 2025. [Google Scholar] [CrossRef]
- Huang, C.; Li, G.; Wu, J.; Liang, J.; Wang, X. Identification of pathogenic variants in cancer genes using base editing screens with editing efficiency correction. Genome Biol. 2021, 22, 80. [Google Scholar] [CrossRef]
- Yuan, B.; Zhang, S.; Song, L.; Chen, J.; Cao, J.; Qiu, J.; Qiu, Z.; Chen, J.; Zhao, X.M.; Cheng, T.L. Engineering of cytosine base editors with DNA damage minimization and editing scope diversification. Nucleic Acids Res. 2023, 51, e105. [Google Scholar] [CrossRef] [PubMed]
- Testa, L.C.; Musunuru, K. Base Editing and Prime Editing: Potential Therapeutic Options for Rare and Common Diseases. BioDrugs 2023, 37, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Terheyden-Keighley, D.; Huhne, M.; Berger, T.; Hiller, B.; Martins, S.; Gamerschlag, A.; Sabour, D.; Meffert, A.; Kislat, A.; Slotta, C.; et al. GMP-compliant iPS cell lines show widespread plasticity in a new set of differentiation workflows for cell replacement and cancer immunotherapy. Stem Cells Transl. Med. 2024, 13, 898–911. [Google Scholar] [CrossRef] [PubMed]
- Bexte, T.; Albinger, N.; Al Ajami, A.; Wendel, P.; Buchinger, L.; Gessner, A.; Alzubi, J.; Sarchen, V.; Vogler, M.; Rasheed, H.M.; et al. CRISPR/Cas9 editing of NKG2A improves the efficacy of primary CD33-directed chimeric antigen receptor natural killer cells. Nat. Commun. 2024, 15, 8439. [Google Scholar] [CrossRef]
- Shankar, K.; Zingler-Hoslet, I.; Tabima, D.M.; Zima, S.; Shi, L.; Gimse, K.; Forsberg, M.H.; Katta, V.; Davis, S.Z.; Maldonado, D.; et al. Virus-free CRISPR knockin of a chimeric antigen receptor into KLRC1 generates potent GD2-specific natural killer cells. Mol. Ther. 2025, 33, 1014–1030. [Google Scholar] [CrossRef]
- Harbottle, J.A. Immunotherapy to get on point with base editing. Drug Discov. Today 2021, 26, 2350–2357. [Google Scholar] [CrossRef]
- Luciani, F.; Safavi, A.; Guruprasad, P.; Chen, L.; Ruella, M. Advancing CAR T-cell Therapies with Artificial Intelligence: Opportunities and Challenges. Blood Cancer Discov. 2025, 6, 159–162. [Google Scholar] [CrossRef]
- Shahzadi, M.; Rafique, H.; Waheed, A.; Naz, H.; Waheed, A.; Zokirova, F.R.; Khan, H. Artificial intelligence for chimeric antigen receptor-based therapies: A comprehensive review of current applications and future perspectives. Ther. Adv. Vaccines Immunother. 2024, 12, 25151355241305856. [Google Scholar] [CrossRef]
- Choudhery, M.S.; Arif, T.; Mahmood, R.; Harris, D.T. CAR-T-Cell-Based Cancer Immunotherapies: Potentials, Limitations, and Future Prospects. J. Clin. Med. 2024, 13, 3202. [Google Scholar] [CrossRef]
- Wang, X.; Byrne, M.E.; Liu, C.; Ma, M.T.; Liu, D. Scalable process development of NK and CAR-NK expansion in a closed bioreactor. Front. Immunol. 2024, 15, 1412378. [Google Scholar] [CrossRef]
- Lu, S.J.; Feng, Q. CAR-NK cells from engineered pluripotent stem cells: Off-the-shelf therapeutics for all patients. Stem Cells Transl. Med. 2021, 10 (Suppl. S2), S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Albinger, N.; Muller, S.; Kostyra, J.; Kuska, J.; Mertlitz, S.; Penack, O.; Zhang, C.; Moker, N.; Ullrich, E. Manufacturing of primary CAR-NK cells in an automated system for the treatment of acute myeloid leukemia. Bone Marrow Transplant. 2024, 59, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, L.V.; Christensen, E.B.; Barnkob, M.B.; Barington, T. The clinical landscape of CAR NK cells. Exp. Hematol. Oncol. 2025, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Pfefferle, A.; Contet, J.; Wong, K.; Chen, C.; Verhoeyen, E.; Slichter, C.K.; Schluns, K.S.; Cursons, J.; Berry, R.; Nikolic, I.; et al. Optimisation of a primary human CAR-NK cell manufacturing pipeline. Clin. Transl. Immunol. 2024, 13, e1507. [Google Scholar] [CrossRef]

{kind=link}
| Editing Target(s) | Immune Barrier(s) Addressed | Mechanism(s) | Key Risks or Limitations | Reference |
|---|---|---|---|---|
| B2M KO + HLA-E OE | T-cell rejection + NK fratricide | Removes classical HLA, restores inhibition via HLA-E | Multiplex editing complexity; off-target effects | [17] |
| HLA-A/B KO + HLA-C retention | T-cell rejection + NK tolerance | Minimizes TCR recognition while preserving NK KIR signals | HLA diversity may limit universality | [34] |
| CD47 OE | Macrophage clearance | Suppresses SIRPα-mediated phagocytosis | Potential tumor immune escape mimicry | [35] |
| HLA-E OE | NK cell inhibition (via NKG2A) | Engages NKG2A to inhibit NK activation | Not effective in NKG2C-dominant NKs | [36] |
| KIR editing | NK autoregulation/activation tuning | Removes inhibitory or adds activating KIRs | Loss of tolerance; tissue toxicity | [37] |
| CISH KO | IL-15 pathway suppression | Boosts IL-15 sensitivity and persistence | Cytokine overstimulation risk | [38] |
| FOXO1 KO | Transcriptional exhaustion | Promotes T-bet/Eomes expression, memory traits | Possible disruption of homeostasis | [39] |
| IL-1R8 KO | Infiltration + activation | Enhances chemokine response and IFN-γ secretion | Auto-inflammation potential | [40] |
| Target Gene | Biological Role | Editing Strategy | Expected Effect(s) | Preclinical Evidence | Risks |
|---|---|---|---|---|---|
| CISH | IL-15 signaling brake | Knockout | Boosts expansion, persistence, and granule release | [38] | Overstimulation, potential bystander toxicity |
| FOXO1 | Transcriptional effector suppression | Knockout or suppression | Enhances effector programming (T-bet, Eomes) | [39] | Disruption of homeostasis, survival defects |
| IL-1R8 | Suppressor of IL-1R signaling and chemotaxis | Knockout | Improves tumor infiltration and IFN-γ | [40] | Autoimmune-like inflammation |
| TIGIT | Checkpoint receptor limiting cytotoxicity | Knockout or antibody blockade | Restores NK activation and tumor killing | [54] | Loss of peripheral tolerance |
| Developer/Trial ID | Cell Source | Gene Editing Targets | CAR Antigen | Indication | Phase | Outcomes/Status |
|---|---|---|---|---|---|---|
| Fate Therapeutics/NCT04555811 | iPSC | B2M KO, HLA-E OE, CD38 KO | CD19 | Non-Hodgkin lymphoma | Phase 1 | ORR: 38%, CRS: 0%, mild cytopenia |
| Nkarta/NCT05020678 | Peripheral blood NK | None (natural KIR mismatch) | CD19 | B-ALL, CLL, NHL | Phase 1 | Ongoing |
| MD Anderson/NCT03056339 | Cord blood | IL-15 OE, iC9 safety switch | CD19 | B-cell malignancies | Phase 1/2 | Recruiting |
| Wugen/NCT05470140 | Donor-derived NK | None yet | None | Acute myeloid leukemia | Phase 1 | Recruiting |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H. Overcoming Immune Barriers in Allogeneic CAR-NK Therapy: From Multiplex Gene Editing to AI-Driven Precision Design. Biomolecules 2025, 15, 935. https://doi.org/10.3390/biom15070935
Kim H. Overcoming Immune Barriers in Allogeneic CAR-NK Therapy: From Multiplex Gene Editing to AI-Driven Precision Design. Biomolecules. 2025; 15(7):935. https://doi.org/10.3390/biom15070935
Chicago/Turabian StyleKim, Hyunyoung. 2025. "Overcoming Immune Barriers in Allogeneic CAR-NK Therapy: From Multiplex Gene Editing to AI-Driven Precision Design" Biomolecules 15, no. 7: 935. https://doi.org/10.3390/biom15070935
APA StyleKim, H. (2025). Overcoming Immune Barriers in Allogeneic CAR-NK Therapy: From Multiplex Gene Editing to AI-Driven Precision Design. Biomolecules, 15(7), 935. https://doi.org/10.3390/biom15070935