
Discovery of Dietary Plant Flavonols as Novel Potent Inhibitors Targeting DYRK1A Kinase



Abstract
1. Introduction
2. Materials and Methods
2.1. Structures of Flavonoid Compounds
2.2. DYRK1A Protein Expression and Purification
2.3. DYRK1A Kinase Inhibition Assay
2.4. Cell Lines and Culture
2.5. Cell Viability Assay
2.6. Cell Morphology Analysis
2.7. Molecular Docking
2.8. Molecular Dynamics Simulation
2.9. Statistical Analysis
3. Results
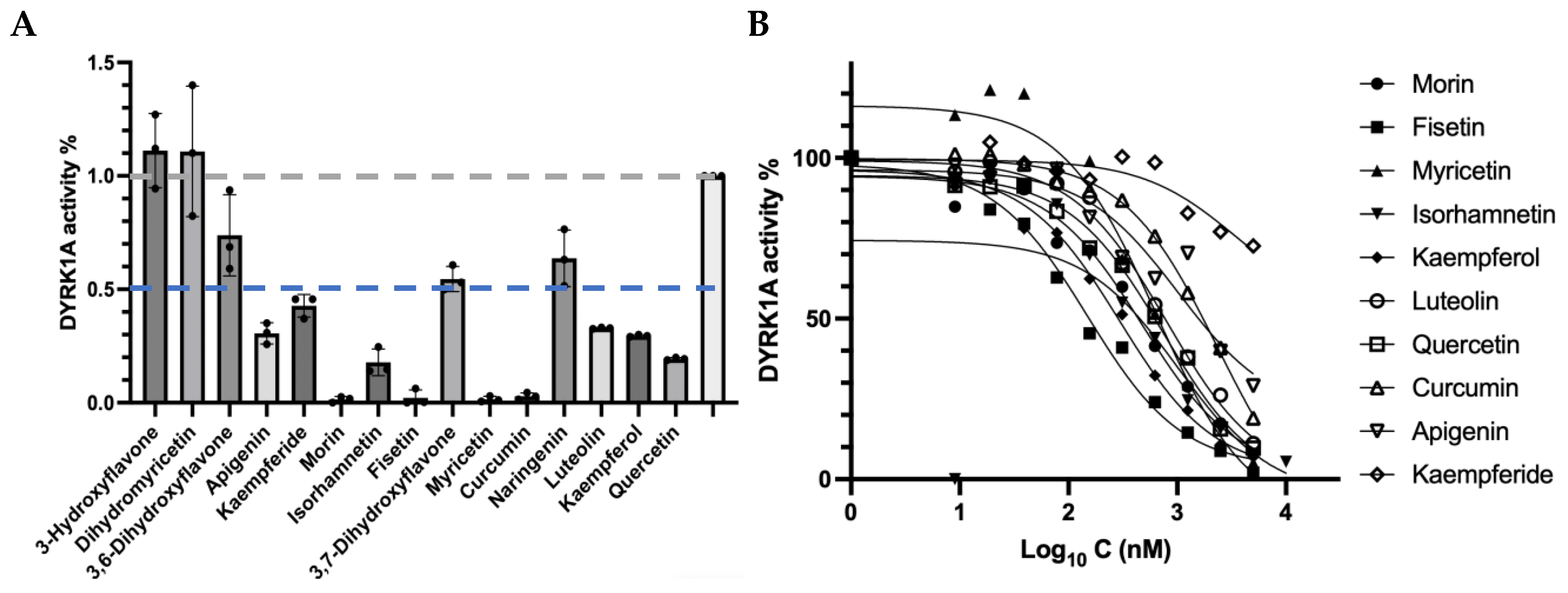
3.1. Identification of New DYRK1A Inhibitors
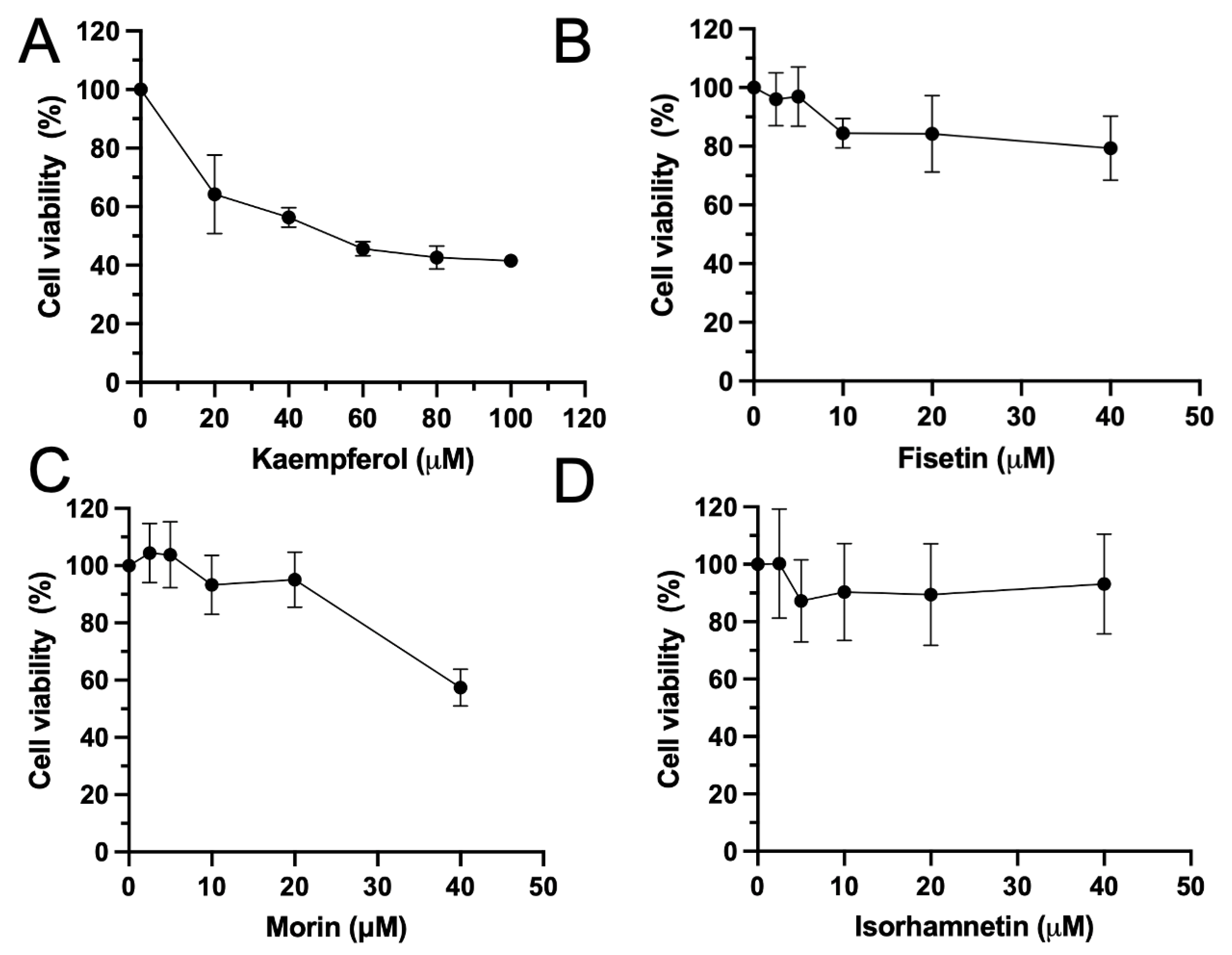
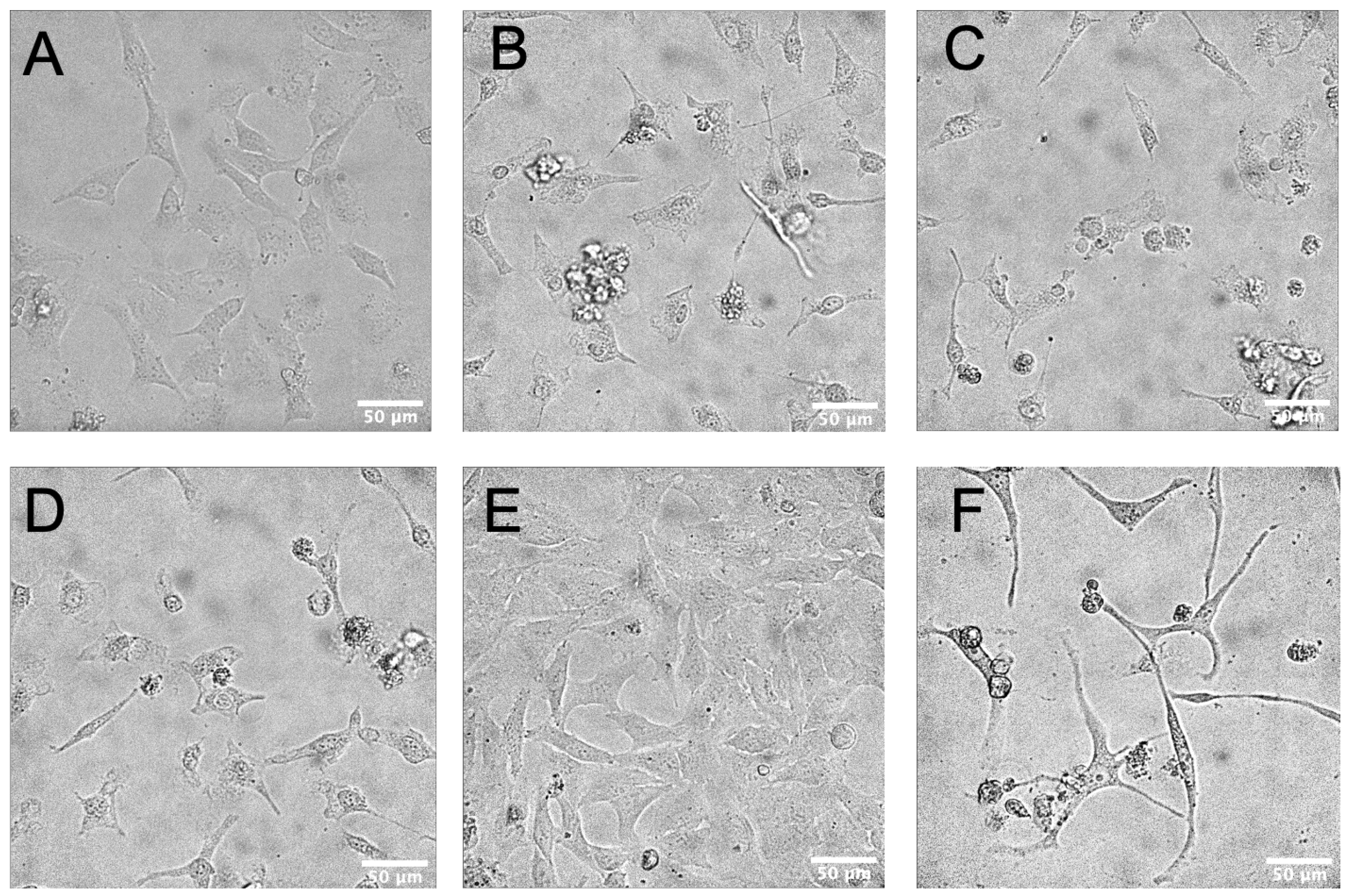
3.2. Flavonoids Induced Cytotoxicity and Morphology Changes on HeLa Cells
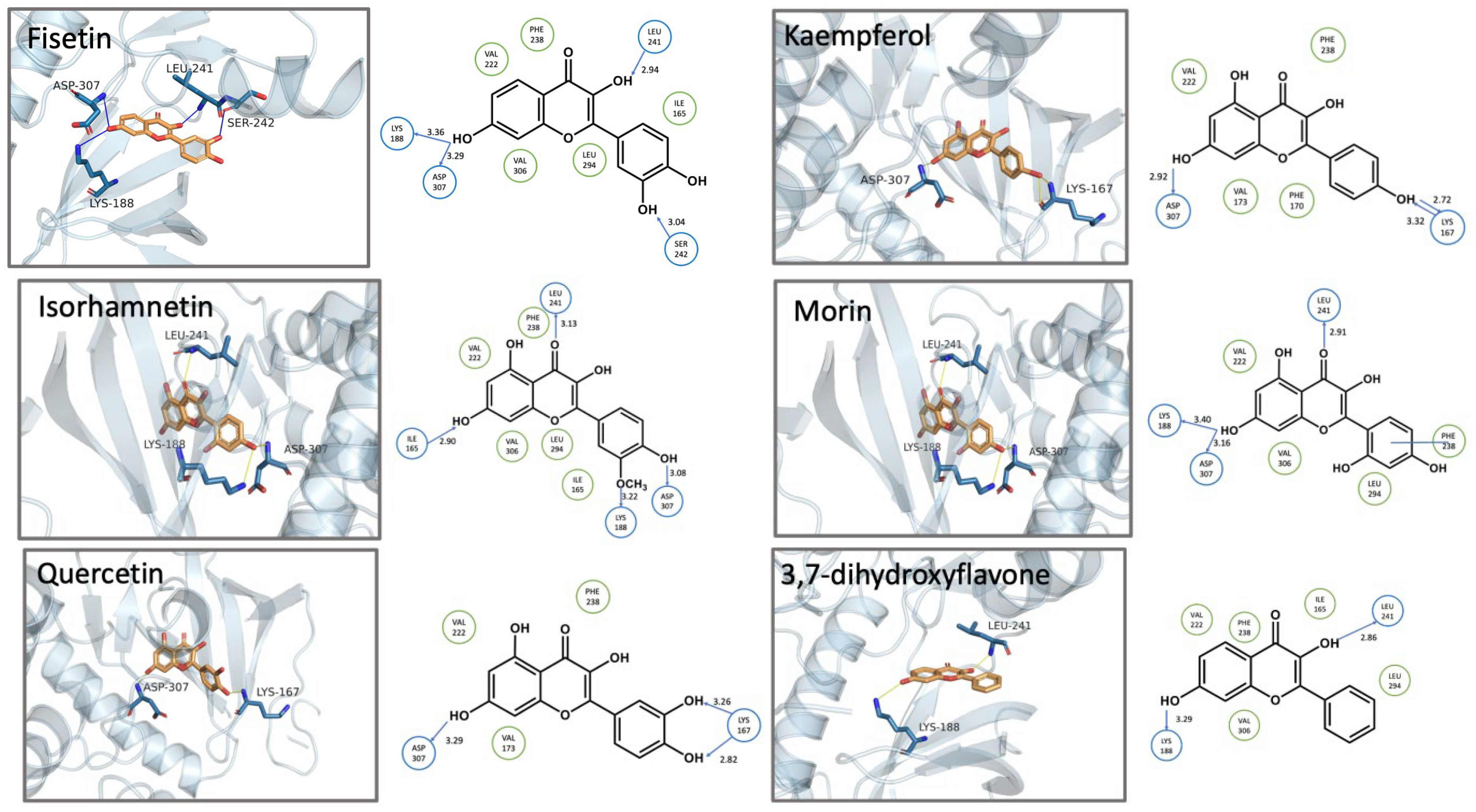
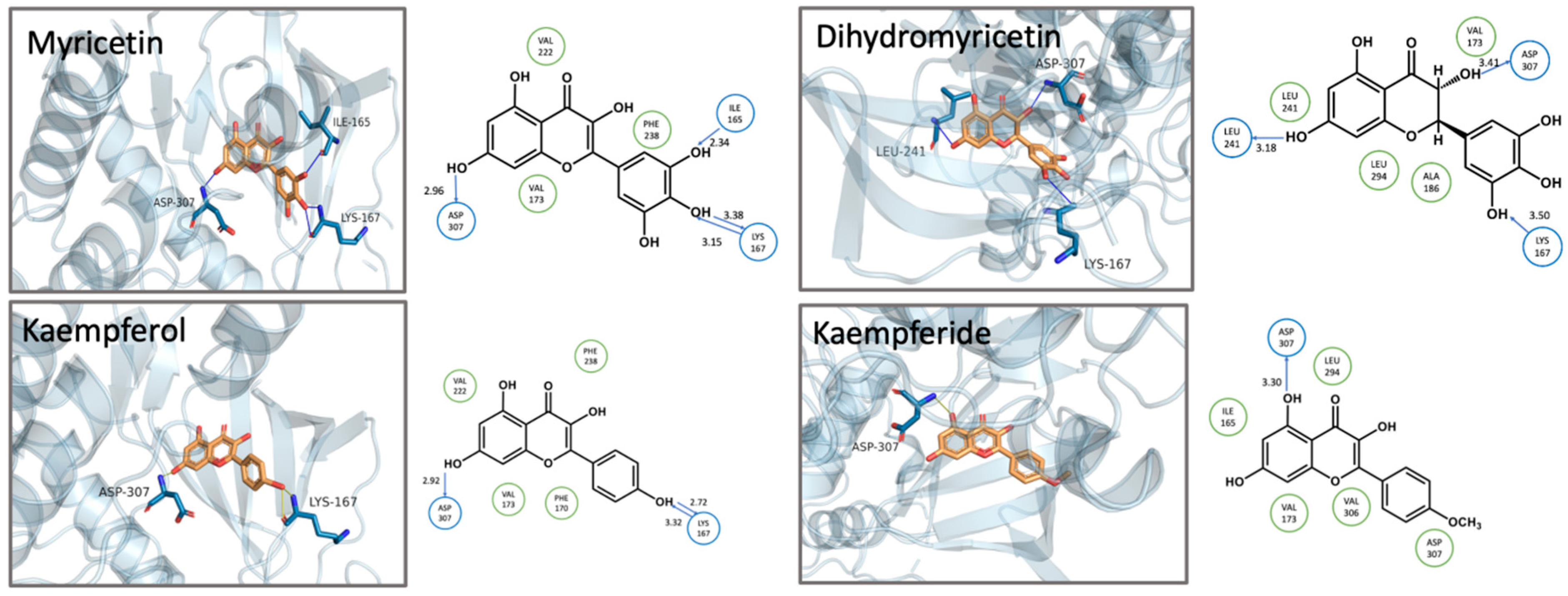
3.3. Structure-Activity Relationship and Molecular Docking
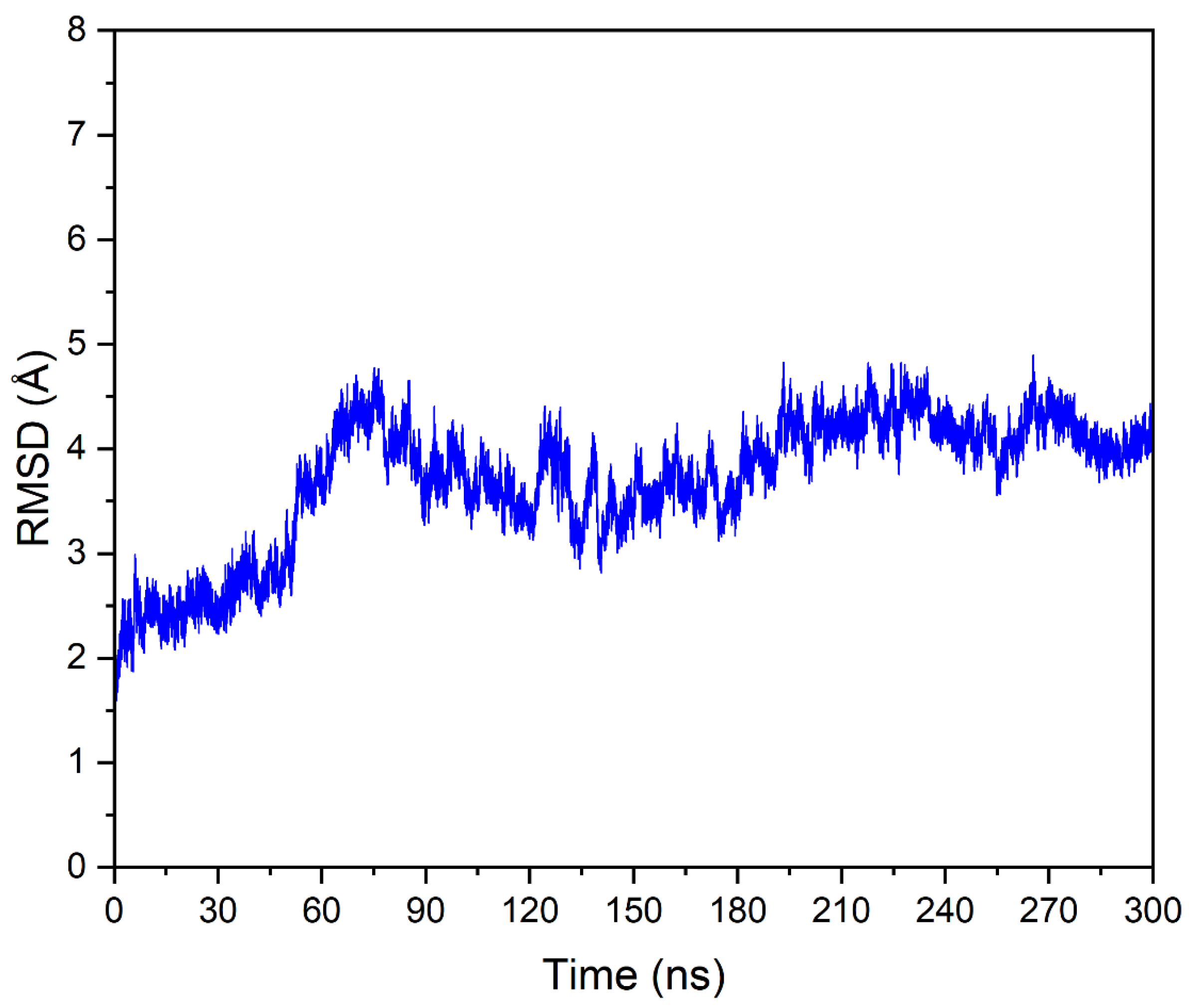
3.4. Molecular Dynamics Simulation of DYRK1A-Fisetin Complex
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Becker, W.; Weber, Y.; Wetzel, K.; Eirmbter, K.; Tejedor, F.J.; Joost, H.-G. Sequence Characteristics, Subcellular Localization, and Substrate Specificity of DYRK-related Kinases, a Novel Family of Dual Specificity Protein Kinases. J. Biol. Chem. 1998, 273, 25893–25902. [Google Scholar] [CrossRef] [PubMed]
- Tejedor, F.J.; Hämmerle, B. MNB/DYRK1A as a multiple regulator of neuronal development. FEBS J. 2011, 278, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Jarhad, D.B.; Mashelkar, K.K.; Kim, H.-R.; Noh, M.; Jeong, L.S. Dual-Specificity Tyrosine Phosphorylation-Regulated Kinase 1A (DYRK1A) Inhibitors as Potential Therapeutics. J. Med. Chem. 2018, 61, 9791–9810. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wang, Y.; Wang, J.; Ren, C.; Chen, H.; Zhang, J. DYRK1A inhibitors for disease therapy: Current status and perspectives. Eur. J. Med. Chem. 2022, 229, 114062. [Google Scholar] [CrossRef]
- Laham, A.J.; El-Awady, R.; Saber-Ayad, M.; Wang, N.; Yan, G.; Boudreault, J.; Ali, S.; Lebrun, J.-J. Targeting the DYRK1A kinase prevents cancer progression and metastasis and promotes cancer cells response to G1/S targeting chemotherapy drugs. NPJ Precis. Oncol. 2024, 8, 128. [Google Scholar] [CrossRef]
- Yang, Y.; Fan, X.; Liu, Y.; Ye, D.; Liu, C.; Yang, H.; Su, Z.; Zhang, Y.; Liu, Y. Function and inhibition of DYRK1A: Emerging roles of treating multiple human diseases. Biochem. Pharmacol. 2023, 212, 115521. [Google Scholar] [CrossRef]
- Becker, W.; Sippl, W. Activation, regulation, and inhibition of DYRK1A. FEBS J. 2011, 278, 246–256. [Google Scholar] [CrossRef]
- Pustelny, K.; Grygier, P.; Barzowska, A.; Pucelik, B.; Matsuda, A.; Mrowiec, K.; Slugocka, E.; Popowicz, G.M.; Dubin, G.; Czarna, A. Binding mechanism and biological effects of flavone DYRK1A inhibitors for the design of new antidiabetics. Sci. Rep. 2023, 13, 18114. [Google Scholar] [CrossRef]
- Göckler, N.; Jofre, G.; Papadopoulos, C.; Soppa, U.; Tejedor, F.J.; Becker, W. Harmine specifically inhibits protein kinase DYRK1A and interferes with neurite formation. FEBS J. 2009, 276, 6324–6337. [Google Scholar] [CrossRef]
- Araldi, G.L.; Hwang, Y.-W. Design, synthesis, and biological evaluation of polyphenol derivatives as DYRK1A inhibitors. The discovery of a potentially promising treatment for Multiple Sclerosis. Bioorganic Med. Chem. Lett. 2022, 64, 128675. [Google Scholar] [CrossRef]
- Bain, J.; Mclauchlan, H.; Elliott, M.; Cohen, P. The specificities of protein kinase inhibitors: An update. Biochem. J. 2003, 371, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Chou, F.; Huang, L.; Yin, F.; Fang, J.; Wang, J.-B.; Jia, Z. Recent insights into oxidative metabolism of quercetin: Catabolic profiles, degradation pathways, catalyzing metalloenzymes and molecular mechanisms. Crit. Rev. Food Sci. Nutr. 2022, 64, 1312–1339. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Pandey, A.K. Chemistry and Biological Activities of Flavonoids: An Overview. Sci. World J. 2013, 2013, 162750. [Google Scholar] [CrossRef] [PubMed]
- Boly, R.; Gras, T.; Lamkami, T.; Guissou, P.; Serteyn, D.; Kiss, R.; Dubois, J. Quercetin inhibits a large panel of kinases implicated in cancer cell biology. Int. J. Oncol. 2011, 38, 833–842. [Google Scholar]
- Hou, D.-X.; Kumamoto, T. Flavonoids as Protein Kinase Inhibitors for Cancer Chemoprevention: Direct Binding and Molecular Modeling. Antioxid. Redox Signal. 2010, 13, 691–719. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Banerjee, S.; Ji, C.; Mayfield, J.E.; Goel, A.; Xiao, J.; Dixon, J.E.; Guo, X. Ancient drug curcumin impedes 26S proteasome activity by direct inhibition of dual-specificity tyrosine-regulated kinase 2. Proc. Natl. Acad. Sci. USA 2018, 115, 8155–8160. [Google Scholar] [CrossRef]
- Shah, S.; Narang, R.; Singh, V.J.; Pilli, G.; Nayak, S.K. A Review on Anticancer Profile of Flavonoids: Sources, Chemistry, Mechanisms, Structure-activity Relationship and Anticancer Activity. Curr. Drug Res. Rev. 2023, 15, 122–148. [Google Scholar]
- Al-Khayri, J.M.; Sahana, G.R.; Nagella, P.; Joseph, B.V.; Alessa, F.M.; Al-Mssallem, M.Q. Flavonoids as Potential Anti-Inflammatory Molecules: A Review. Molecules 2022, 27, 2901. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Ding, K.; Qiao, Y.; Zhang, L.; Zheng, L.; Pan, T.; Zhang, L. Flavonoids Regulate Inflammation and Oxidative Stress in Cancer. Molecules 2020, 25, 5628. [Google Scholar] [CrossRef]
- Zhang, Z.; Shi, J.; Nice, E.C.; Huang, C.; Shi, Z. The Multifaceted Role of Flavonoids in Cancer Therapy: Leveraging Autophagy with a Double-Edged Sword. Antioxidants 2021, 10, 1138. [Google Scholar] [CrossRef]
- Neumann, F.; Gourdain, S.; Albac, C.; Dekker, A.D.; Bui, L.C.; Dairou, J.; Schmitz-Afonso, I.; Hue, N.; Rodrigues-Lima, F.; Delabar, J.M.; et al. DYRK1A inhibition and cognitive rescue in a Down syndrome mouse model are induced by new fluoro-DANDY derivatives. Sci. Rep. 2018, 8, 2859. [Google Scholar] [CrossRef]
- Courraud, J.; Quartier, A.; Drouot, N.; Zapata-Bodalo, I.; Gilet, J.; Benchoua, A.; Mandel, J.-L.; Piton, A. DYRK1A roles in human neural progenitors. Front. Neurosci. 2025, 19, 1533253. [Google Scholar] [CrossRef]
- Rammohan, M.; Harris, E.; Bhansali, R.S.; Zhao, E.; Li, L.S.; Crispino, J.D. The chromosome 21 kinase DYRK1A: Emerging roles in cancer biology and potential as a therapeutic target. Oncogene 2022, 41, 2003–2011. [Google Scholar] [CrossRef]
- Massey, A.J.; Benwell, K.; Burbridge, M.; Kotschy, A.; Walmsley, D.L. Targeting DYRK1A/B kinases to modulate p21-cyclin D1-p27 signalling and induce anti-tumour activity in a model of human glioblastoma. J. Cell. Mol. Med. 2021, 25, 10650–10662. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, N.; Zang, S.; Liu, H.; Wang, P.; Ji, C.; Sun, X.; Rishi, A. Tumor Suppressor DYRK1A Effects on Proliferation and Chemoresistance of AML Cells by Downregulating c-Myc. PLoS ONE 2014, 9, e98853. [Google Scholar] [CrossRef]
- Laham, A.J.; Saber-Ayad, M.; El-Awady, R. DYRK1A: A down syndrome-related dual protein kinase with a versatile role in tumorigenesis. Cell. Mol. Life Sci. 2021, 78, 603–619. [Google Scholar] [CrossRef]
- Youns, M.; Hegazy, W.A.H.; Ahmad, A. The Natural Flavonoid Fisetin Inhibits Cellular Proliferation of Hepatic, Colorectal, and Pancreatic Cancer Cells through Modulation of Multiple Signaling Pathways. PLoS ONE 2017, 12, e0169335. [Google Scholar] [CrossRef]
- Zhou, C.; Huang, Y.; Nie, S.; Zhou, S.; Gao, X.; Chen, G. Biological effects and mechanisms of fisetin in cancer: A promising anti-cancer agent. Eur. J. Med. Res. 2023, 28, 297. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H. Isorhamnetin induces ROS-dependent cycle arrest at G2/M phase and apoptosis in human hepatocarcinoma Hep3B cells. Gen. Physiol. Biophys. 2019, 38, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yin, J.; Rankin, G.O.; Chen, Y.C. Kaempferol Induces G2/M Cell Cycle Arrest via Checkpoint Kinase 2 and Promotes Apoptosis via Death Receptors in Human Ovarian Carcinoma A2780/CP70 Cells. Molecules 2018, 23, 1095. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-A.; Kim, J.-Y.; Lee, J.-Y.; Kang, C.-M.; Kwon, H.-J.; Yoo, Y.-D.; Kim, T.-W.; Lee, Y.-S.; Lee, S.-J. Induction of cell cycle arrest and apoptosis in human breast cancer cells by quercetin. Int. J. Oncol. 2001, 19, 837–844. [Google Scholar] [CrossRef]
- Zhang, X.-H.; Zou, Z.-Q.; Xu, C.-W.; Shen, Y.-Z.; Li, D. Myricetin induces G2/M phase arrest in HepG2 cells by inhibiting the activity of the cyclin B/Cdc2 complex. Mol. Med. Rep. 2011, 4, 273–277. [Google Scholar]
- Maharjan, S.; Kwon, Y.-S.; Lee, M.-G.; Lee, K.-S.; Nam, K.-S. Cell cycle arrest-mediated cell death by morin in MDA-MB-231 triple-negative breast cancer cells. Pharmacol. Rep. 2021, 73, 1315–1327. [Google Scholar] [CrossRef]
- Batra, P.; Sharma, A.K. Anti-cancer potential of flavonoids: Recent trends and future perspectives. 3 Biotech 2013, 3, 439–459. [Google Scholar] [CrossRef]
- Li, C.; Liu, X.G.; Lin, X.; Chen, X. Structure-activity relationship of 7 flavonoids on recombinant human protein kinase CK2 holoenzyme. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2009, 34, 20–26. [Google Scholar]
- Kondža, M.; Brizić, I.; Jokić, S. Flavonoids as CYP3A4 Inhibitors In Vitro. Biomedicines 2024, 12, 644. [Google Scholar] [CrossRef]
- Todorova, T.; Traykov, M.; Tadjer, A.; Velkov, Z. Structure of flavones and flavonols. Part I: Role of substituents on the planarity of the system. Comput. Theor. Chem. 2013, 1017, 85–90. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
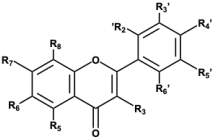 Flavones Flavones | 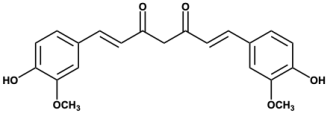 Curcumin Curcumin | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compounds | Flavone’s Substituents | Molecular Properties a | |||||||||
| R3 | R5 | R6 | R7 | R2′ | R3′ | R4′ | R5′ | LogP | HBA/HBD | PSA (Å2) | |
| Apigenin | -H | -OH | -H | -OH | -H | -OH | -H | -H | 3.04 | 5/3 | 87 |
| Dihydromyricetin b | -OH | -OH | -H | -OH | -H | -OH | -OH | -OH | 1.23 | 8/6 | 148 |
| 3,6-Dihydroxyflavone | -OH | -H | -OH | -H | -H | -H | -H | -H | 3.64 | 4/2 | 67 |
| 3,7-Dihydroxyflavone | -OH | -H | -H | -OH | -H | -H | -H | -H | 3.27 | 4/2 | 67 |
| Fisetin | -OH | -H | -H | -OH | -H | -OH | -OH | -H | 2.52 | 6/4 | 107 |
| 3-Hydroxyflavone | -OH | -H | -H | -H | -H | -H | -H | -H | 3.76 | 3/1 | 47 |
| Isorhamnetin | -OH | -OH | -H | -OH | -H | -OCH3 | -OH | -H | 1.76 | 7/4 | 116 |
| Kaempferide | -OH | -OH | -H | -OH | -H | -H | -OCH3 | -H | 2.74 | 6/3 | 96 |
| Kaempferol | -OH | -OH | -H | -OH | -H | -H | -OH | -H | 2.05 | 6/4 | 107 |
| Luteolin | -H | -OH | -H | -OH | -H | -OH | -OH | -H | 2.40 | 6/4 | 107 |
| Morin | -OH | -OH | -H | -OH | -OH | -H | -OH | -H | 1.61 | 7/5 | 127 |
| Myricetin | -OH | -OH | -H | -OH | -H | -OH | -OH | -OH | 2.11 | 8/6 | 148 |
| Naringenin b | -H | -OH | -H | -OH | -H | -H | -OH | -H | 3.19 | 5/3 | 87 |
| Quercetin | -OH | -OH | -H | -OH | -H | -OH | -OH | -H | 2.08 | 7/5 | 127 |
| Curcumin | - | - | - | - | - | - | - | - | 2.92 | 6/2 | 93 |
| Order | Compound | IC50 (nM) | 95% CI | Calculated Affinity (kcal/mol) | |
|---|---|---|---|---|---|
| LogIC50 | IC50 | ||||
| 1 | Fisetin | 149.5 | 2.012 to 2.340 | 102.8 to 219.0 | −9.0 |
| 2 | Kaempferol | 296.3 | 2.312 to 2.633 | 205.1 to 429.1 | −8.3 |
| 3 | Isorhamnetin | 418.0 | 2.421 to 2.823 | 263.9 to 664.8 | −8.5 |
| 4 | Morin | 478.4 | 2.445 to 2.921 | 278.7 to 833.1 | −8.6 |
| 5 | Myricetin | 633.2 | 2.525 to 3.097 | 335.2 to 1250 | −8.3 |
| 6 | Quercetin | 737.9 | 2.683 to 3.058 | 482.2 to 1142 | −8.3 |
| 7 | Luteolin | 797.8 | 2.754 to 3.055 | 568.2 to 1136 | −8.3 |
| 8 | Apigenin | 1019 | 2.386 to 3.798 | 243.3 to 2681 | −8.8 |
| 9 | Curcumin | 2351 | 3.238 to 3.516 | 1730 to 3283 | −7.9 |
| 10 | Kaempferide | 3517 | 2.806 to --a | 640.5 to --a | −8.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, J.; Zhou, Q.; Guo, B.; Jia, Z. Discovery of Dietary Plant Flavonols as Novel Potent Inhibitors Targeting DYRK1A Kinase. Biomolecules 2025, 15, 934. https://doi.org/10.3390/biom15070934
Jin J, Zhou Q, Guo B, Jia Z. Discovery of Dietary Plant Flavonols as Novel Potent Inhibitors Targeting DYRK1A Kinase. Biomolecules. 2025; 15(7):934. https://doi.org/10.3390/biom15070934
Chicago/Turabian StyleJin, Jin, Qihong Zhou, Bin Guo, and Zongchao Jia. 2025. "Discovery of Dietary Plant Flavonols as Novel Potent Inhibitors Targeting DYRK1A Kinase" Biomolecules 15, no. 7: 934. https://doi.org/10.3390/biom15070934
APA StyleJin, J., Zhou, Q., Guo, B., & Jia, Z. (2025). Discovery of Dietary Plant Flavonols as Novel Potent Inhibitors Targeting DYRK1A Kinase. Biomolecules, 15(7), 934. https://doi.org/10.3390/biom15070934