
Targeting Sodium Transport Reveals CHP1 Downregulation as a Novel Molecular Feature of Malignant Progression in Clear Cell Renal Cell Carcinoma: Insights from Integrated Multi-Omics Analyses

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
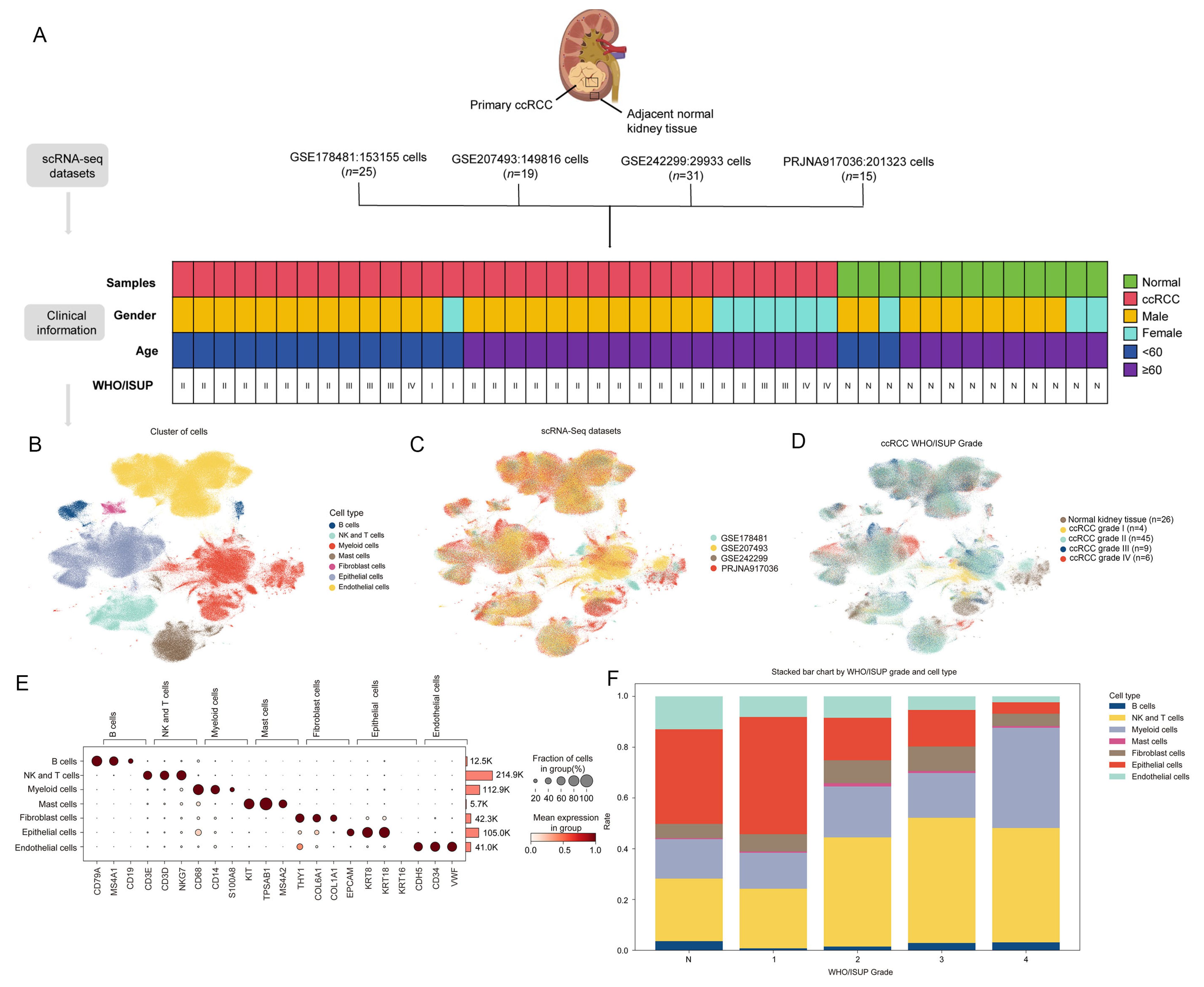
2.1. Collection of Single-Cell Transcriptomic Data
2.2. Preprocessing of scRNA-Seq Data
2.3. Cell Subtype Annotation
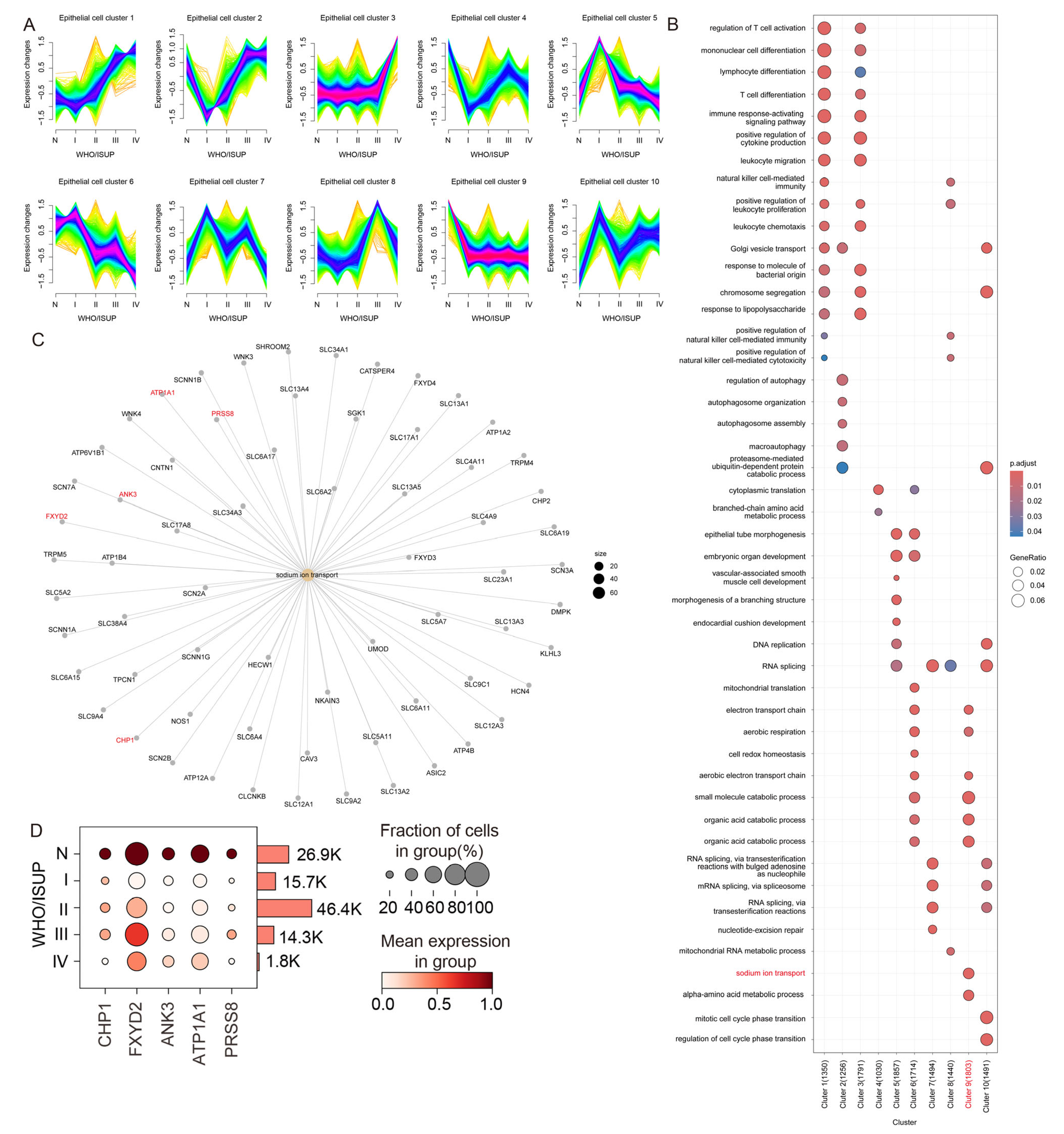
2.4. Mfuzz-Based Expression Pattern Clustering and Functional Enrichment Analysis
2.5. Chromosomal Copy Number Variation (CNV) Analysis
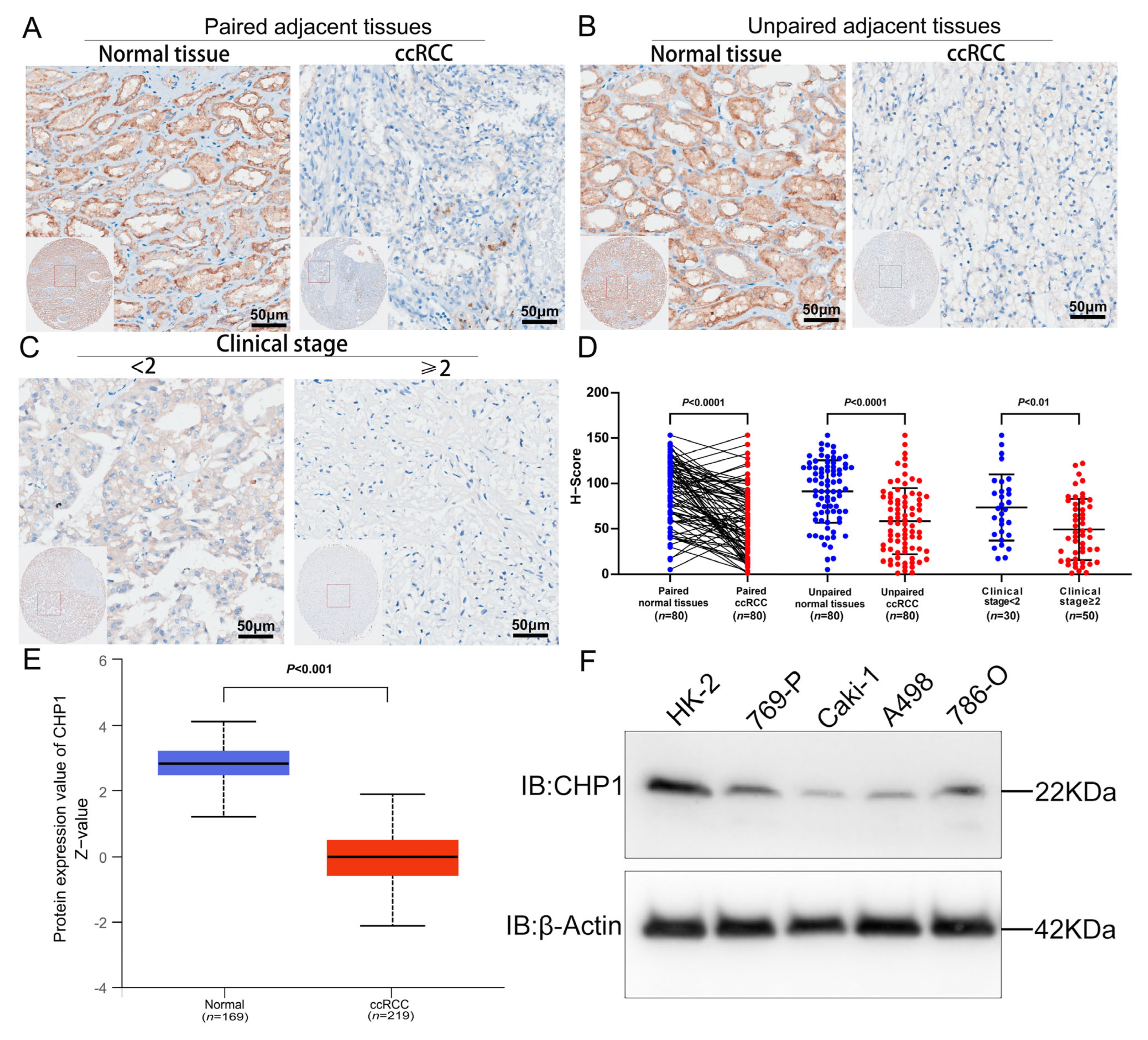
2.6. Public Database Analysis of CHP1 Expression
2.7. Immunohistochemistry
2.8. Cell Culture and Western Blotting
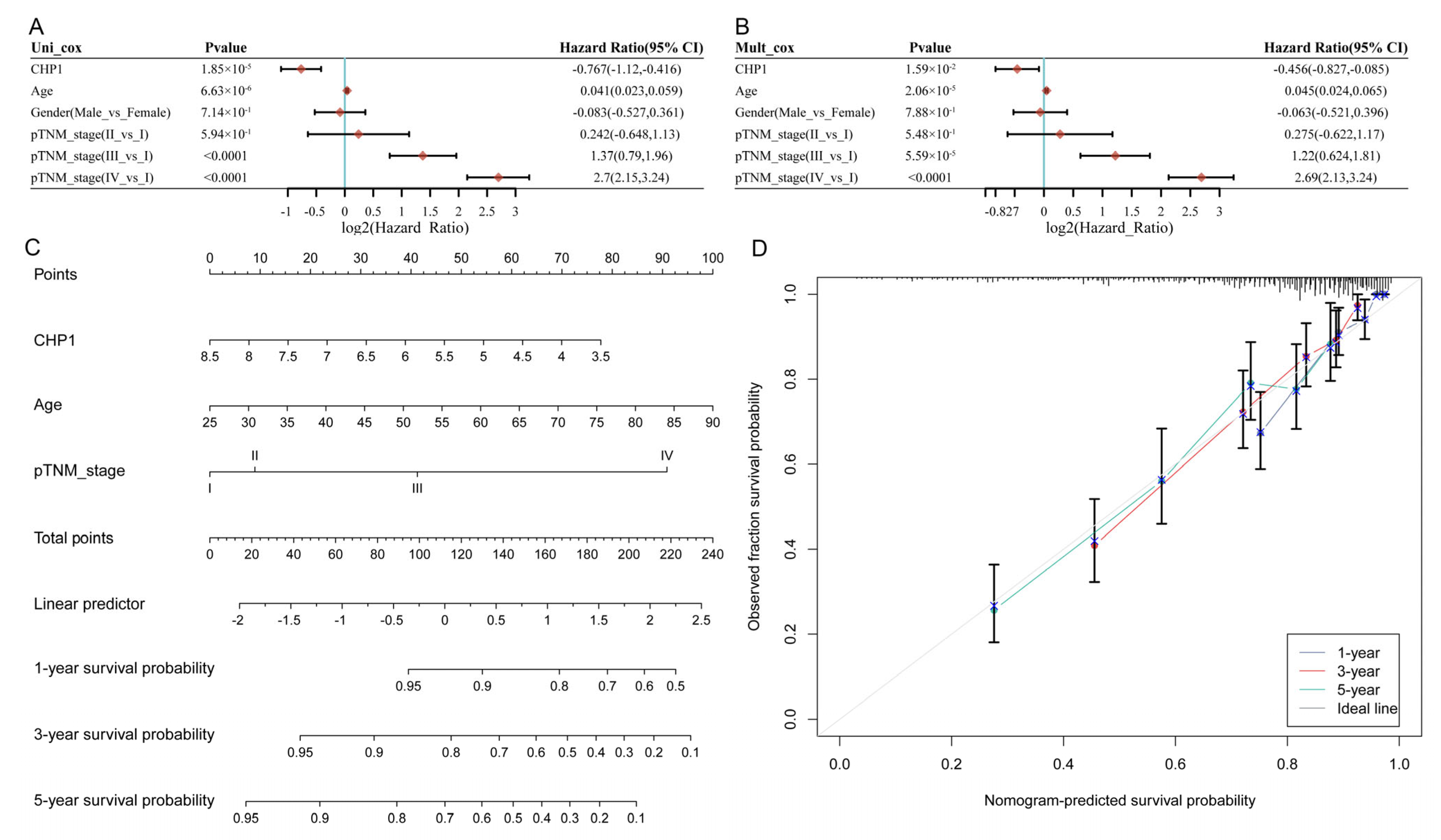
2.9. Prognostic Analysis of CHP1 in ccRCC Patients
2.10. Pseudotime Trajectory Analysis
2.11. CNV Burden Analysis
2.12. Cell–Cell Communication Analysis
2.13. Spatial Co-Localization Analysis
2.14. Differential Gene Expression Analysis
2.15. Protein–Protein Interaction Analysis
2.16. Plasmids and Cell Transfection
2.17. Co-Immunoprecipitation Analysis
2.18. Transcription Factor Prediction
2.19. Dual-Luciferase Reporter Gene Assay
2.20. Virtual Screening and Molecular Docking of FDA-Approved Drugs
2.21. Molecular Dynamics Simulation
2.22. Statistical Analysis
3. Results
3.1. Cellular Atlas of ccRCC
3.2. Gene Expression Profiling of Epithelial Subpopulations During ccRCC Progression
3.3. Sodium Ion Transport-Related Genes Are Significantly Downregulated in Malignant Tumor Cells
3.4. Reduced CHP1 Protein Expression in ccRCC Tissues and Cell Lines
3.5. CHP1 Serves as a Favorable Prognostic Indicator in KIRC
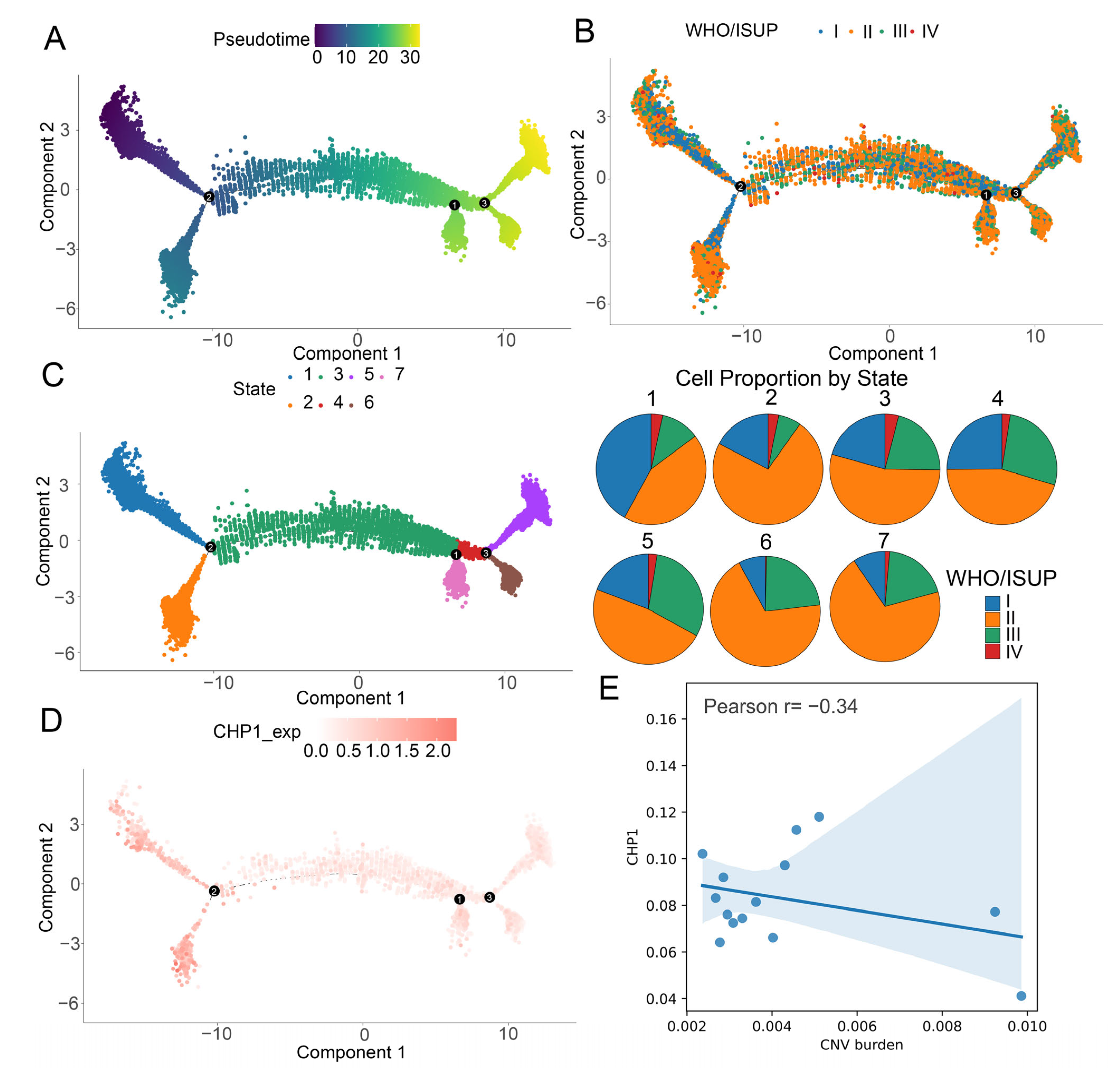
3.6. Gradual Decrease in CHP1 Expression Along the Developmental Trajectory of Tumor Cells
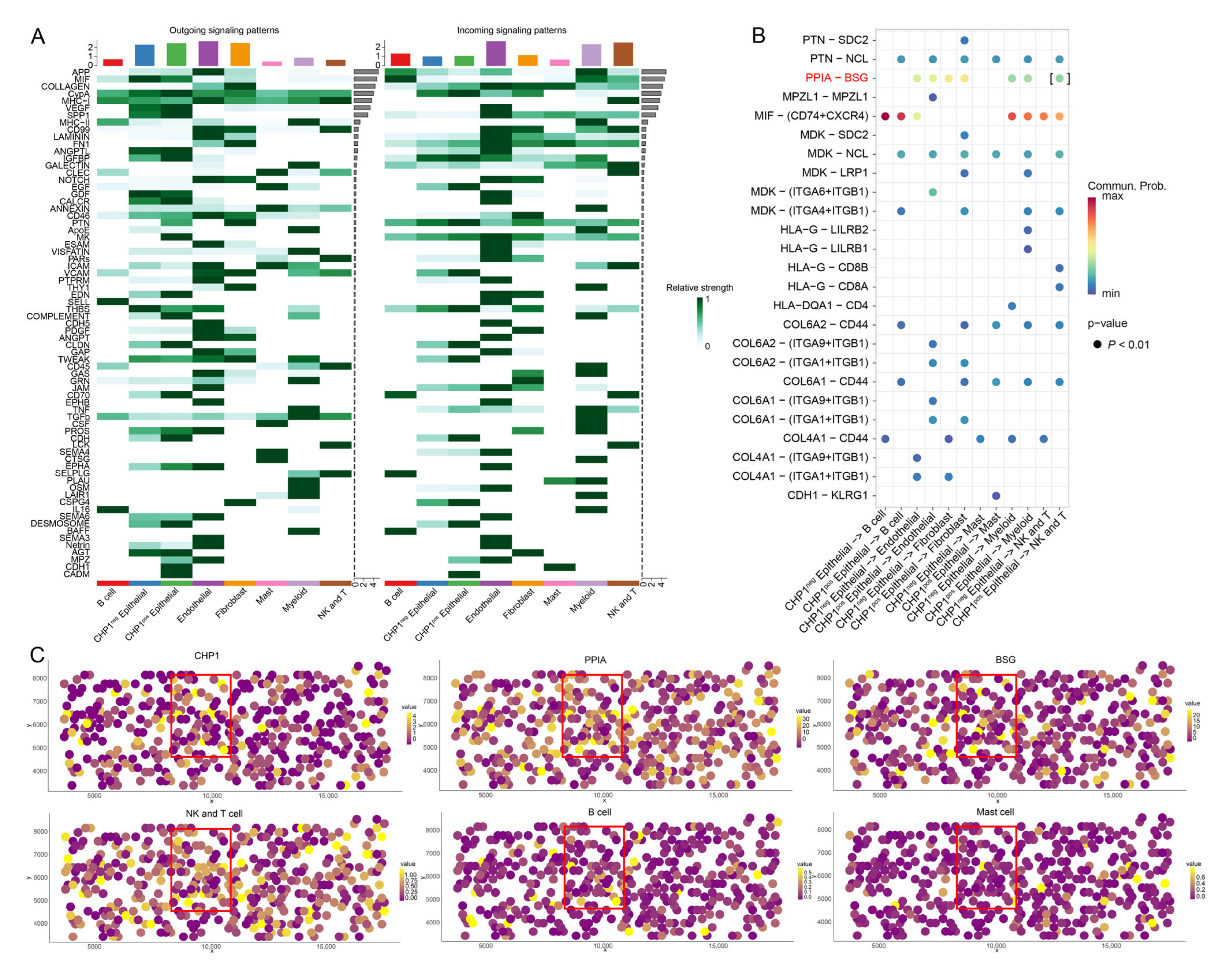
3.7. Impact of CHP1 Expression on Cell–Cell Communication
3.8. CHP1 Expression Affects Amino Acid Transport in Tumor Cells
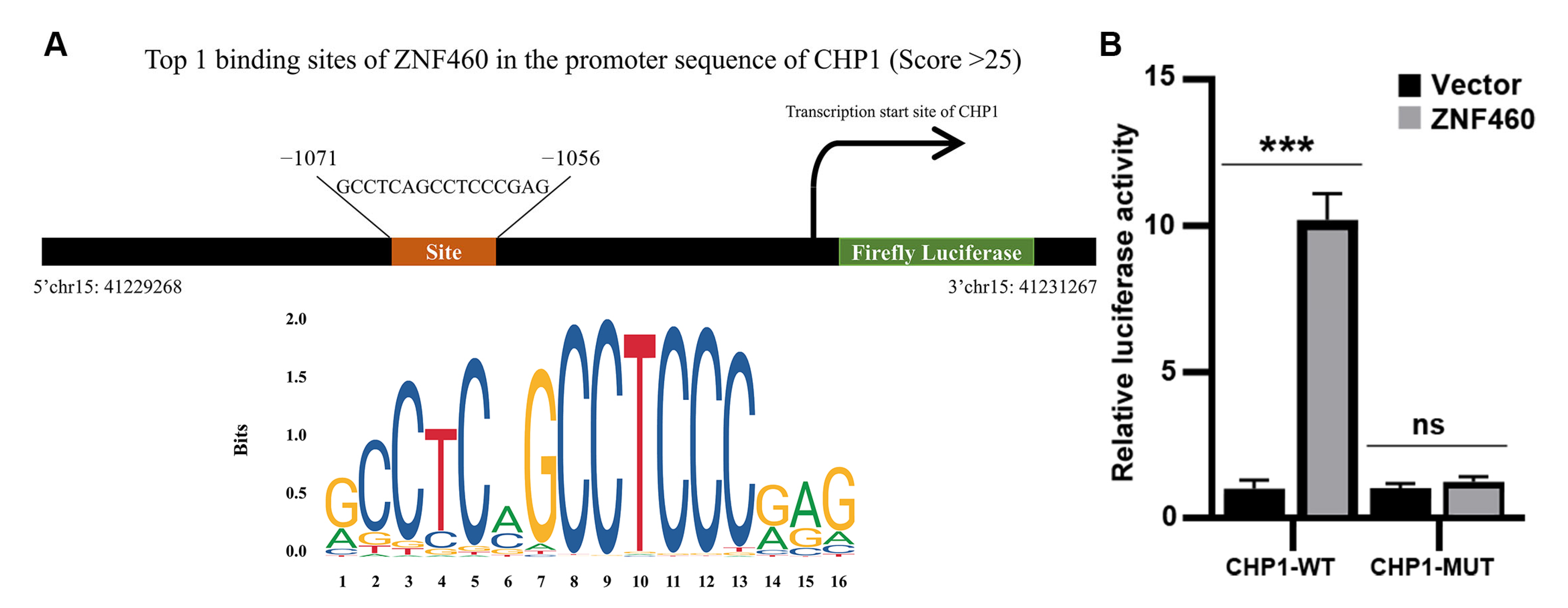
3.9. Prediction of Transcription Factors Involved in Regulating CHP1 Expression
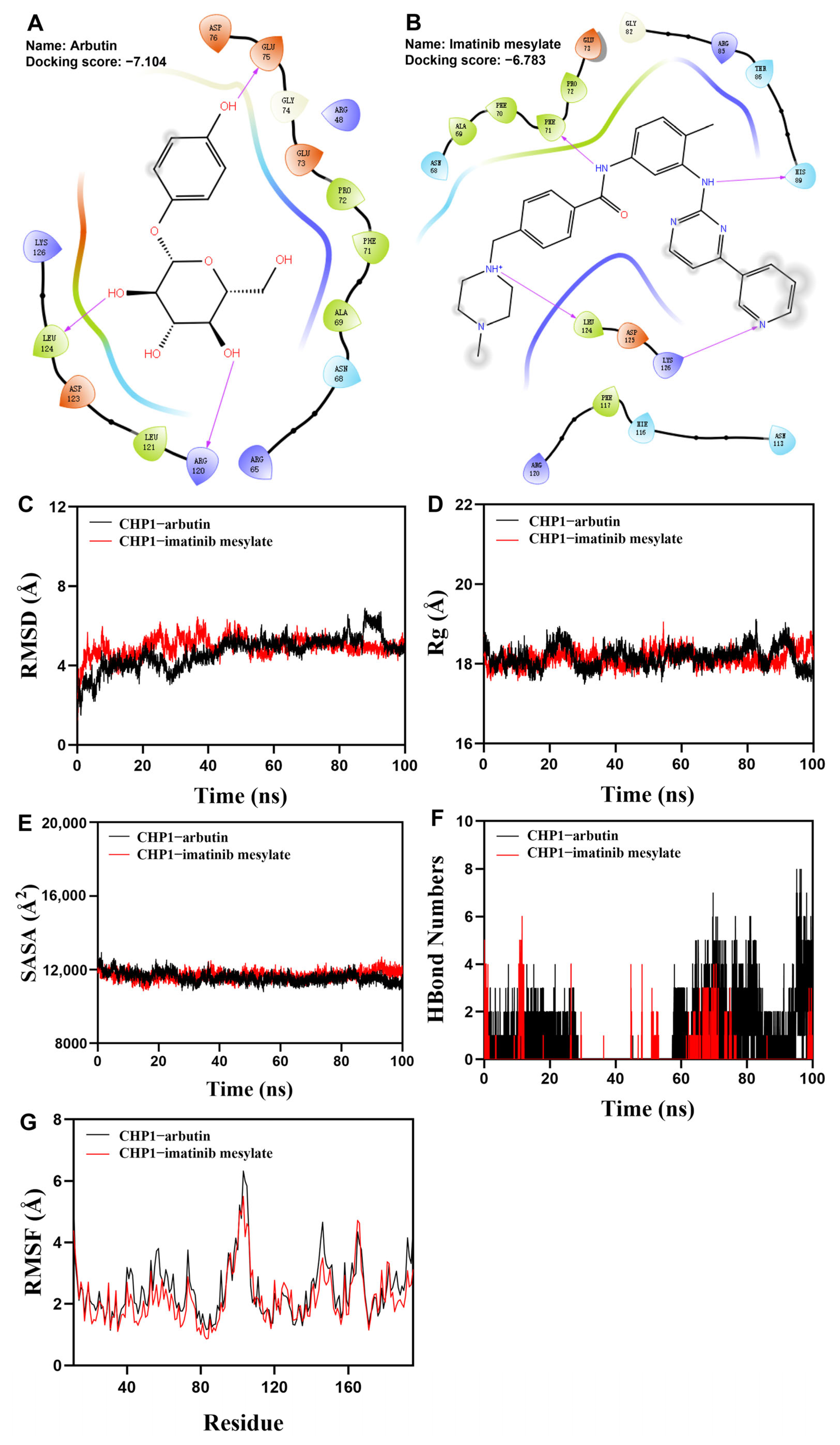
3.10. Virtual Drug Screening Identifies Two Compounds Binding to CHP1 Protein
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Chen, S.; Cao, Z.; Prettner, K.; Kuhn, M.; Yang, J.; Jiao, L.; Wang, Z.; Li, W.; Geldsetzer, P.; Barnighausen, T.; et al. Estimates and Projections of the Global Economic Cost of 29 Cancers in 204 Countries and Territories From 2020 to 2050. JAMA Oncol. 2023, 9, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, D.; Eriksson, P.; Krawczyk, K.; Nilsson, H.; Hansson, J.; Veerla, S.; Sjolund, J.; Hoglund, M.; Johansson, M.E.; Axelson, H. Cell-Type-Specific Gene Programs of the Normal Human Nephron Define Kidney Cancer Subtypes. Cell Rep. 2017, 20, 1476–1489. [Google Scholar] [CrossRef]
- Moch, H.; Amin, M.B.; Berney, D.M.; Comperat, E.M.; Gill, A.J.; Hartmann, A.; Menon, S.; Raspollini, M.R.; Rubin, M.A.; Srigley, J.R.; et al. The 2022 World Health Organization Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours. Eur. Urol. 2022, 82, 458–468. [Google Scholar] [CrossRef]
- Jonasch, E.; Walker, C.L.; Rathmell, W.K. Clear cell renal cell carcinoma ontogeny and mechanisms of lethality. Nat. Rev. Nephrol. 2021, 17, 245–261. [Google Scholar] [CrossRef]
- Zhu, Z.; Jin, Y.; Zhou, J.; Chen, F.; Chen, M.; Gao, Z.; Hu, L.; Xuan, J.; Li, X.; Song, Z.; et al. PD1/PD-L1 blockade in clear cell renal cell carcinoma: Mechanistic insights, clinical efficacy, and future perspectives. Mol. Cancer 2024, 23, 146. [Google Scholar] [CrossRef]
- Hwang, A.; Mehra, V.; Chhetri, J.; Ali, S.; Tran, M.; Roddie, C. Current Treatment Options for Renal Cell Carcinoma: Focus on Cell-Based Immunotherapy. Cancers 2024, 16, 1209. [Google Scholar] [CrossRef]
- Miao, D.; Margolis, C.A.; Gao, W.; Voss, M.H.; Li, W.; Martini, D.J.; Norton, C.; Bosse, D.; Wankowicz, S.M.; Cullen, D.; et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 2018, 359, 801–806. [Google Scholar] [CrossRef]
- Pang, T.; Su, X.; Wakabayashi, S.; Shigekawa, M. Calcineurin homologous protein as an essential cofactor for Na+/H+ exchangers. J. Biol. Chem. 2001, 276, 17367–17372. [Google Scholar] [CrossRef]
- Di Sole, F.; Vadnagara, K.; Moe, O.W.; Babich, V. Calcineurin homologous protein: A multifunctional Ca2+-binding protein family. Am. J. Physiol. Ren. Physiol. 2012, 303, F165–F179. [Google Scholar] [CrossRef]
- Liu, Y.; Zaun, H.C.; Orlowski, J.; Ackerman, S.L. CHP1-mediated NHE1 biosynthetic maturation is required for Purkinje cell axon homeostasis. J. Neurosci. 2013, 33, 12656–12669. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.G.; Nicholson Puthenveedu, S.; Shen, Y.; La, K.; Ozlu, C.; Wang, T.; Klompstra, D.; Gultekin, Y.; Chi, J.; Fidelin, J.; et al. CHP1 Regulates Compartmentalized Glycerolipid Synthesis by Activating GPAT4. Mol. Cell 2019, 74, 45–58.e7. [Google Scholar] [CrossRef] [PubMed]
- Janzen, E.; Mendoza-Ferreira, N.; Hosseinibarkooie, S.; Schneider, S.; Hupperich, K.; Tschanz, T.; Grysko, V.; Riessland, M.; Hammerschmidt, M.; Rigo, F.; et al. CHP1 reduction ameliorates spinal muscular atrophy pathology by restoring calcineurin activity and endocytosis. Brain 2018, 141, 2343–2361. [Google Scholar] [CrossRef] [PubMed]
- Pang, T.; Wakabayashi, S.; Shigekawa, M. Expression of calcineurin B homologous protein 2 protects serum deprivation-induced cell death by serum-independent activation of Na+/H+ exchanger. J. Biol. Chem. 2002, 277, 43771–43777. [Google Scholar] [CrossRef]
- Gutierrez-Ford, C.; Levay, K.; Gomes, A.V.; Perera, E.M.; Som, T.; Kim, Y.M.; Benovic, J.L.; Berkovitz, G.D.; Slepak, V.Z. Characterization of tescalcin, a novel EF-hand protein with a single Ca2+-binding site: Metal-binding properties, localization in tissues and cells, and effect on calcineurin. Biochemistry 2003, 42, 14553–14565. [Google Scholar] [CrossRef]
- Tirosh, I.; Suva, M.L. Cancer cell states: Lessons from ten years of single-cell RNA-sequencing of human tumors. Cancer Cell 2024, 42, 1497–1506. [Google Scholar] [CrossRef]
- Alchahin, A.M.; Mei, S.; Tsea, I.; Hirz, T.; Kfoury, Y.; Dahl, D.; Wu, C.L.; Subtelny, A.O.; Wu, S.; Scadden, D.T.; et al. A transcriptional metastatic signature predicts survival in clear cell renal cell carcinoma. Nat. Commun. 2022, 13, 5747. [Google Scholar] [CrossRef]
- Yu, Z.; Lv, Y.; Su, C.; Lu, W.; Zhang, R.; Li, J.; Guo, B.; Yan, H.; Liu, D.; Yang, Z.; et al. Integrative Single-Cell Analysis Reveals Transcriptional and Epigenetic Regulatory Features of Clear Cell Renal Cell Carcinoma. Cancer Res. 2023, 83, 700–719. [Google Scholar] [CrossRef]
- Zvirblyte, J.; Nainys, J.; Juzenas, S.; Goda, K.; Kubiliute, R.; Dasevicius, D.; Kincius, M.; Ulys, A.; Jarmalaite, S.; Mazutis, L. Single-cell transcriptional profiling of clear cell renal cell carcinoma reveals a tumor-associated endothelial tip cell phenotype. Commun. Biol. 2024, 7, 780. [Google Scholar] [CrossRef]
- Zhang, C.; Du, Z.; Gao, Y.; Lim, K.S.; Zhou, W.; Huang, H.; He, H.; Xiao, J.; Xu, D.; Li, Q. Methionine secreted by tumor-associated pericytes supports cancer stem cells in clear cell renal carcinoma. Cell Metab. 2024, 36, 778–792.e710. [Google Scholar] [CrossRef]
- Van de Sande, B.; Lee, J.S.; Mutasa-Gottgens, E.; Naughton, B.; Bacon, W.; Manning, J.; Wang, Y.; Pollard, J.; Mendez, M.; Hill, J.; et al. Applications of single-cell RNA sequencing in drug discovery and development. Nat. Rev. Drug Discov. 2023, 22, 496–520. [Google Scholar] [CrossRef]
- Meylan, M.; Petitprez, F.; Becht, E.; Bougouin, A.; Pupier, G.; Calvez, A.; Giglioli, I.; Verkarre, V.; Lacroix, G.; Verneau, J.; et al. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity 2022, 55, 527–541.e5. [Google Scholar] [CrossRef]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef] [PubMed]
- Wolock, S.L.; Lopez, R.; Klein, A.M. Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst. 2019, 8, 281–291.e289. [Google Scholar] [CrossRef] [PubMed]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.R.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Healy, J.; McInnes, L. Uniform manifold approximation and projection. Nat. Rev. Methods Primers 2024, 4, 82. [Google Scholar] [CrossRef]
- Traag, V.A.; Waltman, L.; van Eck, N.J. From Louvain to Leiden: Guaranteeing well-connected communities. Sci. Rep. 2019, 9, 5233. [Google Scholar] [CrossRef]
- Hu, C.; Li, T.; Xu, Y.; Zhang, X.; Li, F.; Bai, J.; Chen, J.; Jiang, W.; Yang, K.; Ou, Q.; et al. CellMarker 2.0: An updated database of manually curated cell markers in human/mouse and web tools based on scRNA-seq data. Nucleic Acids Res. 2023, 51, D870–D876. [Google Scholar] [CrossRef]
- Kumar, L.; Futschik, M.E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5–7. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef] [PubMed]
- McClelland, R.A.; Finlay, P.; Walker, K.J.; Nicholson, D.; Robertson, J.F.; Blamey, R.W.; Nicholson, R.I. Automated quantitation of immunocytochemically localized estrogen receptors in human breast cancer. Cancer Res. 1990, 50, 3545–3550. [Google Scholar] [PubMed]
- Qiu, X.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.A.; Trapnell, C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 2017, 14, 979–982. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Plikus, M.V.; Nie, Q. CellChat for systematic analysis of cell–cell communication from single-cell transcriptomics. Nat. Protoc. 2025, 20, 180–219. [Google Scholar] [CrossRef]
- Biancalani, T.; Scalia, G.; Buffoni, L.; Avasthi, R.; Lu, Z.; Sanger, A.; Tokcan, N.; Vanderburg, C.R.; Segerstolpe, A.; Zhang, M.; et al. Deep learning and alignment of spatially resolved single-cell transcriptomes with Tangram. Nat. Methods 2021, 18, 1352–1362. [Google Scholar] [CrossRef]
- Pham, D.; Tan, X.; Balderson, B.; Xu, J.; Grice, L.F.; Yoon, S.; Willis, E.F.; Tran, M.; Lam, P.Y.; Raghubar, A.; et al. Robust mapping of spatiotemporal trajectories and cell-cell interactions in healthy and diseased tissues. Nat. Commun. 2023, 14, 7739. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef]
- Kandasamy, P.; Gyimesi, G.; Kanai, Y.; Hediger, M.A. Amino acid transporters revisited: New views in health and disease. Trends Biochem. Sci. 2018, 43, 752–789. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Yan, Y.; Tao, H.; He, J.; Huang, S.Y. The HDOCK server for integrated protein-protein docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef]
- Mooers, B.H.M. Shortcuts for faster image creation in PyMOL. Protein Sci. 2020, 29, 268–276. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Rauluseviciute, I.; Riudavets-Puig, R.; Blanc-Mathieu, R.; Castro-Mondragon, J.A.; Ferenc, K.; Kumar, V.; Lemma, R.B.; Lucas, J.; Cheneby, J.; Baranasic, D.; et al. JASPAR 2024: 20th anniversary of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2024, 52, D174–D182. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Dong, Y.; Gao, Y.; Ilie, A.; Kim, D.; Boucher, A.; Li, B.; Zhang, X.C.; Orlowski, J.; Zhao, Y. Structure and mechanism of the human NHE1-CHP1 complex. Nat. Commun. 2021, 12, 3474. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, S.A.; Ball, W.J., Jr.; Bander, N.H.; Liu, H.; Pardee, J.D.; Rajasekaran, A.K. Reduced expression of beta-subunit of Na,K-ATPase in human clear-cell renal cell carcinoma. J. Urol. 1999, 162, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Selvakumar, P.; Owens, T.A.; David, J.M.; Petrelli, N.J.; Christensen, B.C.; Lakshmikuttyamma, A.; Rajasekaran, A.K. Epigenetic silencing of Na,K-ATPase beta 1 subunit gene ATP1B1 by methylation in clear cell renal cell carcinoma. Epigenetics 2014, 9, 579–586. [Google Scholar] [CrossRef]
- Krishtal, O. The ASICs: Signaling molecules? Modulators? Trends Neurosci. 2003, 26, 477–483. [Google Scholar] [CrossRef]
- Li, Y.; Xu, G.; Huang, K.; Wang, J.; Zhang, J.; Liu, J.; Wang, Z.; Chen, G. Alteration of ASIC1 expression in clear cell renal cell carcinoma. OncoTargets Ther. 2015, 8, 2121–2127. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, Y.; Li, L.; Yang, W.; Xu, Y.; Zhou, J.; Ma, K.; Zhang, K.; Zhuang, H.; Gong, Y.; et al. Downregulation of FXYD2 Is Associated with Poor Prognosis and Increased Regulatory T Cell Infiltration in Clear Cell Renal Cell Carcinoma. J. Immunol. Res. 2022, 2022, 4946197. [Google Scholar] [CrossRef]
- Moore, P.J.; Tarran, R. The epithelial sodium channel (ENaC) as a therapeutic target for cystic fibrosis lung disease. Expert. Opin. Ther. Targets 2018, 22, 687–701. [Google Scholar] [CrossRef]
- Zheng, Q.; Wang, Y.; Zhao, R.; Han, P.; Zhao, J.; Li, L.; Zhou, X.; Li, P.; Mo, Y.; Pan, X.; et al. Inactivation of epithelial sodium ion channel molecules serves as effective diagnostic biomarkers in clear cell renal cell carcinoma. Genes. Genom. 2023, 45, 855–866. [Google Scholar] [CrossRef]
- Sweadner, K.J.; Wetzel, R.K.; Arystarkhova, E. Genomic organization of the human FXYD2 gene encoding the gamma subunit of the Na,K-ATPase. Biochem. Biophys. Res. Commun. 2000, 279, 196–201. [Google Scholar] [CrossRef]
- Geering, K. Function of FXYD proteins, regulators of Na, K-ATPase. J. Bioenerg. Biomembr. 2005, 37, 387–392. [Google Scholar] [CrossRef]
- Cordeiro, B.M.; Leite Fontes, C.F.; Meyer-Fernandes, J.R. Molecular Basis of Na, K-ATPase Regulation of Diseases: Hormone and FXYD2 Interactions. Int. J. Mol. Sci. 2024, 25, 13398. [Google Scholar] [CrossRef]
- Somsuan, K.; Aluksanasuwan, S. Bioinformatic analyses reveal the prognostic significance and potential role of ankyrin 3 (ANK3) in kidney renal clear cell carcinoma. Genom. Inf. Inform. 2023, 21, e22. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Ko, J.Y.; Kong, H.K.; Lee, M.; Chung, W.; Lim, S.; Son, D.; Oh, S.; Park, J.W.; Kim, D.Y.; et al. Hypomethylation of ATP1A1 Is Associated with Poor Prognosis and Cancer Progression in Triple-Negative Breast Cancer. Cancers 2024, 16, 1666. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Guo, Y.; Yang, Y.; Wei, X.; Zhang, S.; Zhang, Y.; Li, K.; Yuan, M.; Guo, D.; Macias, V.; et al. PRSS8 suppresses colorectal carcinogenesis and metastasis. Oncogene 2019, 38, 497–517. [Google Scholar] [CrossRef] [PubMed]
- Xi, D.; Wang, J.; Yang, Y.; Ji, F.; Li, C.; Yan, X. A novel natural killer-related signature to effectively predict prognosis in hepatocellular carcinoma. BMC Med. Genom. 2023, 16, 211. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, J.; Zhu, R.; Huang, G.; Zeng, J.; Yu, H.; He, Z.; Han, C. Integrating TCGA and Single-Cell Sequencing Data for Hepatocellular Carcinoma: A Novel Glycosylation (GLY)/Tumor Microenvironment (TME) Classifier to Predict Prognosis and Immunotherapy Response. Metabolites 2024, 14, 51. [Google Scholar] [CrossRef]
- Liao, E.C.; Law, C.H.; Chen, H.Y.; Wei, Y.S.; Tsai, Y.T.; Lin, L.H.; Lin, M.W.; Wang, Y.S.; Chou, H.C.; Chan, H.L. PPIA enhances cell growth and metastasis through CD147 in oral cancer. Arch. Biochem. Biophys. 2025, 765, 110328. [Google Scholar] [CrossRef]
- Nyalali, A.M.K.; Leonard, A.U.; Xu, Y.; Li, H.; Zhou, J.; Zhang, X.; Rugambwa, T.K.; Shi, X.; Li, F. CD147: An integral and potential molecule to abrogate hallmarks of cancer. Front. Oncol. 2023, 13, 1238051. [Google Scholar] [CrossRef]
- Zhou, F.F.; Xie, W.; Chen, S.Q.; Wang, X.K.; Liu, Q.; Pan, X.K.; Su, F.; Feng, M.H. SLC38A1 promotes proliferation and migration of human colorectal cancer cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2017, 37, 30–36. [Google Scholar] [CrossRef]
- Liu, L.; Su, S.; Ye, D.; Yu, Z.; Lu, W.; Li, X. Long non-coding RNA OGFRP1 regulates cell proliferation and ferroptosis by miR-299-3p/SLC38A1 axis in lung cancer. Anticancer. Drugs 2022, 33, 826–839. [Google Scholar] [CrossRef]
- Feng, H.G.; Wu, C.X.; Zhong, G.C.; Gong, J.P.; Miao, C.M.; Xiong, B. Integrative analysis reveals that SLC38A1 promotes hepatocellular carcinoma development via PI3K/AKT/mTOR signaling via glutamine mediated energy metabolism. J. Cancer Res. Clin. Oncol. 2023, 149, 15879–15898. [Google Scholar] [CrossRef]
- Wang, K.; Cao, F.; Fang, W.; Hu, Y.; Chen, Y.; Ding, H.; Yu, G. Activation of SNAT1/SLC38A1 in human breast cancer: Correlation with p-Akt overexpression. BMC Cancer 2013, 13, 343. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Z.; Li, Z.; Yang, L.; Liu, J.; Liu, Y.; Liu, Y.; Hou, Y.; Mei, M.; Huang, Y. CENPA promotes glutamine metabolism and tumor progression by up-regulating SLC38A1 in endometrial cancer. Cell. Signal. 2024, 117, 111110. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Xu, L.; Wang, Y.; Jiang, Q.; Liu, Z.; Zhang, J.; Zhou, Q.; Zeng, H.; Tong, S.; Wang, T.; et al. Tumor-associated Macrophage-derived Interleukin-23 Interlinks Kidney Cancer Glutamine Addiction with Immune Evasion. Eur. Urol. 2019, 75, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Nahar, L.; Al-Groshi, A.; Kumar, A.; Sarker, S.D. Arbutin: Occurrence in Plants, and Its Potential as an Anticancer Agent. Molecules 2022, 27, 8786. [Google Scholar] [CrossRef]
- Savage, D.G.; Antman, K.H. Imatinib mesylate--a new oral targeted therapy. N. Engl. J. Med. 2002, 346, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Sliwinska-Hill, U. Interaction of imatinib mesylate with human serum transferrin: The comparative spectroscopic studies. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2017, 173, 468–475. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Zhu, R.-T.; Chen, J.-R.; Liu, X.-M.; Huang, G.-L.; Zeng, J.-C.; Yu, H.-B.; Liu, X.; Han, C.-F. Targeting Sodium Transport Reveals CHP1 Downregulation as a Novel Molecular Feature of Malignant Progression in Clear Cell Renal Cell Carcinoma: Insights from Integrated Multi-Omics Analyses. Biomolecules 2025, 15, 1019. https://doi.org/10.3390/biom15071019
Wu Y, Zhu R-T, Chen J-R, Liu X-M, Huang G-L, Zeng J-C, Yu H-B, Liu X, Han C-F. Targeting Sodium Transport Reveals CHP1 Downregulation as a Novel Molecular Feature of Malignant Progression in Clear Cell Renal Cell Carcinoma: Insights from Integrated Multi-Omics Analyses. Biomolecules. 2025; 15(7):1019. https://doi.org/10.3390/biom15071019
Chicago/Turabian StyleWu, Yun, Ri-Ting Zhu, Jia-Ru Chen, Xiao-Min Liu, Guo-Liang Huang, Jin-Cheng Zeng, Hong-Bing Yu, Xin Liu, and Cui-Fang Han. 2025. "Targeting Sodium Transport Reveals CHP1 Downregulation as a Novel Molecular Feature of Malignant Progression in Clear Cell Renal Cell Carcinoma: Insights from Integrated Multi-Omics Analyses" Biomolecules 15, no. 7: 1019. https://doi.org/10.3390/biom15071019
APA StyleWu, Y., Zhu, R.-T., Chen, J.-R., Liu, X.-M., Huang, G.-L., Zeng, J.-C., Yu, H.-B., Liu, X., & Han, C.-F. (2025). Targeting Sodium Transport Reveals CHP1 Downregulation as a Novel Molecular Feature of Malignant Progression in Clear Cell Renal Cell Carcinoma: Insights from Integrated Multi-Omics Analyses. Biomolecules, 15(7), 1019. https://doi.org/10.3390/biom15071019