
Role of Oral Bacteria in Mediating Gemcitabine Resistance in Pancreatic Cancer

 ,
,  , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Bacterial Strains, Plasmids, and Growth Conditions
2.3. CDD Protein Alignment and Phylogenetic Analysis
2.4. Construction of E. coli cdd Mutant and Complement Strains
2.5. Bacteria and Gemcitabine Co-Incubation
2.6. PDAC Cell Drug Inhibition Assay
2.7. Quantifications of dFdC/dFdU in Bacteria–Gemcitabine Filtrates
2.8. Statistical Analysis
3. Results
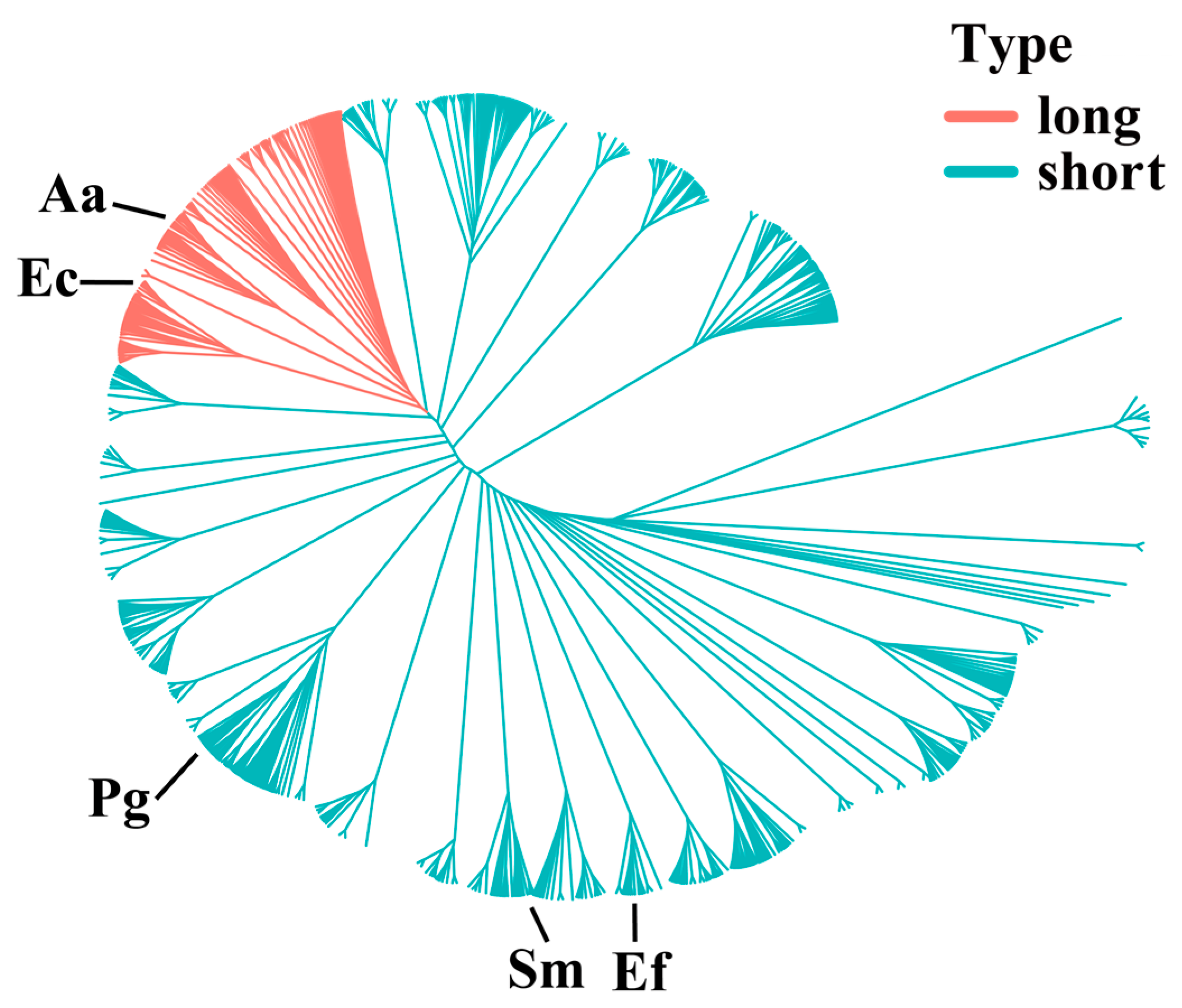
3.1. Alignment of CDD Protein Sequences
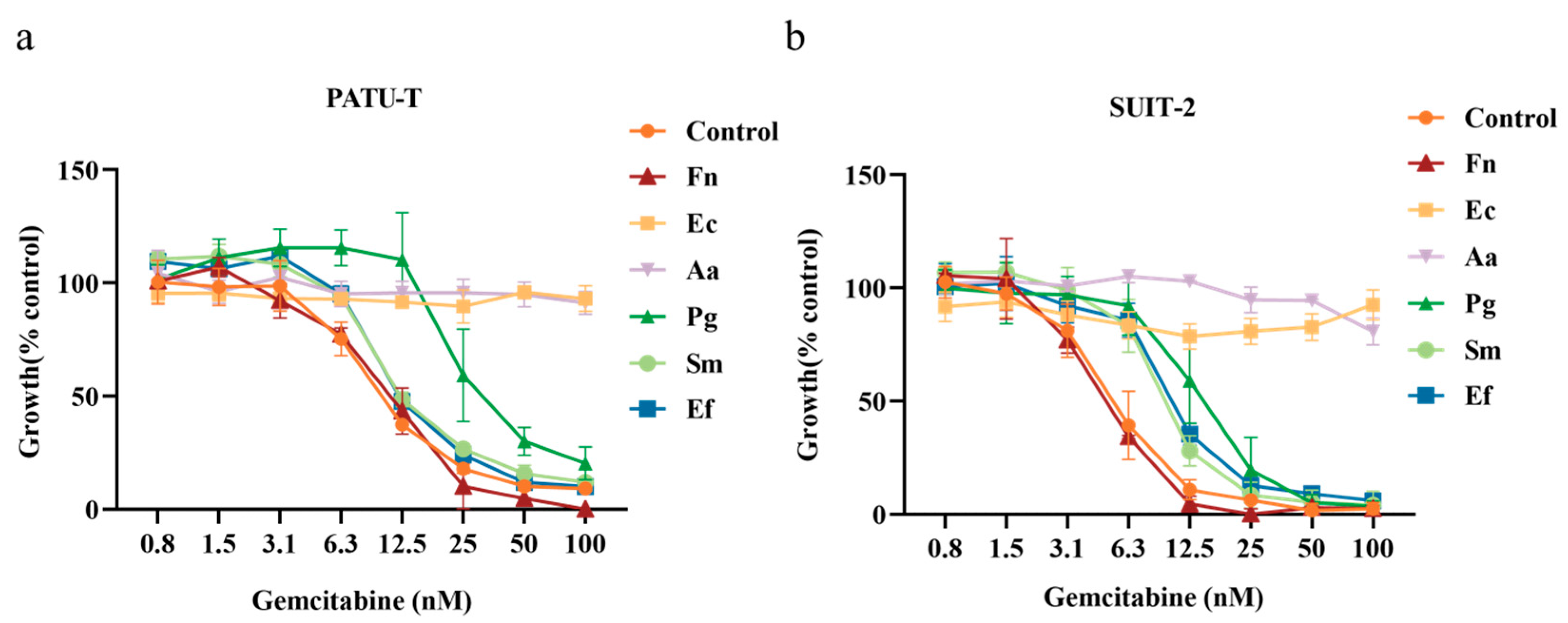
3.2. Growth Inhibition of PDAC Cells by Bacteria–Gemcitabine Filtrates
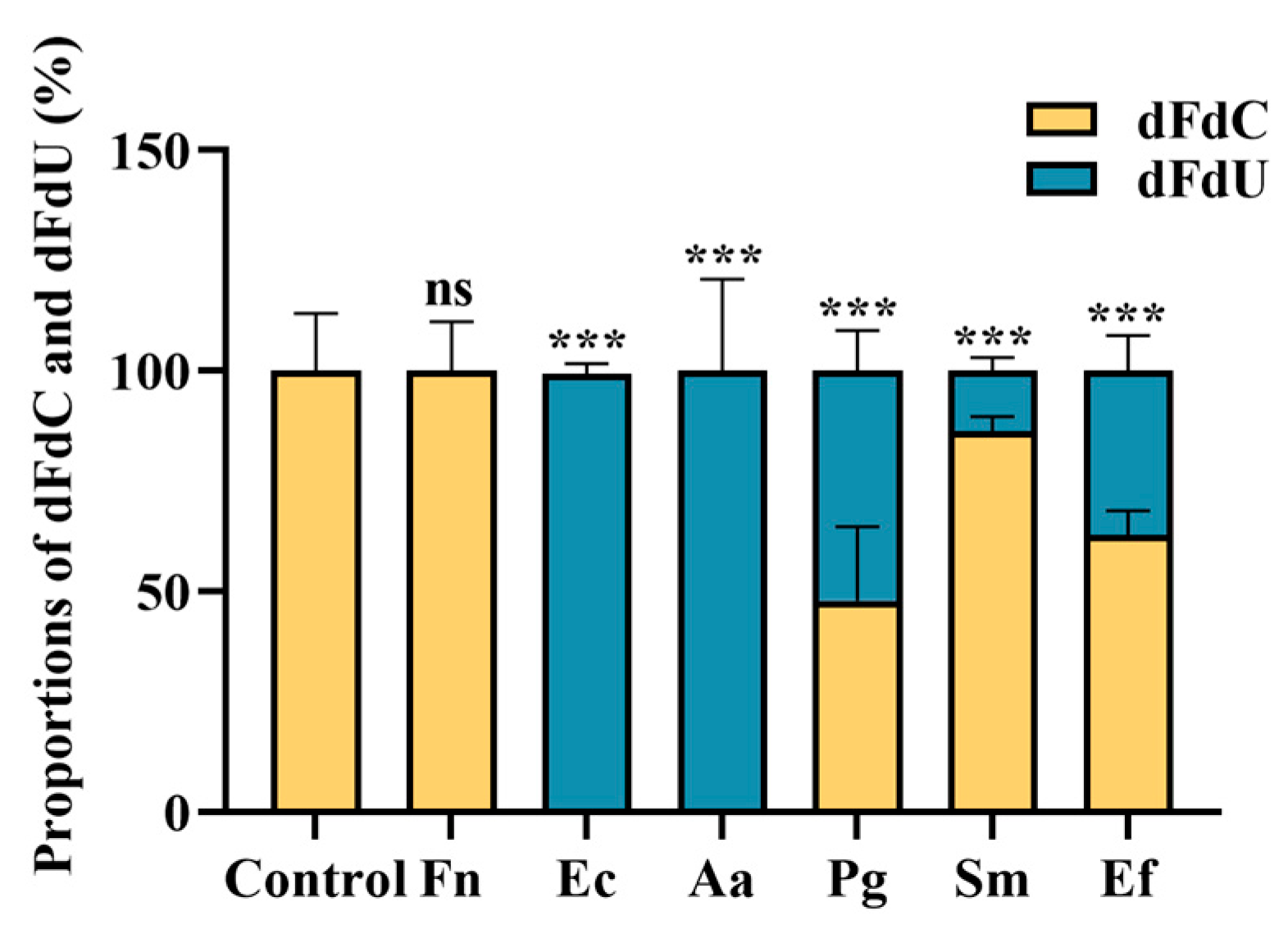
3.3. Quantification of Gemcitabine Degradation in Filtrates
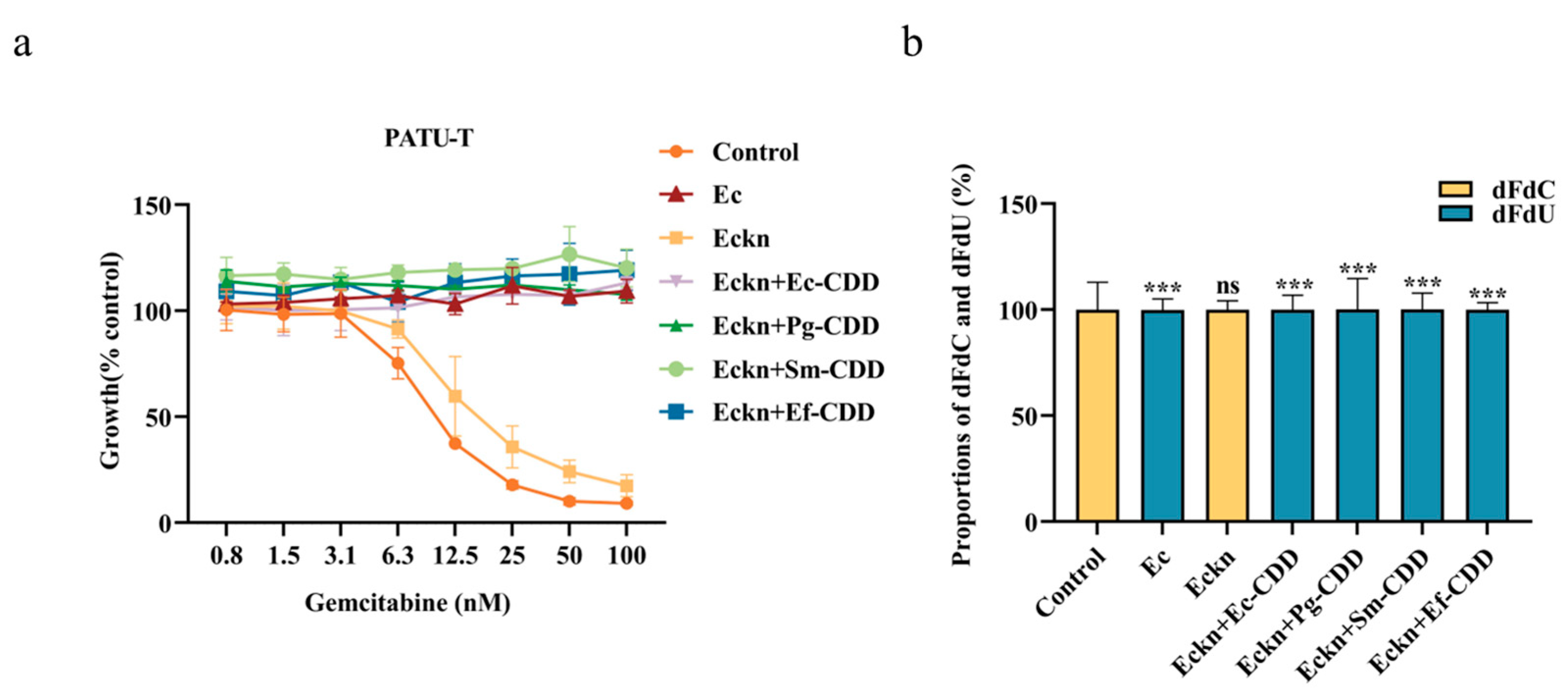
3.4. CDD Function Is Independent of CDD Length
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PDAC | pancreatic ductal adenocarcinoma |
| CDD | cytidine deaminase |
| CDDL | long isoform of CDD |
| CDDS | short isoform of CDD |
| PATU-T | PA-TU-8988T |
| SUIT-2 | SUIT-2-28 |
| SRB | sulforhodamine B |
| Eckn | cdd deletion E. coli strain |
References
- Halbrook, C.J.; Lyssiotis, C.A.; Pasca di Magliano, M.; Maitra, A. Pancreatic cancer: Advances and challenges. Cell 2023, 186, 1729–1754. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Randazzo, O.; Papini, F.; Mantini, G.; Gregori, A.; Parrino, B.; Liu, D.S.K.; Cascioferro, S.; Carbone, D.; Peters, G.J.; Frampton, A.E.; et al. “Open Sesame?”: Biomarker Status of the Human Equilibrative Nucleoside Transporter-1 and Molecular Mechanisms Influencing its Expression and Activity in the Uptake and Cytotoxicity of Gemcitabine in Pancreatic Cancer. Cancers 2020, 12, 3206. [Google Scholar] [CrossRef]
- Beatty, G.L.; Werba, G.; Lyssiotis, C.A.; Simeone, D.M. The biological underpinnings of therapeutic resistance in pancreatic cancer. Genes Dev. 2021, 35, 940–962. [Google Scholar] [CrossRef]
- Capula, M.; Perán, M.; Xu, G.; Donati, V.; Yee, D.; Gregori, A.; Assaraf, Y.G.; Giovannetti, E.; Deng, D. Role of drug catabolism, modulation of oncogenic signaling and tumor microenvironment in microbe-mediated pancreatic cancer chemoresistance. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2022, 64, 100864. [Google Scholar] [CrossRef] [PubMed]
- Wilson, I.D.; Nicholson, J.K. Gut microbiome interactions with drug metabolism, efficacy, and toxicity. Transl. Res. J. Lab. Clin. Med. 2017, 179, 204–222. [Google Scholar] [CrossRef]
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef]
- Ilievski, V.; Toth, P.T.; Valyi-Nagy, K.; Valyi-Nagy, T.; Green, S.J.; Marattil, R.S.; Aljewari, H.W.; Wicksteed, B.; O’Brien-Simpson, N.M.; Reynolds, E.C.; et al. Identification of a periodontal pathogen and bihormonal cells in pancreatic islets of humans and a mouse model of periodontitis. Sci. Rep. 2020, 10, 9976. [Google Scholar] [CrossRef]
- Kinskey, J.C.; Huda, T.I.; Gozlan, E.C.; Quach, J.U.; Arturo, J.F.; Chobrutskiy, A.; Chobrutskiy, B.I.; Blanck, G. The presence of intratumoral Porphyromonas gingivalis correlates with a previously defined pancreatic adenocarcinoma, immune cell expression phenotype and with tumor resident, adaptive immune receptor features. Carcinogenesis 2023, 44, 411–417. [Google Scholar] [CrossRef]
- Huang, Y.; Zhu, N.; Zheng, X.; Liu, Y.; Lu, H.; Yin, X.; Hao, H.; Tan, Y.; Wang, D.; Hu, H.; et al. Intratumor Microbiome Analysis Identifies Positive Association Between Megasphaera and Survival of Chinese Patients with Pancreatic Ductal Adenocarcinomas. Front. Immunol. 2022, 13, 785422. [Google Scholar] [CrossRef] [PubMed]
- Del Castillo, E.; Meier, R.; Chung, M.; Koestler, D.C.; Chen, T.; Paster, B.J.; Charpentier, K.P.; Kelsey, K.T.; Izard, J.; Michaud, D.S. The Microbiomes of Pancreatic and Duodenum Tissue Overlap and Are Highly Subject Specific but Differ between Pancreatic Cancer and Noncancer Subjects. Cancer Epidemiol. Biomark. Prev. 2019, 28, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, K.; Kamada, N. Exploring the oral-gut linkage: Interrelationship between oral and systemic diseases. Mucosal Immunol. 2024, 17, 147–153. [Google Scholar] [CrossRef]
- Schmidt, T.S.; Hayward, M.R.; Coelho, L.P.; Li, S.S.; Costea, P.I.; Voigt, A.Y.; Wirbel, J.; Maistrenko, O.M.; Alves, R.J.; Bergsten, E.; et al. Extensive transmission of microbes along the gastrointestinal tract. eLife 2019, 8, e42693. [Google Scholar] [CrossRef] [PubMed]
- Parahitiyawa, N.B.; Jin, L.J.; Leung, W.K.; Yam, W.C.; Samaranayake, L.P. Microbiology of odontogenic bacteremia: Beyond endocarditis. Clin. Microbiol. Rev. 2009, 22, 46–64. [Google Scholar] [CrossRef]
- Kitamoto, S.; Nagao-Kitamoto, H.; Hein, R.; Schmidt, T.M.; Kamada, N. The Bacterial Connection between the Oral Cavity and the Gut Diseases. J. Dent. Res. 2020, 99, 1021–1029. [Google Scholar] [CrossRef]
- Riquelme, E.; Zhang, Y.; Zhang, L.; Montiel, M.; Zoltan, M.; Dong, W.; Quesada, P.; Sahin, I.; Chandra, V.; San Lucas, A.; et al. Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell 2019, 178, 795–806.e12. [Google Scholar] [CrossRef]
- Pushalkar, S.; Hundeyin, M.; Daley, D.; Zambirinis, C.P.; Kurz, E.; Mishra, A.; Mohan, N.; Aykut, B.; Usyk, M.; Torres, L.E.; et al. The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression. Cancer Discov. 2018, 8, 403–416. [Google Scholar] [CrossRef]
- Saba, E.; Farhat, M.; Daoud, A.; Khashan, A.; Forkush, E.; Menahem, N.H.; Makkawi, H.; Pandi, K.; Angabo, S.; Kawasaki, H.; et al. Oral bacteria accelerate pancreatic cancer development in mice. Gut 2024, 73, 770–786. [Google Scholar] [CrossRef]
- Falconer, J.L.; Rajani, R.; Androshchuk, V.; Yogarajah, A.; Greenbury, R.A.; Ismail, A.; Oh, N.; Nibali, L.; D’Agostino, E.M.; Sousa, V. Exploring links between oral health and infective endocarditis. Front. Oral Health 2024, 5, 1426903. [Google Scholar] [CrossRef]
- Iwai, T. Periodontal bacteremia and various vascular diseases. J. Periodontal Res. 2009, 44, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Baba, Y.; Ishimoto, T.; Tsutsuki, H.; Zhang, T.; Nomoto, D.; Okadome, K.; Yamamura, K.; Harada, K.; Eto, K.; et al. Fusobacterium nucleatum confers chemoresistance by modulating autophagy in oesophageal squamous cell carcinoma. Br. J. Cancer 2021, 124, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 2017, 170, 548–563.e16. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, M.; Chowdhury, N.; Wellslager, B.; Oleinik, N.; Kassir, M.F.; Lee, H.G.; Engevik, M.; Peterson, Y.; Pandruvada, S.; Szulc, Z.M.; et al. Opportunistic pathogen Porphyromonas gingivalis targets the LC3B-ceramide complex and mediates lethal mitophagy resistance in oral tumors. iScience 2024, 27, 109860. [Google Scholar] [CrossRef] [PubMed]
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.; Yu, W.H.; Lakshmanan, A.; Wade, W.G. The human oral microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef]
- Buchholz, M.; Biebl, A.; Neesse, A.; Wagner, M.; Iwamura, T.; Leder, G.; Adler, G.; Gress, T.M. SERPINE2 (protease nexin I) promotes extracellular matrix production and local invasion of pancreatic tumors in vivo. Cancer Res. 2003, 63, 4945–4951. [Google Scholar]
- Ajdić, D.; McShan, W.M.; McLaughlin, R.E.; Savić, G.; Chang, J.; Carson, M.B.; Primeaux, C.; Tian, R.; Kenton, S.; Jia, H.; et al. Genome sequence of Streptococcus mutans UA159, a cariogenic dental pathogen. Proc. Natl. Acad. Sci. USA 2002, 99, 14434–14439. [Google Scholar] [CrossRef]
- Duggan, J.M.; Sedgley, C.M. Biofilm formation of oral and endodontic Enterococcus faecalis. J. Endod. 2007, 33, 815–818. [Google Scholar] [CrossRef]
- Jensen, S.I.; Lennen, R.M.; Herrgård, M.J.; Nielsen, A.T. Seven gene deletions in seven days: Fast generation of Escherichia coli strains tolerant to acetate and osmotic stress. Sci. Rep. 2015, 5, 17874. [Google Scholar] [CrossRef]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef]
- Macrina, F.L.; Tobian, J.A.; Jones, K.R.; Evans, R.P.; Clewell, D.B. A cloning vector able to replicate in Escherichia coli and Streptococcus sanguis. Gene 1982, 19, 345–353. [Google Scholar] [CrossRef] [PubMed]
- van de Guchte, M.; van der Vossen, J.M.; Kok, J.; Venema, G. Construction of a lactococcal expression vector: Expression of hen egg white lysozyme in Lactococcus lactis subsp. lactis. Appl. Environ. Microbiol. 1989, 55, 224–228. [Google Scholar] [CrossRef]
- Kuraku, S.; Zmasek, C.M.; Nishimura, O.; Katoh, K. aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res. 2013, 41, W22–W28. [Google Scholar] [CrossRef]
- Gouveia-Oliveira, R.; Sackett, P.W.; Pedersen, A.G. MaxAlign: Maximizing usable data in an alignment. BMC Bioinform. 2007, 8, 312. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Li, L.; Luo, X.; Chen, M.; Tang, W.; Zhan, L.; Dai, Z.; Lam, T.T.; Guan, Y.; Yu, G. Ggtree: A serialized data object for visualization of a phylogenetic tree and annotation data. iMeta 2022, 1, e56. [Google Scholar] [CrossRef]
- El Hassouni, B.; Mantini, G.; Immordino, B.; Peters, G.J.; Giovannetti, E. CX-5461 Inhibits Pancreatic Ductal Adenocarcinoma Cell Growth, Migration and Induces DNA Damage. Molecules 2019, 24, 4445. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.J.; Honeywell, R.J.; Maulandi, M.; Giovannetti, E.; Losekoot, N.; Etienne-Grimaldi, M.C.; Milano, G.; Serdjebi, C.; Ciccolini, J. Selection of the best blood compartment to measure cytidine deaminase activity to stratify for optimal gemcitabine or cytarabine treatment. Nucleosides Nucleotides Nucleic Acids 2014, 33, 403–412. [Google Scholar] [CrossRef]
- Wei, A.L.; Li, M.; Li, G.Q.; Wang, X.; Hu, W.M.; Li, Z.L.; Yuan, J.; Liu, H.Y.; Zhou, L.L.; Li, K.; et al. Oral microbiome and pancreatic cancer. World J. Gastroenterol. 2020, 26, 7679–7692. [Google Scholar] [CrossRef]
- Geller, L.T.; Straussman, R. Intratumoral bacteria may elicit chemoresistance by metabolizing anticancer agents. Mol. Cell. Oncol. 2018, 5, e1405139. [Google Scholar] [CrossRef]
- Kirstein, M.N.; Hassan, I.; Guire, D.E.; Weller, D.R.; Dagit, J.W.; Fisher, J.E.; Remmel, R.P. High-performance liquid chromatographic method for the determination of gemcitabine and 2′,2′-difluorodeoxyuridine in plasma and tissue culture media. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2006, 835, 136–142. [Google Scholar] [CrossRef]
- Landick, R. Transcriptional Pausing as a Mediator of Bacterial Gene Regulation. Annu. Rev. Microbiol. 2021, 75, 291–314. [Google Scholar] [CrossRef] [PubMed]
- Mascher, T. Past, Present, and Future of Extracytoplasmic Function σ Factors: Distribution and Regulatory Diversity of the Third Pillar of Bacterial Signal Transduction. Annu. Rev. Microbiol. 2023, 77, 625–644. [Google Scholar] [CrossRef] [PubMed]
- Kompaniiets, D.; Wang, D.; Yang, Y.; Hu, Y.; Liu, B. Structure and molecular mechanism of bacterial transcription activation. Trends Microbiol. 2024, 32, 379–397. [Google Scholar] [CrossRef] [PubMed]
- Ricaurte, D.; Huang, Y.; Sheth, R.U.; Gelsinger, D.R.; Kaufman, A.; Wang, H.H. High-throughput transcriptomics of 409 bacteria-drug pairs reveals drivers of gut microbiota perturbation. Nat. Microbiol. 2024, 9, 561–575. [Google Scholar] [CrossRef]
- LaCourse, K.D.; Zepeda-Rivera, M.; Kempchinsky, A.G.; Baryiames, A.; Minot, S.S.; Johnston, C.D.; Bullman, S. The cancer chemotherapeutic 5-fluorouracil is a potent Fusobacterium nucleatum inhibitor and its activity is modified by intratumoral microbiota. Cell Rep. 2022, 41, 111625. [Google Scholar] [CrossRef]
- Sobrero, P.; and Valverde, C. The bacterial protein Hfq: Much more than a mere RNA-binding factor. Crit. Rev. Microbiol. 2012, 38, 276–299. [Google Scholar] [CrossRef]
- Schumacher, M.A.; Pearson, R.F.; Møller, T.; Valentin-Hansen, P.; Brennan, R.G. Structures of the pleiotropic translational regulator Hfq and an Hfq-RNA complex: A bacterial Sm-like protein. EMBO J. 2002, 21, 3546–3556. [Google Scholar] [CrossRef]
- Watkins, D.; Arya, D. Models of Hfq interactions with small non-coding RNA in Gram-negative and Gram-positive bacteria. Front. Cell. Infect. Microbiol. 2023, 13, 1282258. [Google Scholar] [CrossRef]
- Amin, M.B.; Greene, F.L.; Edge, S.B.; Compton, C.C.; Gershenwald, J.E.; Brookland, R.K.; Meyer, L.; Gress, D.M.; Byrd, D.R.; Winchester, D.P. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA A Cancer J. Clin. 2017, 67, 93–99. [Google Scholar] [CrossRef]
- Che, P.P.; Mapanao, A.K.; Gregori, A.; Ermini, M.L.; Zamborlin, A.; Capula, M.; Ngadimin, D.; Slotman, B.J.; Voliani, V.; Sminia, P.; et al. Biodegradable Ultrasmall-in-Nano Architectures Loaded with Cisplatin Prodrug in Combination with Ionizing Radiation Induces DNA Damage and Apoptosis in Pancreatic Ductal Adenocarcinoma. Cancers 2022, 14, 3034. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | Relevant Characteristic(s) | Source of Reference |
|---|---|---|
| E. coli K12 | Wild type | DSMZ |
| E. coli DH5α | F-, endA1deoR, recA1, hsdR17 (rk-,mk+), supE44, thi-1, gyrA96, relA1 | Invitrogen |
| A. actinomycetem-comitans Y4 | Wild type | Our own strain collection |
| S. mutans UA159 | Wild type | (Ajdić et al., 2002) [27] |
| E. faecalis V583 | Wild type | (Duggan and Sedgley, 2007) [28] |
| P. gingivalis ATCC33277 | Wild type | ATCC |
| F. nucleatum ATCC10953 | Wild type | ATCC |
| E. coli Δcdd (Eckn) | E. coli K12 with deletion of cdd | This study |
| Plasmids | ||
| pSIJ8 | Temperature-sensitive plasmid containing the lambda Red recombineering genes and a flippase recombinase, Ampr | (Jensen et al., 2015) [29] |
| pKD4 | E. coli knockout vector, Ampr, Kanr | (Datsenko and Wanner, 2000) [30] |
| pVA838 | E. coli-streptococcal shuttle vector; Er | (Macrina et al., 1982) [31] |
| pMG36e | E. coli-lactococcal expression vector, containing constitutive lactococcal P32 promoter; Emr | (van de Guchte et al., 1989) [32] |
| pJV1 | pVA838 containing P32 and cdd gene from E. coli K12 | This study |
| pJV2 | pVA838 containing P32 and cdd gene from S. mutans UA159 | This study |
| pJV9 | pVA838 containing P32 and cdd gene from E. faecalis V583 | This study |
| pJV12 | pVA838 containing P32 and cdd gene from P. gingivalis ATCC33277 | This study |
| Primers | Oligonucleotide and sequence (5′ to 3′) | |
| cdd_fw | ACGGGTTCGTAAACTGTTATCCCATTACATGATTATGAGGCAACGCCATGTGTAGGCTGGAGCTGCTTC | |
| cdd_rv | AAGGCGTTCACGCCGCATCCGGCACCAGGCTTAAGCGAGAAGCACTCGGTGCTTGCATATGAATATCCTCCTTAG | |
| cdd_seq_fw | CCGAGCTGGATTATCAGGAAGG | |
| cdd_seq_rv | GGACTAACAGGCTGAGGAACAC | |
| cdd_Ec_fw | ATGCATCCACGTTTTCAAACC | |
| cdd_Ec_rv | TTAAGCGAGAAGCACTCGGTCGAT | |
| cdd_Sm_fw | ATGGTGGTTATTGATTTAATCAG | |
| cdd_Sm_rv | TTACTTTAACTCTCTAAAGGAATAAG | |
| cdd_Ef_fw | ATGACAGTAAAACAAGAATGGCTTGAT | |
| cdd_Ef_rv | TTAAAAATCTTTATCTGTAAATG | |
| cdd_Pg_fw | ATGCTCCGTAAAACTCTTTC | |
| cdd_Pg_rv | CTATCGTTCCAAGTCGCTACCG |
| Tested Samples | PATU-T | SUIT-2 | ||
|---|---|---|---|---|
| 1 h | 4 h | 1 h | 4 h | |
| Gem | 11.6 ± 1.6 | 5.1 ± 2.1 | ||
| Gem-Fn | 11.9 ± 0.6 | 11.1 ± 1.1 | 4.9 ± 1.3 | 5.1 ± 0.8 |
| Gem-Ec | NI | – | NI | – |
| Gem-Aa | NI | – | NI | – |
| Gem-Pg | 51.1 ± 18.4 *** | – | 15.5 ± 4.3 *** | – |
| Gem-Ef | 17.3 ± 2.1 | 17.1 ± 1.1 ** | 10.1 ± 3.6 * | 11.4 ± 1.7 *** |
| Gem-Sm | 15.2 ± 0.2 | 18.1 ± 1.7 ** | 8.6 ± 3.1 | 10.8 ± 1.6 *** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, G.; Jiang, Y.; Sun, C.; Brandt, B.W.; Nazmi, K.; Morelli, L.; Lencioni, G.; Giovannetti, E.; Deng, D. Role of Oral Bacteria in Mediating Gemcitabine Resistance in Pancreatic Cancer. Biomolecules 2025, 15, 1018. https://doi.org/10.3390/biom15071018
Xu G, Jiang Y, Sun C, Brandt BW, Nazmi K, Morelli L, Lencioni G, Giovannetti E, Deng D. Role of Oral Bacteria in Mediating Gemcitabine Resistance in Pancreatic Cancer. Biomolecules. 2025; 15(7):1018. https://doi.org/10.3390/biom15071018
Chicago/Turabian StyleXu, Geng, Yaling Jiang, Chen Sun, Bernd W. Brandt, Kamran Nazmi, Luca Morelli, Giulia Lencioni, Elisa Giovannetti, and Dongmei Deng. 2025. "Role of Oral Bacteria in Mediating Gemcitabine Resistance in Pancreatic Cancer" Biomolecules 15, no. 7: 1018. https://doi.org/10.3390/biom15071018
APA StyleXu, G., Jiang, Y., Sun, C., Brandt, B. W., Nazmi, K., Morelli, L., Lencioni, G., Giovannetti, E., & Deng, D. (2025). Role of Oral Bacteria in Mediating Gemcitabine Resistance in Pancreatic Cancer. Biomolecules, 15(7), 1018. https://doi.org/10.3390/biom15071018