

Gene Fusions as Potential Therapeutic Targets in Soft Tissue Sarcomas

Abstract
1. Introduction
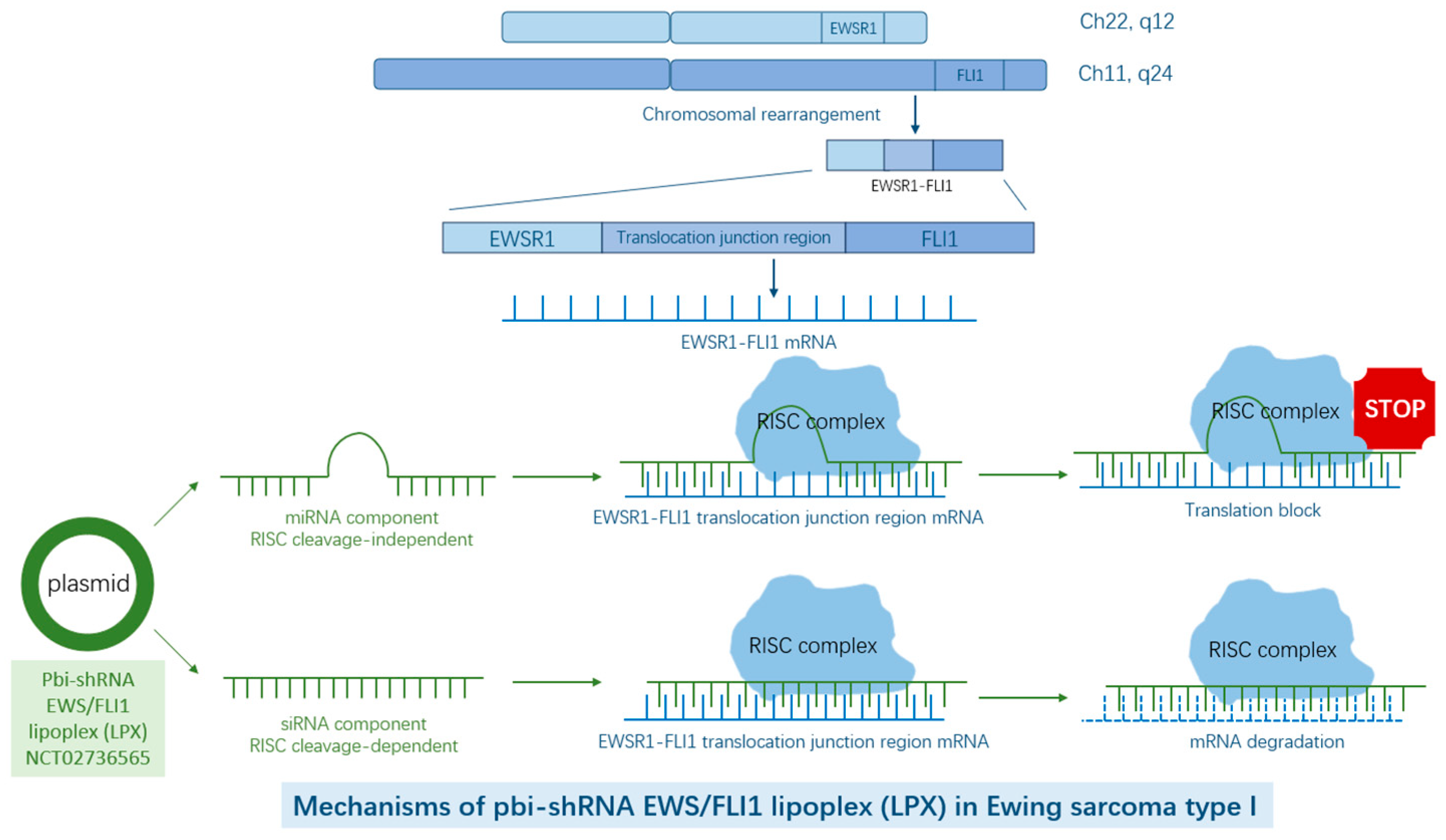
2. EWSR1–FLI1/ERG as Targets in Ewing Sarcoma
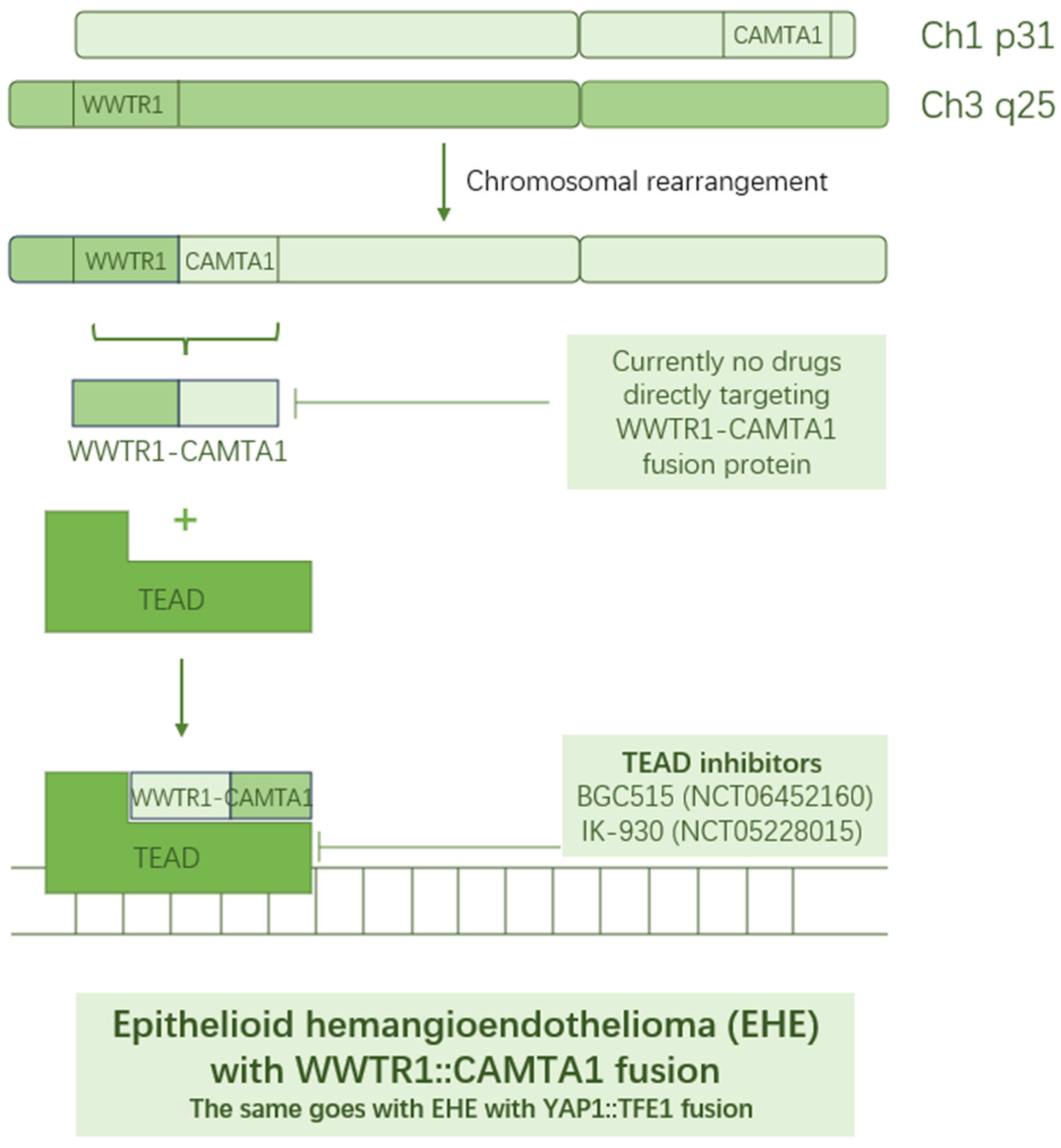
3. WWTR1–CAMTA1/YAP1–TFE1 as Targets in Epithelioid Hemangioendothelioma
4. FUS/EWSR1–DDIT3 as Targets in Myxoid Liposarcoma
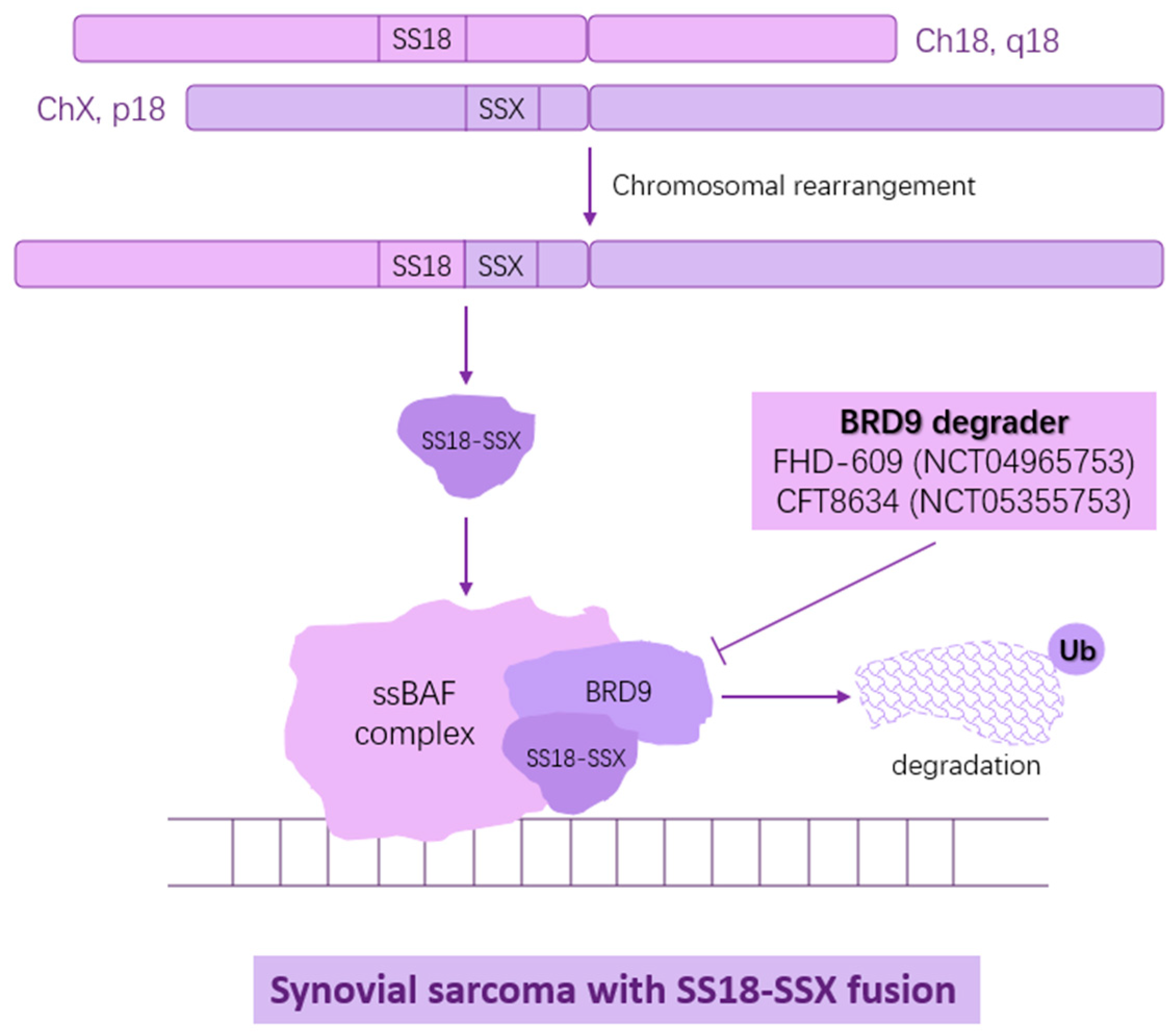
5. SS18-SSX1/2/4 as Targets in Synovial Sarcoma
6. COL6A3-CSF1 as Target in Malignant Tenosynovial Giant Cell Tumor

7. PAX3/7–FOXO1 as Targets in Alveolar Rhabdomyosarcoma
8. Other Rare Soft Tissue Sarcomas with Hallmark Fusion Proteins and Their Potential Targeted Therapeutics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Trials | NCT Number | Trial Phase | Status | Interventions |
|---|---|---|---|---|
| Myxoid Liposarcoma | ||||
| Clinical Trial of SP-2577 (Seclidemstat) in Patients with Relapsed or Refractory Ewing or Ewing-related Sarcomas | NCT03600649 | Phase I | Active, not recruiting | Seclidemstat, a LSD1 inhibitor |
| Cixutumumab and Doxorubicin Hydrochloride in Treating Patients with Unresectable, Locally Advanced, or Metastatic Soft Tissue Sarcoma | NCT00720174 | Phase I | Completed | Cixutumumab, IGF-1R inhibitor, in combination with chemotherapy drugs |
| Vismodegib and Gamma-Secretase/Notch Signalling Pathway Inhibitor RO4929097 in Treating Patients with Advanced or Metastatic Sarcoma | NCT01154452 | Phase I/II | Completed | Vismodegib, Hedgehog signaling inhibitor, together with RO4929097, Notch signaling inhibitor |
| Spearhead 1 Study in Subjects with Advanced Synovial Sarcoma or Myxoid/Round-Cell Liposarcoma | NCT04044768 | Phase II | Recruiting | Car-T cell targeting MAGE-A4 |
| MAGE-A4c1o32T for Multi-Tumor | NCT03132922 | Phase I | Active, not recruiting | Car-T cell targeting MAGE-A4 |
| Letetresgene Autoleucel Engineered T Cells in NY-ESO-1 Positive Participants with Advanced Myxoid/Round-Cell Liposarcoma | NCT02992743 | Pilot study | Completed | Car-T cell targeting NY-ESO-1 |
| NY-ESO-1-redirected CRISPR (TCRendo and PD1) Edited T Cells (NYCE T Cells) | NCT03399448 | Phase I | Completed | Car-T cell targeting NY-ESO-1 |
| Malignant tenosynovial giant cell tumor (mTSGCT) | ||||
| Study of Vimseltinib (DCC-3014) in Patients with Advanced Tumors and Tenosynovial Giant Cell Tumor | NCT03069469 | Phase I/II | Active, not recruiting | Vimseltinib, CSF1R inhibitor |
| Clinical Study of HMPL-653 in Treatment of Advanced Malignant Solid Tumors and TGCT | NCT05277454 | Phase I | Recruiting | HMPL-653, CSF1R inhibitor |
| Study of Emactuzumab for Tenosynovial Giant Cell Tumor (TGCT) (TANGENT) | NCT05417789 | Phase III | Recruiting | Emactuzumab, CSF1R inhibitor |
| Study of Cabiralizumab in Patients with Pigmented Villonodular Synovitis/Diffuse Type Tenosynovial Giant Cell Tumor (FPA008-002) | NCT02471716 | Phase I/II | Completed | Cabiralizumab, CSF1R inhibitor |
| MCS110 in Patients with Pigmented Villonodular Synovitis (PVNS) | NCT01643850 | Phase II | Completed | MCS110, CSF1 inhibitor |
| Epithelioid hemangioendothelioma | ||||
| A Study of BGC515 Capsules in Subjects with Advanced Solid Tumors | NCT06452160 | Phase I | Recruiting | BGC515, TEAD inhibitor |
| Oral TEAD Inhibitor Targeting the Hippo Pathway in Subjects with Advanced Solid Tumors | NCT05228015 | Phase I | Active, not recruiting | IK-930, TEAD inhibitor |
| Trametinib in Treating Patients with Epithelioid Hemangioendothelioma That is Metastatic, Locally Advanced, or Cannot Be Removed by Surgery | NCT03148275 | Phase II | Completed | Trametinib, MEK inhibitor |
| Alveolar rhabdomyosarcoma | ||||
| Insulin-like Growth Factor 1 Receptor (IGF-1R) Antibody AMG479 (Ganitumab) in Combination with the Src-Family Kinase (SFK) Inhibitor Dasatinib in People with Embryonal and Alveolar Rhabdomyosarcoma | NCT03041701 | Phase I/II | Terminated | Ganitumab, IGF-1R inhibitor, in combination with Dasatinib, Src-family kinase inhibitor |
| Temozolomide, Cixutumumab, and Combination Chemotherapy in Treating Patients with Metastatic Rhabdomyosarcoma | NCT01055314 | Pilot study | Completed | Cixutumumab, IGF-1R inhibitor, in combination with chemotherapy drugs |
| Cixutumumab and Doxorubicin Hydrochloride in Treating Patients with Unresectable, Locally Advanced, or Metastatic Soft Tissue Sarcoma | NCT00720174 | Phase I | Completed | Cixutumumab, IGF-1R inhibitor, in combination with chemotherapy drugs |
| Cixutumumab and Temsirolimus in Treating Younger Patients with Recurrent or Refractory Sarcoma | NCT01614795 | Phase II | Completed | Cixutumumab, IGF-1R inhibitor, in combination with Temsirolimus, mTOR inhibitor |
| Phosphaturic mesenchymal tumor | ||||
| BGJ398 for Patients with Tumors with FGFR Genetic Alterations (CBGJ398XUS04) | NCT02160041 | Phase II | Terminated | BGJ398, FGFR inhibitor |
| BGJ398 for the Treatment of Tumor-Induced Osteomalacia | NCT03510455 | Phase II | Terminated | BGJ398, FGFR inhibitor |
| Study to Assess the Safety, Pharmacokinetics and Efficacy of KRN23 in Adult Chinese Patients With TIO | NCT05357573 | Phase IV | Completed | KRN23, FGF23 monoclonal antibody |
| A Study of KRN23 in Subjects with Tumor-Induced Osteomalacia or Epidermal Nevus Syndrome | NCT02722798 | Phase II | Completed | KRN23, FGF23 monoclonal antibody |
| Desmoplastic small round cell tumor | ||||
| Clinical Trial of SP-2577 (Seclidemstat) in Patients with Relapsed or Refractory Ewing or Ewing-related Sarcomas | NCT03600649 | Phase I | Active, not recruiting | Seclidemstat, LSD1 inhibitor |
| Synovial sarcoma | ||||
| First in Man Study Investigating the Biodistribution, the Safety and Optimal Recommended Dose of a New Radiolabelled Monoclonal Antibody Targeting Frizzled Homolog 10 (SYNFRIZZ) | NCT01469975 | Phase I | Terminated | Yttrium 90-radiolabeled OTSA101 (OTSA101-DTPA-90Y), radiolabeled FZD10 inhibitor |
| Phase I Study of Radiolabeled OTSA101-DTPA in Patients with Relapsed or Refractory Synovial Sarcoma | NCT04176016 | Phase I | Terminated | Yttrium 90-radiolabeled OTSA101 (OTSA101-DTPA-90Y), radiolabeled FZD10 inhibitor |
| FHD-609 in Subjects with Advanced Synovial Sarcoma or Advanced SMARCB1-Loss Tumors | NCT04965753 | Phase I | Terminated | FDH-609, sSWI/SNF component BRD9 inhibitor |
| A Study to Assess the Safety and Tolerability of CFT8634 in Locally Advanced or Metastatic SMARCB1-Perturbed Cancers, Including Synovial Sarcoma and SMARCB1-Null Tumors | NCT05355753 | Phase I/II | Terminated | CFT8634, BRD9 degrader |
| Alveolar soft part sarcoma | ||||
| Sunitinib or Cediranib for Alveolar Soft Part Sarcoma | NCT01391962 | Phase II | Active, not recruiting | Sunitinib or cediranib, multi-RTK inihibitors |
| Phase II Study of Cediranib (AZD2171) in Patients with Alveolar Soft Part Sarcoma | NCT00942877 | Phase II | Active, not recruiting | Cediranib, a pan-VEGFR inhibitor |
| A Trial of Cediranib in the Treatment of Patients with Alveolar Soft Part Sarcoma (CASPS) (CASPS) | NCT01337401 | Phase II | Unknown status | Cediranib, a pan-VEGFR inhibitor |
| Extraskeletal myxoid chondrosarcoma | ||||
| Clinical Trial of SP-2577 (Seclidemstat) in Patients with Relapsed or Refractory Ewing or Ewing-related Sarcomas | NCT03600649 | Phase I | Active, not recruiting | Seclidemstat, LSD1 inhibitor |
| Trial of Pazopanib in Patients with Solitary Fibrous Tumor and Extraskeletal Myxoid Chondrosarcoma | NCT02066285 | Phase II | Completed | Paopanib, multi-kinase inhibitor |
| Ewing sarcoma | ||||
| A Clinical Study of TK216 in Patients with Relapsed or Refractory Ewing’s Sarcoma | NCT05046314 | Phase II | Recruiting | TK216, a ETS binding drug |
| TK216 in Patients with Relapsed or Refractory Ewing Sarcoma | NCT02657005 | Phase I/II | Terminated | TK216, a ETS binding drug |
| A Study of INCB059872 in Relapsed or Refractory Ewing Sarcoma | NCT03514407 | Phase I | Terminated | INCB059872, a LSD1 inhibitor |
| Clinical Trial of SP-2577 (Seclidemstat) in Patients with Relapsed or Refractory Ewing or Ewing-related Sarcomas | NCT03600649 | Phase I | Active, not recruiting | Seclidemstat, a LSD1 inhibitor |
| Pbi-shRNA™ EWS/FLI1 Type 1 LPX in Subjects with Advanced Ewing’s Sarcoma | NCT02736565 | Phase I | Completed | Pbi-shRNA EWS/FLI1 lipoplex, a plasmid construct that breaks junction between EWS–FLI1 and target mRNAs |
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CSF1R | Colony-stimulating factor 1. |
| MLPS | Myxoid liposarcoma. |
| LSD1 | Lysine specific demethylase 1. |
| SFT | Solitary fibrous tumor. |
| ASO | Antisense oligonucleotide. |
| UTR | Untranslated region. |
| TSGCT | Tenosynovial giant cell tumor. |
| EHE | Epithelioid hemangioendothelioma. |
| ARMS | Alveolar rhabdomyosarcoma. |
| mTOR | Mammalian target of rapamycin. |
| SpRMS | Spindle cell rhabdomyosarcoma. |
| PMT | Phosphaturic mesenchymal tumor. |
| FGF | Fibroblast growth factor. |
| EMC | Extraskeletal myxoid chondrosarcoma. |
| DSRCT | Desmoplastic small round-cell tumor. |
| OFMT | Ossifying fibromyxoid tumor. |
| MECA | Myoepithelial carcinoma. |
References
- Taylor, B.S.; Barretina, J.; Maki, R.G.; Antonescu, C.R.; Singer, S.; Ladanyi, M. Advances in sarcoma genomics and new therapeutic targets. Nat. Rev. Cancer 2011, 11, 541–557. [Google Scholar] [CrossRef] [PubMed]
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer 2015, 15, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Marcus, L.; Donoghue, M.; Aungst, S.; Myers, C.E.; Helms, W.S.; Shen, G.; Zhao, H.; Stephens, O.; Keegan, P.; Pazdur, R. FDA Approval Summary: Entrectinib for the Treatment of NTRK gene Fusion Solid Tumors. Clin. Cancer Res. 2021, 27, 928–932. [Google Scholar] [CrossRef]
- Duke, E.S.; Bradford, D.; Marcovitz, M.; Amatya, A.K.; Mishra-Kalyani, P.S.; Nguyen, E.; Price, L.S.; Fourie Zirkelbach, J.; Li, Y.; Bi, Y.; et al. FDA Approval Summary: Selpercatinib for the Treatment of Advanced RET Fusion-Positive Solid Tumors. Clin. Cancer Res. 2023, 29, 3573–3578. [Google Scholar] [CrossRef]
- Grunewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Alava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing sarcoma. Nat. Rev. Dis. Primers 2018, 4, 5. [Google Scholar] [CrossRef]
- Sorensen, P.H.; Lessnick, S.L.; Lopez-Terrada, D.; Liu, X.F.; Triche, T.J.; Denny, C.T. A second Ewing’s sarcoma translocation, t (21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat. Genet. 1994, 6, 146–151. [Google Scholar] [CrossRef]
- Italiano, A.; Mir, O.; Mathoulin-Pelissier, S.; Penel, N.; Piperno-Neumann, S.; Bompas, E.; Chevreau, C.; Duffaud, F.; Entz-Werlé, N.; Saada, E.; et al. Cabozantinib in patients with advanced Ewing sarcoma or osteosarcoma (CABONE): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 446–455. [Google Scholar] [CrossRef]
- Attia, S.; Bolejack, V.; Ganjoo, K.N.; George, S.; Agulnik, M.; Rushing, D.; Loggers, E.T.; Livingston, M.B.; Wright, J.; Chawla, S.P.; et al. A phase II trial of regorafenib in patients with advanced Ewing sarcoma and related tumors of soft tissue and bone: SARC024 trial results. Cancer Med. 2023, 12, 1532–1539. [Google Scholar] [CrossRef]
- Duffaud, F.; Blay, J.Y.; Le Cesne, A.; Chevreau, C.; Boudou-Rouquette, P.; Kalbacher, E.; Penel, N.; Perrin, C.; Laurence, V.; Bompas, E.; et al. Regorafenib in patients with advanced Ewing sarcoma: Results of a non-comparative, randomised, double-blind, placebo-controlled, multicentre Phase II study. Br. J. Cancer 2023, 129, 1940–1948. [Google Scholar] [CrossRef]
- Meyers, P.A.; Federman, N.; Daw, N.; Anderson, P.M.; Davis, L.E.; Kim, A.; Macy, M.E.; Pietrofeso, A.; Ratan, R.; Riedel, R.F.; et al. Open-Label, Multicenter, Phase I/II, First-in-Human Trial of TK216: A First-Generation EWS: FLI1 Fusion Protein Antagonist in Ewing Sarcoma. J. Clin. Oncol. 2024, 42, 3725–3734. [Google Scholar] [CrossRef] [PubMed]
- Toretsky, J.A.; Erkizan, V.; Levenson, A.; Abaan, O.D.; Parvin, J.D.; Cripe, T.P.; Rice, A.M.; Lee, S.B.; Uren, A. Oncoprotein EWS-FLI1 activity is enhanced by RNA helicase A. Cancer Res. 2006, 66, 5574–5581. [Google Scholar] [CrossRef] [PubMed]
- Erkizan, H.V.; Kong, Y.L.; Merchant, M.; Schlottmann, S.; Barber-Rotenberg, J.S.; Yuan, L.S.; Abaan, O.D.; Chou, T.H.; Dakshanamurthy, S.; Brown, M.L.; et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat. Med. 2009, 15, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Rask, G.C.; Taslim, C.; Bayanjargal, A.; Cannon, M.V.; Selich-Anderson, J.; Crow, J.C.; Duncan, A.; Theisen, E.R. Seclidemstat blocks the transcriptional function of multiple FET-fusion oncoproteins. bioRxiv 2024. preprint. [Google Scholar]
- Sankar, S.; Theisen, E.R.; Bearss, J.; Mulvihill, T.; Hoffman, L.M.; Sorna, V.; Beckerle, M.C.; Sharma, S.; Lessnick, S.L. Reversible LSD1 Inhibition Interferes with Global EWS/ETS Transcriptional Activity and Impedes Ewing Sarcoma Tumor Growth. Clin. Cancer Res. 2014, 20, 4584–4597. [Google Scholar] [CrossRef]
- Rao, D.D.; Jay, C.; Wang, Z.H.; Luo, X.Q.; Kumar, P.; Eysenbach, H.; Ghisoli, M.; Senzer, N.; Nemunaitis, J. Preclinical Justification of pbi-shRNA EWS/FLI1 Lipoplex (LPX) Treatment for Ewing’s Sarcoma. Mol. Ther. 2016, 24, 1412–1422. [Google Scholar] [CrossRef]
- Rosenberg, A.; Agulnik, M. Epithelioid Hemangioendothelioma: Update on Diagnosis and Treatment. Curr. Treat. Options Oncol. 2018, 19, 19. [Google Scholar] [CrossRef]
- Weiss, S.W.; Enzinger, F.M. Epithelioid Hemangioendothelioma—A Vascular Tumor Often Mistaken for a Carcinoma. Cancer 1982, 50, 970–981. [Google Scholar] [CrossRef]
- Driskill, J.H.; Zheng, Y.G.; Wu, B.K.; Wang, L.; Cai, J.; Rakheja, D.; Dellinger, M.; Pan, D. WWTR1(TAZ)-CAMTA1 reprograms endothelial cells to drive epithelioid hemangioendothelioma. Genes Dev. 2021, 35, 495–511. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Miah, A.B.; Frezza, A.M.; Messiou, C.; Morosi, C.; Caraceni, A.; Antonescu, C.R.; Bajpai, J.; Baldini, E.; Bauer, S.; et al. Enithelioid hemangioendothelioma, an ultra-rare cancer: A consensus paper from the community of experts. ESMO Open 2021, 6, 100170. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Le Loarer, F.; Mosquera, J.M.; Sboner, A.; Zhang, L.; Chen, C.L.; Chen, H.W.; Pathan, N.; Krausz, T.; Dickson, B.C.; et al. Novel YAP1-TFE3 fusion defines a distinct subset of epithelioid hemangioendothelioma. Genes Chromosomes Cancer 2013, 52, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Park, J.; Feng, A.; Awasthi, P.; Wang, Z.Y.; Chen, Q.M.; Iglesias-Bartolome, R. YAP1/TAZ-TEAD transcriptional networks maintain skin homeostasis by regulating cell proliferation and limiting KLF4 activity. Nat. Commun. 2020, 11, 1472. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.T.J.; Thway, K.; Huang, P.H.; Jones, R.L. Clinical and Molecular Spectrum of Liposarcoma. J. Clin. Oncol. 2018, 36, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.S.E.; Colborne, S.; Hughes, C.S.; Morin, G.B.; Nielsen, T.O. The FUS-DDIT3 Interactome in Myxoid Liposarcoma. Neoplasia 2019, 21, 740–751. [Google Scholar] [CrossRef]
- Zullow, H.J.; Sankar, A.; Ingram, D.R.; Same Guerra, D.D.; D’Avino, A.R.; Collings, C.K.; Lazcano, R.; Wang, W.L.; Liang, Y.; Qi, J.; et al. The FUS:: DDIT3 fusion oncoprotein inhibits BAF complex targeting and activity in myxoid liposarcoma. Mol. Cell 2022, 82, 1737–1750. [Google Scholar] [CrossRef]
- Trautmann, M.; Menzel, J.; Bertling, C.; Cyra, M.; Isfort, I.; Steinestel, K.; Elges, S.; Grünewald, I.; Altvater, B.; Rossig, C.; et al. FUS-DDIT3 Fusion Protein-Driven IGF-IR Signaling is a Therapeutic Target in Myxoid Liposarcoma. Clin. Cancer Res. 2017, 23, 6227–6238. [Google Scholar] [CrossRef]
- Trautmann, M.; Cheng, Y.Y.; Jensen, P.; Azoitei, N.; Brunner, I.; Hullein, J.; Slabicki, M.; Isfort, I.; Cyra, M.; Berthold, R.; et al. Requirement for YAP1 signaling in myxoid liposarcoma. EMBO Mol. Med. 2019, 11, e9889. [Google Scholar] [CrossRef]
- Gazendam, A.M.; Popovic, S.; Munir, S.; Parasu, N.; Wilson, D.; Ghert, M. Synovial Sarcoma: A Clinical Review. Curr. Oncol. 2021, 28, 1909–1920. [Google Scholar] [CrossRef]
- Kadoch, C.; Crabtree, G.R. Reversible Disruption of mSWI/SNF (BAF) Complexes by the SS18-SSX Oncogenic Fusion in Synovial Sarcoma. Cell 2013, 153, 71–85. [Google Scholar] [CrossRef]
- McBride, M.J.; Pulice, J.L.; Beird, H.C.; Ingram, D.R.; D’Avino, A.R.; Shern, J.F.; Charville, G.W.; Hornick, J.L.; Nakayama, R.T.; Garcia-Rivera, E.M.; et al. The SS18-SSX Fusion Oncoprotein Hijacks BAF Complex Targeting and Function to Drive Synovial Sarcoma. Cancer Cell 2018, 33, 1128–1141. [Google Scholar] [CrossRef]
- Brien, G.L.; Remillard, D.; Shi, J.W.; Hemming, M.L.; Chabon, J.; Wynne, K.; Dillon, E.T.; Cagney, G.; Van Mierlo, G.; Baltissen, M.P.; et al. Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. eLife 2018, 7, e41305. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, M.; Sievers, E.; Aretz, S.; Kindler, D.; Michels, S.; Friedrichs, N.; Renner, M.; Kirfel, J.; Steiner, S.; Huss, S.; et al. SS18-SSX fusion protein-induced Wnt/β-catenin signaling is a therapeutic target in synovial sarcoma. Oncogene 2014, 33, 5006–5016. [Google Scholar] [CrossRef] [PubMed]
- Giraudet, A.L.; Cassier, P.A.; Iwao-Fukukawa, C.; Garin, G.; Badel, J.N.; Kryza, D.; Chabaud, S.; Gilles-Afchain, L.; Clapisson, G.; Desuzinges, C.; et al. A first-in-human study investigating biodistribution, safety and recommended dose of a new radiolabeled MAb targeting in metastatic synovial sarcoma patients. BMC Cancer 2018, 18, 646. [Google Scholar] [CrossRef] [PubMed]
- Al-Ibraheemi, A.; Ahrens, W.A.; Fritchie, K.; Dong, J.; Oliveira, A.M.; Balzer, B.; Folpe, A.L. Malignant Tenosynovial Giant Cell Tumor: The True “Synovial Sarcoma?” A Clinicopathologic, Immunohistochemical, and Molecular Cytogenetic Study of 10 Cases, Supporting Origin from Synoviocytes. Mod. Pathol. 2019, 32, 242–251. [Google Scholar] [CrossRef]
- West, R.B.; Rubin, B.P.; Miller, M.A.; Subramanian, S.; Kaygusuz, G.; Montgomery, K.; Zhu, S.; Marinelli, R.J.; De Luca, A.; Downs-Kelly, E.; et al. A landscape effect in tenosynovial giant-cell tumor from activation of CSF1 expression by a translocation in a minority of tumor cells. Proc. Natl. Acad. Sci. USA 2006, 103, 690–695. [Google Scholar] [CrossRef]
- Parham, D.M.; Barr, F.G. Classification of Rhabdomyosarcoma and Its Molecular Basis. Adv. Anat. Pathol. 2013, 20, 387–397. [Google Scholar] [CrossRef]
- Azorsa, D.O.; Bode, P.K.; Wachtel, M.; Cheuk, A.T.C.; Meltzer, P.S.; Vokuhl, C.; Camenisch, U.; Khov, H.L.; Bode, B.; Schäfer, B.W.; et al. Immunohistochemical detection of PAX-FOXO1 fusion proteins in alveolar rhabdomyosarcoma using breakpoint specific monoclonal antibodies. Mod. Pathol. 2021, 34, 748–757. [Google Scholar] [CrossRef]
- Barr, F.G. Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma. Oncogene 2001, 20, 5736–5746. [Google Scholar] [CrossRef]
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef]
- Singh, S.; Abu-Zaid, A.; Jin, H.J.; Fang, J.; Wu, Q.; Wang, T.T.; Feng, H.; Quarni, W.; Shao, Y.; Maxham, L.; et al. Targeting KDM4 for treating PAX3-FOXO1-driven alveolar rhabdomyosarcoma. Sci. Transl. Med. 2022, 14, eabq2096. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.S.; Olmos, D.; Missiaglia, E.; Shipley, J. Targeting the insulin-like growth factor pathway in rhabdomyosarcomas: Rationale and future perspectives. Sarcoma 2011, 2011, 209736. [Google Scholar] [CrossRef] [PubMed]
- Akshintala, S.; Bernstein, D.; Glod, J.; Kaplan, R.N.; Shern, J.F.; Yohe, M.E.; Gross, A.M.; Derdak, J.; Dombi, E.; Palacio-Yance, I.; et al. Results of a phase I trial of ganitumab plus dasatinib in patients with rhabdomyosarcoma (RMS). J. Clin. Oncol. 2022, 40, 11561. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.M.; Kalyana-Sundaram, S.; Cao, X.; Lonigro, R.J.; Sung, Y.S.; Chen, C.L.; Zhang, L.; Wang, R.; Su, F.; et al. Identification of recurrent NAB2-STAT6 gene fusions in solitary fibrous tumor by integrative sequencing. Nat. Genet. 2013, 45, 180–185. [Google Scholar] [CrossRef]
- Li, Y.; Mondaza-Hernandez, J.L.; Moura, D.S.; Revenko, A.S.; Tolentino, A.; Nguyen, J.T.; Tran, N.; Meyer, C.A.; Merino-Garcia, J.; Ramos, R.; et al. STAT6-targeting antisense oligonucleotides against solitary fibrous tumor. Mol. Ther. Nucleic Acids 2024, 35, 102154. [Google Scholar] [CrossRef]
- Ladanyi, M.; Lui, M.Y.; Antonescu, C.R.; Krause-Boehm, A.; Meindl, A.; Mertens, F.; Mandahl, N.; van den Berghe, H.; Sciot, R.; Dal Cin, P.; et al. The der (17) t (X; 17) (p11; q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Am. J. Hum. Genet. 2000, 67, 30. [Google Scholar] [CrossRef]
- Tanaka, M.; Homme, M.; Yamazaki, Y.; Shimizu, R.; Takazawa, Y.; Nakamura, T. Modeling Alveolar Soft Part Sarcoma Unveils Novel Mechanisms of Metastasis. Cancer Res. 2017, 77, 897–907. [Google Scholar] [CrossRef]
- Tsuda, M.; Davis, I.J.; Argani, P.; Shukla, N.; McGill, G.G.; Nagai, M.; Saito, T.; Laé, M.; Fisher, D.E.; Ladanyi, M. TFE3 fusions activate MET signaling by transcriptional up-regulation, defining another class of tumors as candidates for therapeutic MET inhibition. Cancer Res. 2007, 67, 919–929. [Google Scholar] [CrossRef]
- Alaggio, R.; Zhang, L.; Sung, Y.S.; Huang, S.C.; Chen, C.L.; Bisogno, G.; Zin, A.; Agaram, N.P.; LaQuaglia, M.P.; Wexler, L.H.; et al. A Molecular Study of Pediatric Spindle and Sclerosing Rhabdomyosarcoma Identification of Novel and Recurrent: VGLL2-: Related Fusions in Infantile Cases. Am. J. Surg. Pathol. 2016, 40, 224–235. [Google Scholar] [CrossRef]
- Yamaguchi, N. Multiple Roles of Vestigial-Like Family Members in Tumor Development. Front. Oncol. 2020, 10, 1266. [Google Scholar] [CrossRef]
- Agaram, N.P.; LaQuaglia, M.P.; Alaggio, R.; Zhang, L.; Fujisawa, Y.; Ladanyi, M.; Wexler, L.H.; Antonescu, C.R. MYOD1-mutant spindle cell and sclerosing rhabdomyosarcoma: An aggressive subtype irrespective of age. A reappraisal for molecular classification and risk stratification. Mod. Pathol. 2019, 32, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Folpe, A.L. Phosphaturic mesenchymal tumors: A review and update. Semin. Diagn. Pathol. 2019, 36, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Hartley, I.R.; Miller, C.B.; Papadakis, G.Z.; Bergwitz, C.; del Rivero, J.; Blau, J.E.; Florenzano, P.; Berglund, J.A.; Tassone, J.; Roszko, K.L.; et al. Targeted FGFR Blockade for the Treatment of Tumor-Induced Osteomalacia. N. Engl. J. Med. 2020, 383, 1387–1389. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.J.; Kim, J.J.; Ozsolak, F.; Widlund, H.R.; Rozenblatt-Rosen, O.; Granter, S.R.; Du, J.; Fletcher, J.A.; Denny, C.T.; Lessnick, S.L.; et al. Oncogenic MITF dysregulation in clear cell sarcoma: Defining the MiT family of human cancers. Cancer Cell 2006, 9, 473–484. [Google Scholar] [CrossRef]
- Davis, I.J.; McFadden, A.W.; Zhang, Y.X.; Coxon, A.; Burgess, T.L.; Wagner, A.J.; Fisher, D.E. Identification of the Receptor Tyrosine Kinase c-Met and Its Ligand, Hepatocyte Growth Factor, as Therapeutic Targets in Clear Cell Sarcoma. Cancer Res. 2010, 70, 639–645. [Google Scholar] [CrossRef]
- Mae, H.; Outani, H.; Imura, Y.; Chijimatsu, R.; Inoue, A.; Kotani, Y.; Yasuda, N.; Nakai, S.; Nakai, T.; Takenaka, S.; et al. Targeting the Clear Cell Sarcoma Oncogenic Driver Fusion Gene by HDAC Inhibition. Cancer Res. Commun. 2023, 3, 1152–1165. [Google Scholar] [CrossRef]
- Magrath, J.W.; Sampath, S.S.; Flinchum, D.A.; Hartono, A.B.; Goldberg, I.N.; Boehling, J.R.; Savkovic, S.D.; Lee, S.B. Comprehensive Transcriptomic Analysis of EWSR1:: WT1 Targets Identifies CDK4/6 Inhibitors as an Effective Therapy for Desmoplastic Small Round Cell Tumors. Cancer Res. 2024, 84, 1426–1442. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Baldi, G.G.; Morosi, C.; Gronchi, A.; Maestro, R. Extraskeletal Myxoid Chondrosarcoma: State of the Art and Current Research on Biology and Clinical Management. Cancers 2020, 12, 2703. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, Q.; Wang, T.; Zou, Z.; Ma, W.; Dong, Z.; Zhong, J.; Liu, W.; Xu, Y.; Hu, T.; Sun, W.; et al. Gene Fusions as Potential Therapeutic Targets in Soft Tissue Sarcomas. Biomolecules 2025, 15, 904. https://doi.org/10.3390/biom15060904
Zheng Q, Wang T, Zou Z, Ma W, Dong Z, Zhong J, Liu W, Xu Y, Hu T, Sun W, et al. Gene Fusions as Potential Therapeutic Targets in Soft Tissue Sarcomas. Biomolecules. 2025; 15(6):904. https://doi.org/10.3390/biom15060904
Chicago/Turabian StyleZheng, Qiongdan, Tong Wang, Zijian Zou, Wenjie Ma, Zirui Dong, Jingqin Zhong, Wanlin Liu, Yu Xu, Tu Hu, Wei Sun, and et al. 2025. "Gene Fusions as Potential Therapeutic Targets in Soft Tissue Sarcomas" Biomolecules 15, no. 6: 904. https://doi.org/10.3390/biom15060904
APA StyleZheng, Q., Wang, T., Zou, Z., Ma, W., Dong, Z., Zhong, J., Liu, W., Xu, Y., Hu, T., Sun, W., & Chen, Y. (2025). Gene Fusions as Potential Therapeutic Targets in Soft Tissue Sarcomas. Biomolecules, 15(6), 904. https://doi.org/10.3390/biom15060904