
Cycloadditions as a Sweet Route to ‘Double C-Glycosylation’

 , , , ,
, , , ,  , and
, and
Abstract
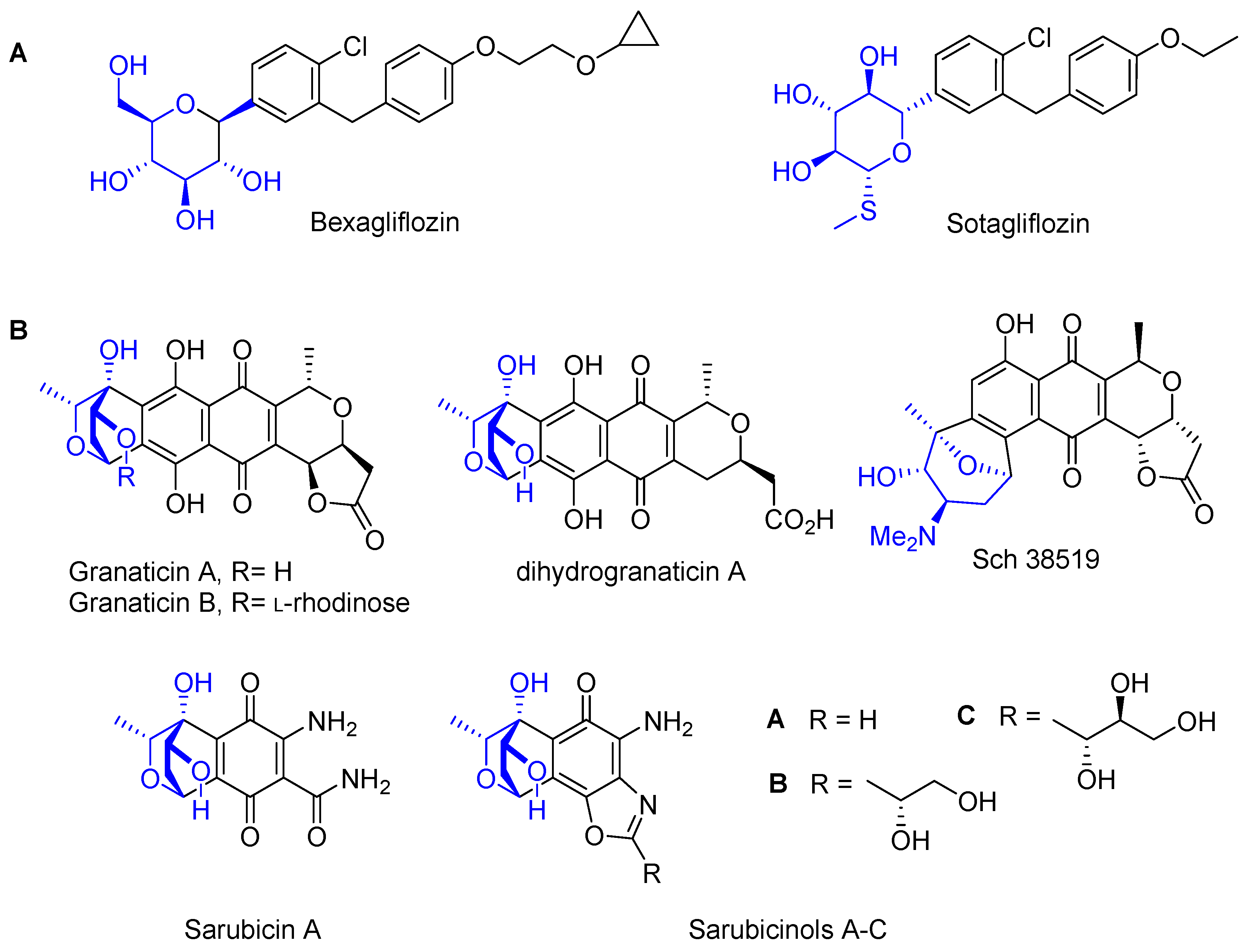
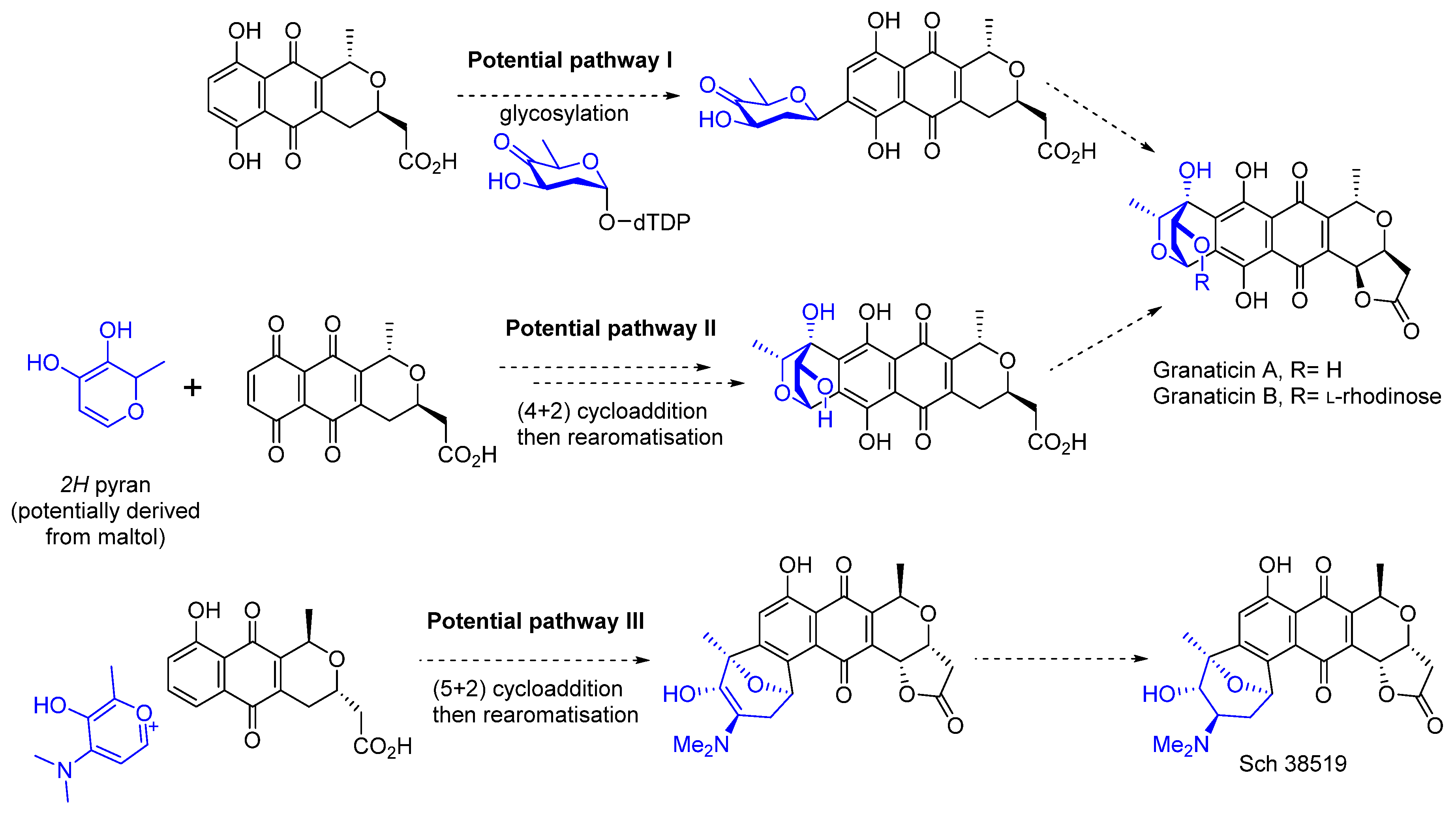
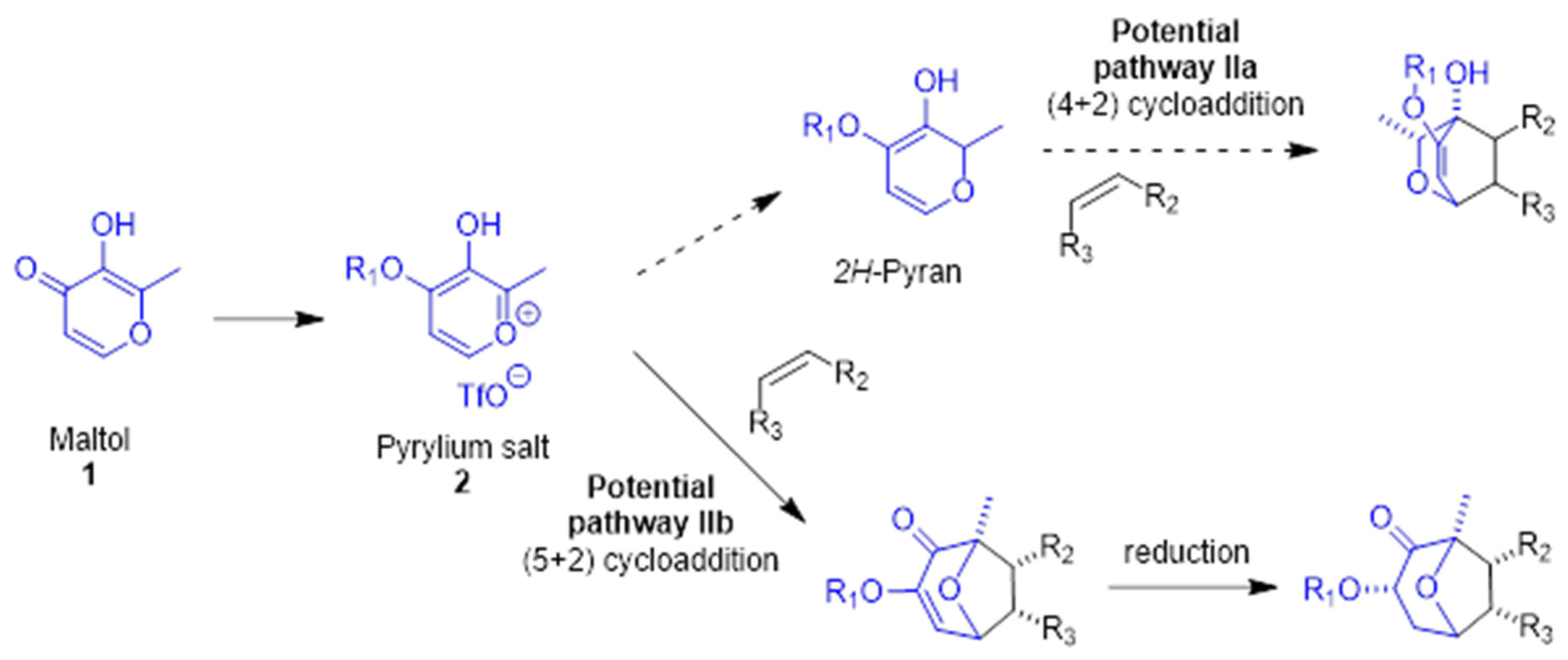
1. Introduction
2. Materials and Methods
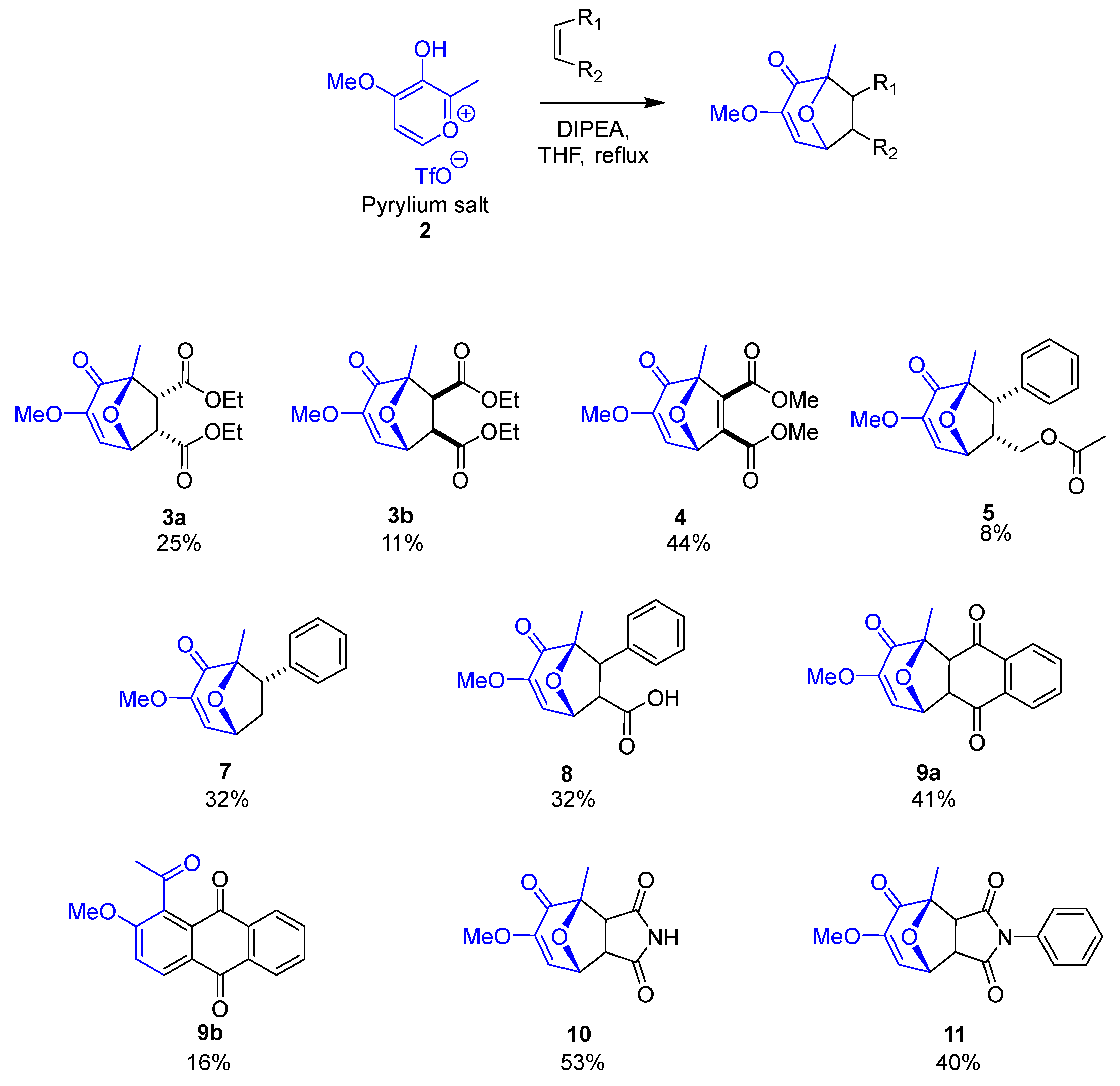
2.1. Preparation of Pyrylium Salt (2)
2.2. General Procedure or Preparation of Cycloadducts
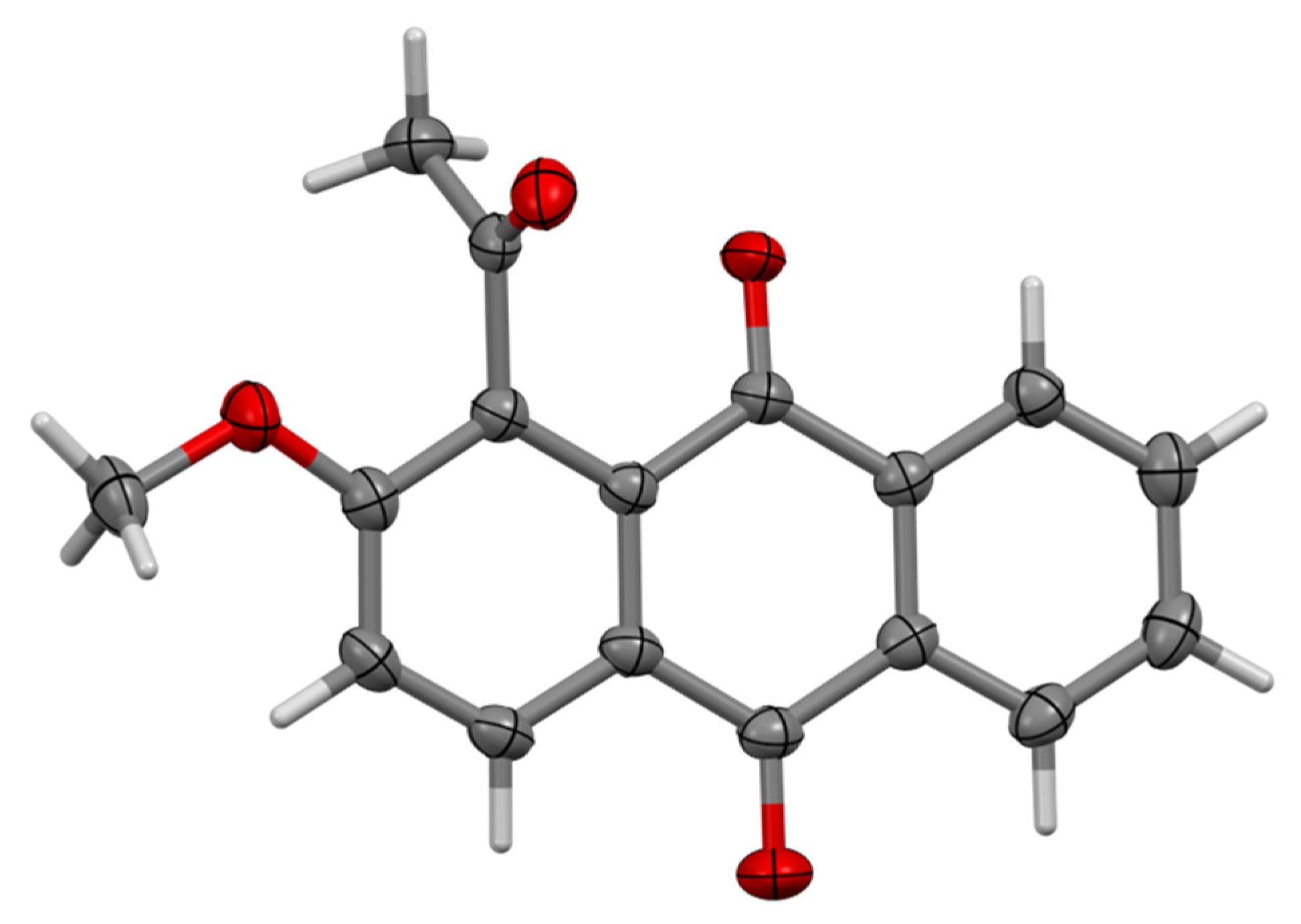
2.3. Single Crystal X-Ray Diffraction
3. Results and Discussion
3.1. Initial Exploration of the Cycloaddition Reaction
3.2. Solvent Screening
3.3. Base Screening
3.4. Exploring Substrate Scope
3.5. Assessment of Antibiotic Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eckardt, K.; Tresselt, D.; Ihn, W.; Kajtar, M.; Angyan, J.; Radics, L.; Hollosi, M. Constitution and absolute stereochemistry of the antibiotic sarubicin A. J. Antibiot. 1983, 36, 976–979. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, G.; Bradler, G.; Eckardt, K.; Tresselt, D.; Ihn, W. Isolation and characterization of sarubicin A, a new antibiotic. J. Antibiot. 1980, 33, 787–790. [Google Scholar] [CrossRef]
- Wang, H.; Qi, H.; Zhang, S.-Y.; Song, W.-S.; Zhang, L.-Q.; Xiang, W.-S.; Wang, J.-D. Sarubicinols A–C, Cytotoxic benzoxazoles from a Streptomyces. J. Nat. Prod. 2022, 85, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Floss, H.G.; Soong, P.; Chang, C.T. Identity of the antitumor antibiotic litmomycin with granaticin A. J. Antibiot. 1975, 28, 156. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.R.; Guo, J.; Li, X.; Zhu, C.H.; Zhu, H.H. Granaticins and their biosynthetic gene cluster from Streptomyces vietnamensis: Evidence of horizontal gene transfer. Antonie Van Leeuwenhoek 2011, 100, 607–617. [Google Scholar] [CrossRef]
- Snipes, C.E.; Chang, C.-J.; Floss, H.G. Biosynthesis of the antibiotic granaticin. J. Am. Chem. Soc. 1979, 101, 701–706. [Google Scholar] [CrossRef]
- Nomura, K.; Okazaki, K.; Hori, K.; Yoshii, E. Total synthesis of (.+-.)-granaticin. J. Am. Chem. Soc. 1987, 109, 3402–3408. [Google Scholar] [CrossRef]
- Hegde, V.R.; King, A.H.; Patel, M.G.; Puar, M.S.; McPhail, A.T. A new isochromanequinone antibiotic, SCH 38519, with a novel sugar linkage. Tetrahedron Lett. 1987, 28, 4485–4488. [Google Scholar] [CrossRef]
- Ichinose, K.; Bedford, D.J.; Tornus, D.; Bechthold, A.; Bibb, M.J.; Revill, W.P.; Floss, H.G.; Hopwood, D.A. The granaticin biosynthetic gene cluster of Streptomyces violaceoruber Tü22: Sequence analysis and expression in a heterologous host. Chem. Biol. 1998, 5, 647–659. [Google Scholar] [CrossRef]
- Draeger, G.; Park, S.-H.; Floss, H.G. Mechanism of the 2-deoxygenation step in the biosynthesis of the deoxyhexose moieties of the antibiotics granaticin and oleandomycin. J. Am. Chem. Soc. 1999, 121, 2611–2612. [Google Scholar] [CrossRef]
- Brasholz, M.; Reissig, H.U. Oxidative cleavage of 3-alkoxy-2, 5-dihydrofurans and its application to the De Novo synthesis of rare monosaccharides as exemplified by l-Cymarose. Angew. Chem. Int. Ed. 2007, 46, 1634–1637. [Google Scholar] [CrossRef] [PubMed]
- CrystalClear-SM Expert, v2.1.; Rigaku Americas: The Woodlands, TX, USA; Rigaku Corporation: Tokyo, Japan, 2013–2014.
- CrysAlisPro, v1.171.42.83a.; Rigaku Oxford Diffraction; Rigaku Corporation: Oxford, UK, 2023.
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G.; Spagna, R. SIR2011: A new package for crystal structure determination and refinement. J. Appl. Crystallogr. 2012, 45, 357–361. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Singh, V.; Murali Krishna, U.; Vikrant; Trivedi, G.K. Cycloaddition of oxidopyrylium species in organic synthesis. Tetrahedron 2008, 64, 3405–3428. [Google Scholar] [CrossRef]
- Gao, K.; Zhang, Y.-G.; Wang, Z.; Ding, H. Recent development on the [5+2] cycloadditions and their application in natural product synthesis. Chem. Commun. 2019, 55, 1859–1878. [Google Scholar] [CrossRef]
- Rokey, S.N.; Simanis, J.A.; Law, C.M.; Pohani, S.; Behrends, S.W.; Bulandr, J.J.; Ferrence, G.M.; Goodell, J.R.; Mitchell, T.A. Intramolecular asymmetric oxidopyrylium-based [5+2] cycloadditions. Tetrahedron Lett. 2020, 61, 152377. [Google Scholar] [CrossRef]
- Bejcek, L.P.; Murelli, R.P. Oxidopyrylium [5+2] cycloaddition chemistry: Historical perspective and recent advances (2008–2018). Tetrahedron 2018, 74, 2501–2521. [Google Scholar] [CrossRef] [PubMed]
- Meck, C.; Mohd, N.; Murelli, R.P. An oxidopyrylium cyclization/ring-opening route to polysubstituted α-hydroxytropolones. Org. Lett. 2012, 14, 5988–5991. [Google Scholar] [CrossRef]
- Wender, P.A.; Mascareñas, J.L. Preparation and cycloadditions of a 4-methoxy-3-oxidopyrylium ylid: A reagent for the synthesis of highly substituted seven-membered rings and tetrahydrofurans. Tetrahedron Lett. 1992, 33, 2115–2118. [Google Scholar] [CrossRef]
- Bejcek, L.P.; Garimallaprabhakaran, A.K.; Suyabatmaz, D.M.; Greer, A.; Hersh, W.H.; Greer, E.M.; Murelli, R.P. Maltol- and Allomaltol-derived oxidopyrylium ylides: Methyl substitution pattern kinetically influences [5 + 3] dimerization versus [5 + 2] cycloaddition reactions. J. Org. Chem. 2019, 84, 14670–14678. [Google Scholar] [CrossRef] [PubMed]
- Schiavone, D.V.; Kapkayeva, D.M.; Murelli, R.P. Investigations into a stoichiometrically equivalent intermolecular oxidopyrylium [5 + 2] cycloaddition reaction leveraging 3-hydroxy-4-pyrone-based oxidopyrylium dimers. J. Org. Chem. 2021, 86, 3826–3835. [Google Scholar] [CrossRef] [PubMed]
- D’Erasmo, M.P.; Meck, C.; Lewis, C.A.; Murelli, R.P. Discovery and development of a three-component oxidopyrylium [5 + 2] cycloaddition. J. Org. Chem. 2016, 81, 3744–3751. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.H.; Sethna, S. Hydroxyanthracene series Part IV. Synthesis of some anthraquinone derivatives. J. Chem. Soc. 1961, 4682–4684. [Google Scholar]
- Pyrek, J.S.; Achmatowicz, O.; Zamojski, A. Naphto- and anthraquinones of Streptomyces thermoviolaceusWR-141. Structures and model syntheses. Tetrahedron 1977, 33, 673–680. [Google Scholar] [CrossRef]
- Sammes, P.G.; Street, L.J. The preparation and some reactions of 3-oxidopyrylium. J. Chem. Soc. Perkin Trans. 1 1983, 1261–1265. [Google Scholar] [CrossRef]
- Bejcek, L.P.; Murelli, R.P. Synthesis of aryl-substituted 2-methoxyphenol derivatives from maltol-derived oxidopyrylium cycloadducts through an acid-mediated ring contraction cascade. Chem. Commun. 2020, 56, 3203–3205. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 6 | 9b | |
|---|---|---|
| formula | C14H16O6 | C17H12O4 |
| fw | 280.27 | 280.27 |
| crystal description | Colourless plate | Yellow prism |
| crystal size [mm3] | 0.24 × 0.18 × 0.02 | 0.12 × 0.09 × 0.05 |
| space group | P | P |
| a [Å] | 9.2341 (3) | 7.884 (3) |
| b [Å] | 10.6447 (5) | 8.055 (3) |
| c [Å] | 14.4081 (6) | 10.842 (3) |
| α [°] | 77.390 (4) | 92.509 (17) |
| β [°] | 74.942 (4) | 108.668 (19) |
| γ [°] | 89.491 (3) | 95.313 (13) |
| vol [Å]3 | 1332.89 (10) | 647.5 (4) |
| Z | 4 | 2 |
| ρ (calc) [g/cm3] | 1.397 | 1.437 |
| μ [mm−1] | 0.928 | 0.850 |
| F(000) | 592 | 292 |
| reflections collected | 12704 | 6785 |
| independent reflections (Rint) | 4770 (0.0308) | 2308 (0.0385) |
| parameters, restraints | 369, 0 | 193, 0 |
| GoF on F2 | 1.072 | 1.129 |
| R1 [I > 2σ(I)] | 0.0428 | 0.0644 |
| wR2 (all data) | 0.1270 | 0.1358 |
| largest diff. peak/hole [e/Å3] | 0.220, −0.241 | 0.512, −0.6620 |
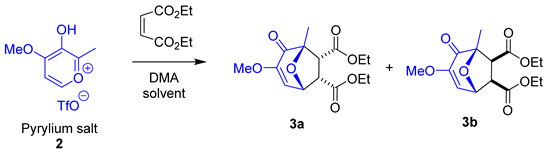 | |||
| Entry | Solvent | Isolated Yield of Diastereoisomer 3a (%) | Isolated Yield of Diastereoisomer 3b (%) |
|---|---|---|---|
| 1 | Neat | 7 | - |
| 2 | Dichloromethane | 10 | - |
| 3 | Toluene | 6 | - |
| 4 | Chloroform | 14 | - |
| 5 | 1,4-Dioxane | 7 | - |
| 6 | Methanol | 5 | - |
| 7 | Acetonitrile | 12 | - |
| 8 | Tetrahydrofuran | 17 | 7 |
| 9 | Ethyl Acetate | 9 | - |
| 10 | Hexane | 9 | - |
| 11 | Diethylether | 17 | - |
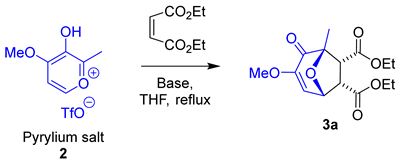 | ||||
| Entry | Base | Equivalents | pKa | Yield 3a (%) |
|---|---|---|---|---|
| 1 | N,N-Dimethylaniline | 1.5 | 5.2 | 17 |
| 2 | 3 | 14 | ||
| 3 | 2,6-Lutidine | 1.5 | 6.7 | - |
| 4 | 3 | - | ||
| 5 | Pyridine | 1.5 | 5.3 | - |
| 6 | 3 | 23 | ||
| 7 | N,N-Diisopropylethylamine | 1.5 | 10.6 | 25 |
| 8 | 3 | 13 | ||
| 9 | 1,8-Diazabicyclo[5.4.0]undec-7-ene | 1.5 | 12.5 | - |
| 10 | 3 | - | ||
| 11 | 4-(Dimethylamino)pyridine | 1.5 | 9.7 | 34 † |
| 12 | 3 | 37 † | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahoney, K.P.P.; Lynch, R.; Bown, R.T.; Sharma, S.V.; Chueakwon, P.; Stephenson, G.R.; Cordes, D.B.; Slawin, A.M.Z.; Goss, R.J.M. Cycloadditions as a Sweet Route to ‘Double C-Glycosylation’. Biomolecules 2025, 15, 905. https://doi.org/10.3390/biom15060905
Mahoney KPP, Lynch R, Bown RT, Sharma SV, Chueakwon P, Stephenson GR, Cordes DB, Slawin AMZ, Goss RJM. Cycloadditions as a Sweet Route to ‘Double C-Glycosylation’. Biomolecules. 2025; 15(6):905. https://doi.org/10.3390/biom15060905
Chicago/Turabian StyleMahoney, Kevin P. P., Rosemary Lynch, Rhea T. Bown, Sunil V. Sharma, Piyasiri Chueakwon, G. Richard Stephenson, David B. Cordes, Alexandra M. Z. Slawin, and Rebecca J. M. Goss. 2025. "Cycloadditions as a Sweet Route to ‘Double C-Glycosylation’" Biomolecules 15, no. 6: 905. https://doi.org/10.3390/biom15060905
APA StyleMahoney, K. P. P., Lynch, R., Bown, R. T., Sharma, S. V., Chueakwon, P., Stephenson, G. R., Cordes, D. B., Slawin, A. M. Z., & Goss, R. J. M. (2025). Cycloadditions as a Sweet Route to ‘Double C-Glycosylation’. Biomolecules, 15(6), 905. https://doi.org/10.3390/biom15060905