


Protein–Protein Interactions in Base Excision Repair

Abstract
1. Introduction
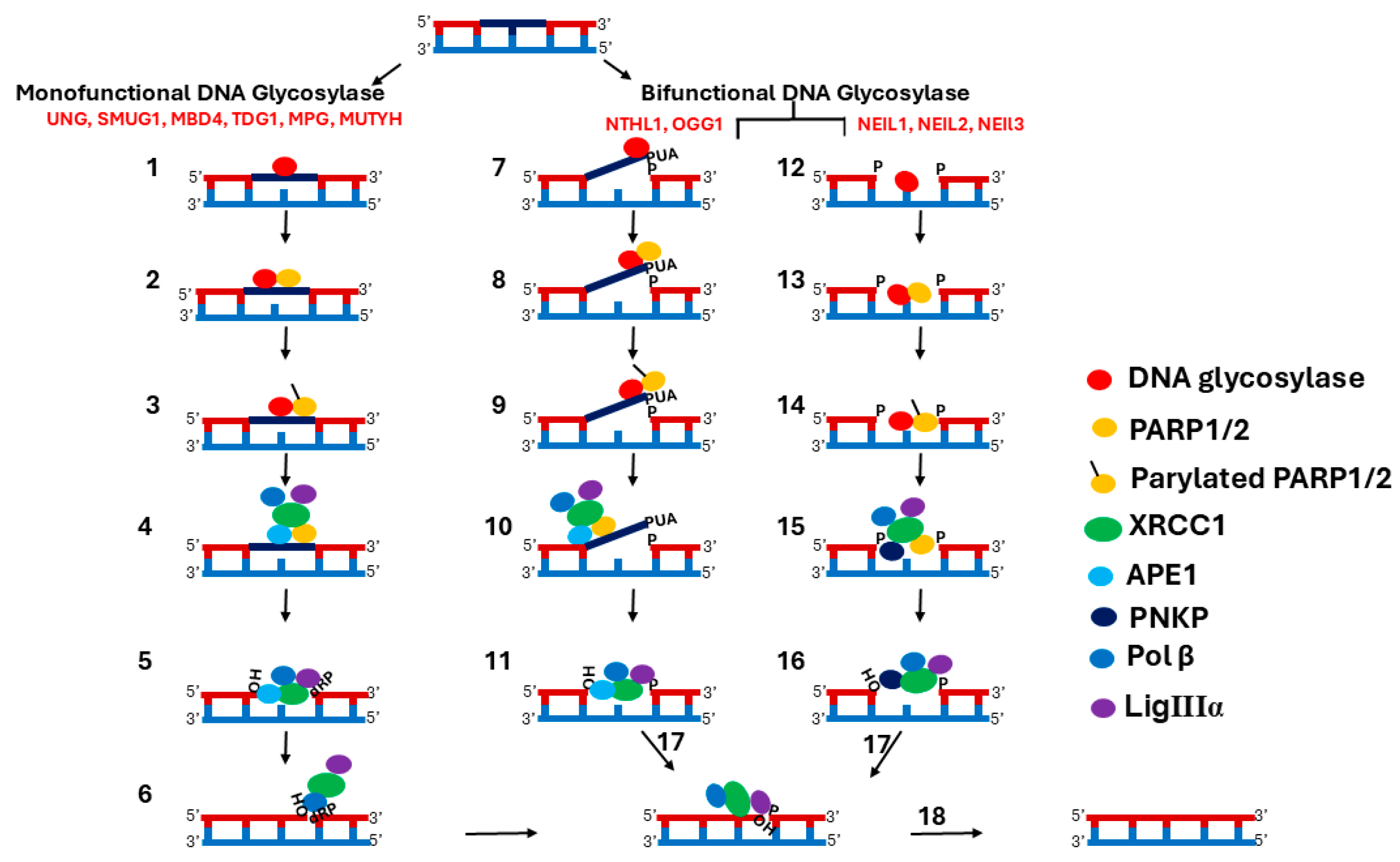
2. Single Nucleotide BER (SN-BER) Pathway
3. Interaction Between SN-BER Components
3.1. DNA Glycosylase—PARP1 Interaction
3.2. Interaction of DNA Glycosylases with Downstream BER Components
3.3. Interaction Between Downstream Components of SN-BER
4. Interaction of BER Components with Other Proteins
4.1. Uracil DNA Glycosylases
4.2. MBD4
4.3. TDG
4.4. MPG/AAG
4.5. MUTYH
4.6. NTHL1
4.7. OGG1
4.8. NEILs
4.9. XRCC1
4.10. APE1
4.11. PNKP
4.12. DNA Pol β
4.13. LigIIIα
4.14. APTX
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Wallace, S.S.; Murphy, D.L.; Sweasy, J.B. Base excision repair and cancer. Cancer Lett. 2012, 327, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Hindi, N.N.; Elsakrmy, N.; Ramotar, D. The base excision repair process: Comparison between higher and lower eukaryotes. Cell. Mol. Life Sci. 2021, 78, 7943–7965. [Google Scholar] [CrossRef]
- Dianov, G.L.; Hubscher, U. Mammalian base excision repair: The forgotten archangel. Nucleic Acids Res. 2013, 41, 3483–3490. [Google Scholar] [CrossRef]
- Prasad, R.; Shock, D.D.; Beard, W.A.; Wilson, S.H. Substrate channeling in mammalian base excision repair pathways: Passing the baton. J. Biol. Chem. 2010, 285, 40479–40488. [Google Scholar] [CrossRef] [PubMed]
- Abbotts, R.; Wilson, D.M., 3rd. Coordination of DNA single strand break repair. Free Radic. Biol. Med. 2017, 107, 228–244. [Google Scholar] [CrossRef]
- Caldecott, K.W. Mammalian DNA base excision repair: Dancing in the moonlight. DNA Repair 2020, 93, 102921. [Google Scholar] [CrossRef]
- Wilson, S.H.; Kunkel, T.A. Passing the baton in base excision repair. Nat. Struct. Biol. 2000, 7, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Mol, C.D.; Izumi, T.; Mitra, S.; Tainer, J.A. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination [corrected]. Nature 2000, 403, 451–456. [Google Scholar] [CrossRef]
- Hegde, M.L.; Hegde, P.M.; Bellot, L.J.; Mandal, S.M.; Hazra, T.K.; Li, G.M.; Boldogh, I.; Tomkinson, A.E.; Mitra, S. Prereplicative repair of oxidized bases in the human genome is mediated by NEIL1 DNA glycosylase together with replication proteins. Proc. Natl. Acad. Sci. USA 2013, 110, E3090–E3099. [Google Scholar] [CrossRef]
- Hegde, P.M.; Dutta, A.; Sengupta, S.; Mitra, J.; Adhikari, S.; Tomkinson, A.E.; Li, G.M.; Boldogh, I.; Hazra, T.K.; Mitra, S.; et al. The C-terminal Domain (CTD) of Human DNA Glycosylase NEIL1 Is Required for Forming BERosome Repair Complex with DNA Replication Proteins at the Replicating Genome: DOMINANT NEGATIVE FUNCTION OF THE CTD. J. Biol. Chem. 2015, 290, 20919–20933. [Google Scholar] [CrossRef]
- Limpose, K.L.; Corbett, A.H.; Doetsch, P.W. BERing the burden of damage: Pathway crosstalk and posttranslational modification of base excision repair proteins regulate DNA damage management. DNA Repair 2017, 56, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Moor, N.A.; Lavrik, O.I. Protein-Protein Interactions in DNA Base Excision Repair. Biochemistry 2018, 83, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Almeida, K.H.; Sobol, R.W. A unified view of base excision repair: Lesion-dependent protein complexes regulated by post-translational modification. DNA Repair 2007, 6, 695–711. [Google Scholar] [CrossRef] [PubMed]
- Saville, K.M.; Clark, J.; Wilk, A.; Rogers, G.D.; Andrews, J.F.; Koczor, C.A.; Sobol, R.W. NAD+-mediated regulation of mammalian base excision repair. DNA Repair 2020, 93, 102930. [Google Scholar] [CrossRef]
- Esadze, A.; Stivers, J.T. Facilitated Diffusion Mechanisms in DNA Base Excision Repair and Transcriptional Activation. Chem. Rev. 2018, 118, 11298–11323. [Google Scholar] [CrossRef]
- Kim, Y.J.; Wilson, D.M., 3rd. Overview of base excision repair biochemistry. Curr. Mol. Pharmacol. 2012, 5, 3–13. [Google Scholar] [CrossRef]
- Gallinari, P.; Jiricny, J. A new class of uracil-DNA glycosylases related to human thymine-DNA glycosylase. Nature 1996, 383, 735–738. [Google Scholar] [CrossRef]
- Lindahl, T. An N-glycosidase from Escherichia coli that releases free uracil from DNA containing deaminated cytosine residues. Proc. Natl. Acad. Sci. USA 1974, 71, 3649–3653. [Google Scholar] [CrossRef]
- Slupphaug, G.; Markussen, F.H.; Olsen, L.C.; Aasland, R.; Aarsaether, N.; Bakke, O.; Krokan, H.E.; Helland, D.E. Nuclear and mitochondrial forms of human uracil-DNA glycosylase are encoded by the same gene. Nucleic Acids Res. 1993, 21, 2579–2584. [Google Scholar] [CrossRef]
- Haushalter, K.A.; Todd Stukenberg, M.W.; Kirschner, M.W.; Verdine, G.L. Identification of a new uracil-DNA glycosylase family by expression cloning using synthetic inhibitors. Curr. Biol. 1999, 9, 174–185. [Google Scholar] [CrossRef]
- Boorstein, R.J.; Cummings, A., Jr.; Marenstein, D.R.; Chan, M.K.; Ma, Y.; Neubert, T.A.; Brown, S.M.; Teebor, G.W. Definitive identification of mammalian 5-hydroxymethyluracil DNA N-glycosylase activity as SMUG1. J. Biol. Chem. 2001, 276, 41991–41997. [Google Scholar] [CrossRef] [PubMed]
- Hendrich, B.; Bird, A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell. Biol. 1998, 18, 6538–6547. [Google Scholar] [CrossRef] [PubMed]
- Bellacosa, A.; Cicchillitti, L.; Schepis, F.; Riccio, A.; Yeung, A.T.; Matsumoto, Y.; Golemis, E.A.; Genuardi, M.; Neri, G. MED1, a novel human methyl-CpG-binding endonuclease, interacts with DNA mismatch repair protein MLH1. Proc. Natl. Acad. Sci. USA 1999, 96, 3969–3974. [Google Scholar] [CrossRef]
- Neddermann, P.; Gallinari, P.; Lettieri, T.; Schmid, D.; Truong, O.; Hsuan, J.J.; Wiebauer, K.; Jiricny, J. Cloning and expression of human G/T mismatch-specific thymine-DNA glycosylase. J. Biol. Chem. 1996, 271, 12767–12774. [Google Scholar] [CrossRef]
- Hardeland, U.; Bentele, M.; Jiricny, J.; Schar, P. The versatile thymine DNA-glycosylase: A comparative characterization of the human, Drosophila and fission yeast orthologs. Nucleic Acids Res. 2003, 31, 2261–2271. [Google Scholar] [CrossRef]
- O’Connor, T.R.; Laval, F. Isolation and structure of a cDNA expressing a mammalian 3-methyladenine-DNA glycosylase. EMBO J. 1990, 9, 3337–3342. [Google Scholar] [CrossRef]
- McGoldrick, J.P.; Yeh, Y.C.; Solomon, M.; Essigmann, J.M.; Lu, A.L. Characterization of a mammalian homolog of the Escherichia coli MutY mismatch repair protein. Mol. Cell. Biol. 1995, 15, 989–996. [Google Scholar] [CrossRef]
- Hilbert, T.P.; Chaung, W.; Boorstein, R.J.; Cunningham, R.P.; Teebor, G.W. Cloning and expression of the cDNA encoding the human homologue of the DNA repair enzyme, Escherichia coli endonuclease III. J. Biol. Chem. 1997, 272, 6733–6740. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Nash, H.M.; Verdine, G.L. A mammalian DNA repair enzyme that excises oxidatively damaged guanines maps to a locus frequently lost in lung cancer. Curr. Biol. 1997, 7, 397–407. [Google Scholar] [CrossRef]
- Hazra, T.K.; Izumi, T.; Boldogh, I.; Imhoff, B.; Kow, Y.W.; Jaruga, P.; Dizdaroglu, M.; Mitra, S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad. Sci. USA 2002, 99, 3523–3528. [Google Scholar] [CrossRef]
- Hazra, T.K.; Kow, Y.W.; Hatahet, Z.; Imhoff, B.; Boldogh, I.; Mokkapati, S.K.; Mitra, S.; Izumi, T. Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J. Biol. Chem. 2002, 277, 30417–30420. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Bandaru, V.; Bond, J.P.; Jaruga, P.; Zhao, X.; Christov, P.P.; Burrows, C.J.; Rizzo, C.J.; Dizdaroglu, M.; Wallace, S.S. The mouse ortholog of NEIL3 is a functional DNA glycosylase In Vitro and In Vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 4925–4930. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, G.K.; Yin, Y.W. The Role of Poly(ADP-ribose) Polymerase 1 in Nuclear and Mitochondrial Base Excision Repair. Biomolecules 2023, 13, 1195. [Google Scholar] [CrossRef]
- McCullough, A.K.; Sanchez, A.; Dodson, M.L.; Marapaka, P.; Taylor, J.S.; Lloyd, R.S. The reaction mechanism of DNA glycosylase/AP lyases at abasic sites. Biochemistry 2001, 40, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Latham, K.A.; Dodson, M.L.; Lloyd, R.S. Studies on the catalytic mechanism of five DNA glycosylases. Probing for enzyme-DNA imino intermediates. J. Biol. Chem. 1995, 270, 19501–19508. [Google Scholar] [CrossRef]
- Zharkov, D.O.; Rieger, R.A.; Iden, C.R.; Grollman, A.P. NH2-terminal proline acts as a nucleophile in the glycosylase/AP-lyase reaction catalyzed by Escherichia coli formamidopyrimidine-DNA glycosylase (Fpg) protein. J. Biol. Chem. 1997, 272, 5335–5341. [Google Scholar] [CrossRef]
- Vidal, A.E.; Hickson, I.D.; Boiteux, S.; Radicella, J.P. Mechanism of stimulation of the DNA glycosylase activity of hOGG1 by the major human AP endonuclease: Bypass of the AP lyase activity step. Nucleic Acids Res. 2001, 29, 1285–1292. [Google Scholar] [CrossRef]
- McNeill, D.R.; Whitaker, A.M.; Stark, W.J.; Illuzzi, J.L.; McKinnon, P.J.; Freudenthal, B.D.; Wilson, D.M., 3rd. Functions of the major abasic endonuclease (APE1) in cell viability and genotoxin resistance. Mutagenesis 2020, 35, 27–38. [Google Scholar] [CrossRef]
- Maher, R.L.; Wallace, S.S.; Pederson, D.S. The lyase activity of bifunctional DNA glycosylases and the 3′-diesterase activity of APE1 contribute to the repair of oxidized bases in nucleosomes. Nucleic Acids Res. 2019, 47, 2922–2931. [Google Scholar] [CrossRef]
- Satoh, M.S.; Lindahl, T. Role of poly(ADP-ribose) formation in DNA repair. Nature 1992, 356, 356–358. [Google Scholar] [CrossRef]
- Schwarz, S.D.; Xu, J.; Gunasekera, K.; Schurmann, D.; Vagbo, C.B.; Ferrari, E.; Slupphaug, G.; Hottiger, M.O.; Schar, P.; Steinacher, R. Covalent PARylation of DNA base excision repair proteins regulates DNA demethylation. Nat. Commun. 2024, 15, 184. [Google Scholar] [CrossRef] [PubMed]
- Hanzlikova, H.; Gittens, W.; Krejcikova, K.; Zeng, Z.; Caldecott, K.W. Overlapping roles for PARP1 and PARP2 in the recruitment of endogenous XRCC1 and PNKP into oxidized chromatin. Nucleic Acids Res. 2017, 45, 2546–2557. [Google Scholar] [CrossRef] [PubMed]
- Lavrik, O.I. PARPs’ impact on base excision DNA repair. DNA Repair 2020, 93, 102911. [Google Scholar] [CrossRef]
- Zhang, H.; Zha, S. The dynamics and regulation of PARP1 and PARP2 in response to DNA damage and during replication. DNA Repair 2024, 140, 103690. [Google Scholar] [CrossRef]
- Ronson, G.E.; Piberger, A.L.; Higgs, M.R.; Olsen, A.L.; Stewart, G.S.; McHugh, P.J.; Petermann, E.; Lakin, N.D. PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1-dependent Rad51 regulation. Nat. Commun. 2018, 9, 746. [Google Scholar] [CrossRef] [PubMed]
- Langelier, M.F.; Planck, J.L.; Roy, S.; Pascal, J.M. Crystal structures of poly(ADP-ribose) polymerase-1 (PARP-1) zinc fingers bound to DNA: Structural and functional insights into DNA-dependent PARP-1 activity. J. Biol. Chem. 2011, 286, 10690–10701. [Google Scholar] [CrossRef]
- Langelier, M.F.; Riccio, A.A.; Pascal, J.M. PARP-2 and PARP-3 are selectively activated by 5′ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res. 2014, 42, 7762–7775. [Google Scholar] [CrossRef]
- Riccio, A.A.; Cingolani, G.; Pascal, J.M. PARP-2 domain requirements for DNA damage-dependent activation and localization to sites of DNA damage. Nucleic Acids Res. 2016, 44, 1691–1702. [Google Scholar] [CrossRef]
- Khodyreva, S.N.; Prasad, R.; Ilina, E.S.; Sukhanova, M.V.; Kutuzov, M.M.; Liu, Y.; Hou, E.W.; Wilson, S.H.; Lavrik, O.I. Apurinic/apyrimidinic (AP) site recognition by the 5′-dRP/AP lyase in poly(ADP-ribose) polymerase-1 (PARP-1). Proc. Natl. Acad. Sci. USA 2010, 107, 22090–22095. [Google Scholar] [CrossRef]
- Lebedeva, N.A.; Dyrkheeva, N.S.; Rechkunova, N.I.; Lavrik, O.I. Apurinic/apyrimidinic endonuclease 1 has major impact in prevention of suicidal covalent DNA-protein crosslink with apurinic/apyrimidinic site in cellular extracts. IUBMB Life 2024, 76, 987–996. [Google Scholar] [CrossRef]
- Demin, A.A.; Hirota, K.; Tsuda, M.; Adamowicz, M.; Hailstone, R.; Brazina, J.; Gittens, W.; Kalasova, I.; Shao, Z.; Zha, S.; et al. XRCC1 prevents toxic PARP1 trapping during DNA base excision repair. Mol. Cell 2021, 81, 3018–3030.e5. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, A.M.; Schaich, M.A.; Smith, M.R.; Flynn, T.S.; Freudenthal, B.D. Base excision repair of oxidative DNA damage: From mechanism to disease. Front. Biosci. 2017, 22, 1493–1522. [Google Scholar]
- Wei, H.; Yu, X. Functions of PARylation in DNA Damage Repair Pathways. Genom. Proteom. Bioinform. 2016, 14, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Breslin, C.; Hornyak, P.; Ridley, A.; Rulten, S.L.; Hanzlikova, H.; Oliver, A.W.; Caldecott, K.W. The XRCC1 phosphate-binding pocket binds poly (ADP-ribose) and is required for XRCC1 function. Nucleic Acids Res. 2015, 43, 6934–6944. [Google Scholar] [CrossRef]
- Ferro, A.M.; Olivera, B.M. Poly(ADP-ribosylation) In Vitro. Reaction parameters and enzyme mechanism. J. Biol. Chem. 1982, 257, 7808–7813. [Google Scholar] [CrossRef]
- Zahradka, P.; Ebisuzaki, K. A shuttle mechanism for DNA-protein interactions. The regulation of poly(ADP-ribose) polymerase. Eur. J. Biochem. 1982, 127, 579–585. [Google Scholar] [CrossRef]
- Masson, M.; Niedergang, C.; Schreiber, V.; Muller, S.; Menissier-de Murcia, J.; de Murcia, G. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol. Cell. Biol. 1998, 18, 3563–3571. [Google Scholar] [CrossRef]
- Akbari, M.; Solvang-Garten, K.; Hanssen-Bauer, A.; Lieske, N.V.; Pettersen, H.S.; Pettersen, G.K.; Wilson, D.M., 3rd; Krokan, H.E.; Otterlei, M. Direct interaction between XRCC1 and UNG2 facilitates rapid repair of uracil in DNA by XRCC1 complexes. DNA Repair 2010, 9, 785–795. [Google Scholar] [CrossRef]
- Campalans, A.; Marsin, S.; Nakabeppu, Y.; O’Connor, T.R.; Boiteux, S.; Radicella, J.P. XRCC1 interactions with multiple DNA glycosylases: A model for its recruitment to base excision repair. DNA Repair 2005, 4, 826–835. [Google Scholar] [CrossRef]
- Marsin, S.; Vidal, A.E.; Sossou, M.; Menissier-de Murcia, J.; Le Page, F.; Boiteux, S.; de Murcia, G.; Radicella, J.P. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. J. Biol. Chem. 2003, 278, 44068–44074. [Google Scholar] [CrossRef]
- Wiederhold, L.; Leppard, J.B.; Kedar, P.; Karimi-Busheri, F.; Rasouli-Nia, A.; Weinfeld, M.; Tomkinson, A.E.; Izumi, T.; Prasad, R.; Wilson, S.H.; et al. AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell 2004, 15, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Marintchev, A.; Mullen, M.A.; Maciejewski, M.W.; Pan, B.; Gryk, M.R.; Mullen, G.P. Solution structure of the single-strand break repair protein XRCC1 N-terminal domain. Nat. Struct. Biol. 1999, 6, 884–893. [Google Scholar] [PubMed]
- London, R.E. The structural basis of XRCC1-mediated DNA repair. DNA Repair 2015, 30, 90–103. [Google Scholar] [CrossRef]
- Almohdar, D.; Murcia, D.; Tang, Q.; Ortiz, A.; Martinez, E.; Parwal, T.; Kamble, P.; Caglayan, M. Impact of DNA ligase 1 and IIIalpha interactions with APE1 and polbeta on the efficiency of base excision repair pathway at the downstream steps. J. Biol. Chem. 2024, 300, 107355. [Google Scholar] [CrossRef]
- Koczor, C.A.; Thompson, M.K.; Sharma, N.; Prakash, A.; Sobol, R.W. Polbeta/XRCC1 heterodimerization dictates DNA damage recognition and basal Polbeta protein levels without interfering with mouse viability or fertility. DNA Repair 2023, 123, 103452. [Google Scholar] [CrossRef] [PubMed]
- Kubota, Y.; Nash, R.A.; Klungland, A.; Schar, P.; Barnes, D.E.; Lindahl, T. Reconstitution of DNA base excision-repair with purified human proteins: Interaction between DNA polymerase beta and the XRCC1 protein. EMBO J. 1996, 15, 6662–6670. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, C.J.; Taylor, R.M.; Thistlethwaite, A.; Zhang, H.; Karimi-Busheri, F.; Lasko, D.D.; Weinfeld, M.; Caldecott, K.W. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell 2001, 104, 107–117. [Google Scholar] [CrossRef]
- Yamamori, T.; DeRicco, J.; Naqvi, A.; Hoffman, T.A.; Mattagajasingh, I.; Kasuno, K.; Jung, S.B.; Kim, C.S.; Irani, K. SIRT1 deacetylates APE1 and regulates cellular base excision repair. Nucleic Acids Res. 2010, 38, 832–845. [Google Scholar] [CrossRef]
- Demple, B.; Harrison, L. Repair of oxidative damage to DNA: Enzymology and biology. Annu. Rev. Biochem. 1994, 63, 915–948. [Google Scholar] [CrossRef]
- Singhal, R.K.; Prasad, R.; Wilson, S.H. DNA polymerase beta conducts the gap-filling step in uracil-initiated base excision repair in a bovine testis nuclear extract. J. Biol. Chem. 1995, 270, 949–957. [Google Scholar] [CrossRef]
- Rass, U.; Ahel, I.; West, S.C. Defective DNA repair and neurodegenerative disease. Cell 2007, 130, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Ahel, I.; Rass, U.; El-Khamisy, S.F.; Katyal, S.; Clements, P.M.; McKinnon, P.J.; Caldecott, K.W.; West, S.C. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature 2006, 443, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Rass, U.; Ahel, I.; West, S.C. Actions of aprataxin in multiple DNA repair pathways. J. Biol. Chem. 2007, 282, 9469–9474. [Google Scholar] [CrossRef]
- Noren Hooten, N.; Fitzpatrick, M.; Kompaniez, K.; Jacob, K.D.; Moore, B.R.; Nagle, J.; Barnes, J.; Lohani, A.; Evans, M.K. Coordination of DNA repair by NEIL1 and PARP-1: A possible link to aging. Aging 2012, 4, 674–685. [Google Scholar] [CrossRef]
- Noren Hooten, N.; Kompaniez, K.; Barnes, J.; Lohani, A.; Evans, M.K. Poly(ADP-ribose) polymerase 1 (PARP-1) binds to 8-oxoguanine-DNA glycosylase (OGG1). J. Biol. Chem. 2011, 286, 44679–44690. [Google Scholar] [CrossRef]
- Waters, T.R.; Gallinari, P.; Jiricny, J.; Swann, P.F. Human thymine DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. J. Biol. Chem. 1999, 274, 67–74. [Google Scholar] [CrossRef]
- Baldwin, M.R.; O’Brien, P.J. Human AP endonuclease 1 stimulates multiple-turnover base excision by alkyladenine DNA glycosylase. Biochemistry 2009, 48, 6022–6033. [Google Scholar] [CrossRef]
- Baldwin, M.R.; O’Brien, P.J. Nonspecific DNA binding and coordination of the first two steps of base excision repair. Biochemistry 2010, 49, 7879–7891. [Google Scholar] [CrossRef] [PubMed]
- Esadze, A.; Rodriguez, G.; Cravens, S.L.; Stivers, J.T. AP-Endonuclease 1 Accelerates Turnover of Human 8-Oxoguanine DNA Glycosylase by Preventing Retrograde Binding to the Abasic-Site Product. Biochemistry 2017, 56, 1974–1986. [Google Scholar] [CrossRef]
- Fitzgerald, M.E.; Drohat, A.C. Coordinating the initial steps of base excision repair. Apurinic/apyrimidinic endonuclease 1 actively stimulates thymine DNA glycosylase by disrupting the product complex. J. Biol. Chem. 2008, 283, 32680–32690. [Google Scholar] [CrossRef]
- Hill, J.W.; Hazra, T.K.; Izumi, T.; Mitra, S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: Potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 2001, 29, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Luncsford, P.J.; Manvilla, B.A.; Patterson, D.N.; Malik, S.S.; Jin, J.; Hwang, B.J.; Gunther, R.; Kalvakolanu, S.; Lipinski, L.J.; Yuan, W.; et al. Coordination of MYH DNA glycosylase and APE1 endonuclease activities via physical interactions. DNA Repair 2013, 12, 1043–1052. [Google Scholar] [CrossRef]
- Marenstein, D.R.; Chan, M.K.; Altamirano, A.; Basu, A.K.; Boorstein, R.J.; Cunningham, R.P.; Teebor, G.W. Substrate specificity of human endonuclease III (hNTH1). Effect of human APE1 on hNTH1 activity. J. Biol. Chem. 2003, 278, 9005–9012. [Google Scholar] [CrossRef] [PubMed]
- Sidorenko, V.S.; Nevinsky, G.A.; Zharkov, D.O. Specificity of stimulation of human 8-oxoguanine-DNA glycosylase by AP endonuclease. Biochem. Biophys. Res. Commun. 2008, 368, 175–179. [Google Scholar] [CrossRef]
- Das, A.; Wiederhold, L.; Leppard, J.B.; Kedar, P.; Prasad, R.; Wang, H.; Boldogh, I.; Karimi-Busheri, F.; Weinfeld, M.; Tomkinson, A.E.; et al. NEIL2-initiated, APE-independent repair of oxidized bases in DNA: Evidence for a repair complex in human cells. DNA Repair 2006, 5, 1439–1448. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.L.; Hegde, P.M.; Arijit, D.; Boldogh, I.; Mitra, S. Human DNA Glycosylase NEIL1’s Interactions with Downstream Repair Proteins Is Critical for Efficient Repair of Oxidized DNA Base Damage and Enhanced Cell Survival. Biomolecules 2012, 2, 564–578. [Google Scholar] [CrossRef]
- Braithwaite, E.K.; Kedar, P.S.; Stumpo, D.J.; Bertocci, B.; Freedman, J.H.; Samson, L.D.; Wilson, S.H. DNA polymerases beta and lambda mediate overlapping and independent roles in base excision repair in mouse embryonic fibroblasts. PLoS ONE 2010, 5, e12229. [Google Scholar] [CrossRef]
- Bakman, A.S.; Boichenko, S.S.; Kuznetsova, A.A.; Ishchenko, A.A.; Saparbaev, M.; Kuznetsov, N.A. The Impact of Human DNA Glycosylases on the Activity of DNA Polymerase beta toward Various Base Excision Repair Intermediates. Int. J. Mol. Sci. 2023, 24, 9594. [Google Scholar] [CrossRef]
- Vidal, A.E.; Boiteux, S.; Hickson, I.D.; Radicella, J.P. XRCC1 coordinates the initial and late stages of DNA abasic site repair through protein-protein interactions. EMBO J. 2001, 20, 6530–6539. [Google Scholar] [CrossRef]
- Prasad, R.; Dyrkheeva, N.; Williams, J.; Wilson, S.H. Mammalian Base Excision Repair: Functional Partnership between PARP-1 and APE1 in AP-Site Repair. PLoS ONE 2015, 10, e0124269. [Google Scholar] [CrossRef]
- Wong, D.; DeMott, M.S.; Demple, B. Modulation of the 3′→5′-exonuclease activity of human apurinic endonuclease (Ape1) by its 5′-incised Abasic DNA product. J. Biol. Chem. 2003, 278, 36242–36249. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, A.M.; Flynn, T.S.; Freudenthal, B.D. Molecular snapshots of APE1 proofreading mismatches and removing DNA damage. Nat. Commun. 2018, 9, 399. [Google Scholar] [CrossRef]
- Liu, Y.; Prasad, R.; Beard, W.A.; Kedar, P.S.; Hou, E.W.; Shock, D.D.; Wilson, S.H. Coordination of steps in single-nucleotide base excision repair mediated by apurinic/apyrimidinic endonuclease 1 and DNA polymerase beta. J. Biol. Chem. 2007, 282, 13532–13541. [Google Scholar] [CrossRef] [PubMed]
- Bakman, A.S.; Boichenko, S.S.; Kuznetsova, A.A.; Ishchenko, A.A.; Saparbaev, M.; Kuznetsov, N.A. Coordination between human DNA polymerase beta and apurinic/apyrimidinic endonuclease 1 in the course of DNA repair. Biochimie 2024, 216, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Fairlamb, M.S.; Spies, M.; Washington, M.T.; Freudenthal, B.D. Visualizing the coordination of apurinic/apyrimidinic endonuclease (APE1) and DNA polymerase beta during base excision repair. J. Biol. Chem. 2023, 299, 104636. [Google Scholar] [CrossRef]
- Moor, N.A.; Vasil’eva, I.A.; Anarbaev, R.O.; Antson, A.A.; Lavrik, O.I. Quantitative characterization of protein-protein complexes involved in base excision DNA repair. Nucleic Acids Res. 2015, 43, 6009–6022. [Google Scholar] [CrossRef]
- Berquist, B.R.; Singh, D.K.; Fan, J.; Kim, D.; Gillenwater, E.; Kulkarni, A.; Bohr, V.A.; Ackerman, E.J.; Tomkinson, A.E.; Wilson, D.M., 3rd. Functional capacity of XRCC1 protein variants identified in DNA repair-deficient Chinese hamster ovary cell lines and the human population. Nucleic Acids Res. 2010, 38, 5023–5035. [Google Scholar] [CrossRef]
- Fang, Q.; Inanc, B.; Schamus, S.; Wang, X.H.; Wei, L.; Brown, A.R.; Svilar, D.; Sugrue, K.F.; Goellner, E.M.; Zeng, X.; et al. HSP90 regulates DNA repair via the interaction between XRCC1 and DNA polymerase beta. Nat. Commun. 2014, 5, 5513. [Google Scholar] [CrossRef]
- Nash, R.A.; Caldecott, K.W.; Barnes, D.E.; Lindahl, T. XRCC1 protein interacts with one of two distinct forms of DNA ligase III. Biochemistry 1997, 36, 5207–5211. [Google Scholar] [CrossRef]
- Taylor, R.M.; Wickstead, B.; Cronin, S.; Caldecott, K.W. Role of a BRCT domain in the interaction of DNA ligase III-alpha with the DNA repair protein XRCC1. Curr. Biol. 1998, 8, 877–880. [Google Scholar] [CrossRef]
- Dulic, A.; Bates, P.A.; Zhang, X.; Martin, S.R.; Freemont, P.S.; Lindahl, T.; Barnes, D.E. BRCT domain interactions in the heterodimeric DNA repair protein XRCC1-DNA ligase III. Biochemistry 2001, 40, 5906–5913. [Google Scholar] [CrossRef] [PubMed]
- Gueven, N.; Becherel, O.J.; Kijas, A.W.; Chen, P.; Howe, O.; Rudolph, J.H.; Gatti, R.; Date, H.; Onodera, O.; Taucher-Scholz, G.; et al. Aprataxin, a novel protein that protects against genotoxic stress. Hum. Mol. Genet. 2004, 13, 1081–1093. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Chan, D.W.; Yang, T.; Rodriguez, M.; Chen, B.P.; Leng, M.; Mu, J.J.; Chen, D.; Songyang, Z.; Wang, Y.; et al. A new XRCC1-containing complex and its role in cellular survival of methyl methanesulfonate treatment. Mol. Cell. Biol. 2004, 24, 8356–8365. [Google Scholar] [CrossRef]
- Rashid, I.; Hammel, M.; Sverzhinsky, A.; Tsai, M.S.; Pascal, J.M.; Tainer, J.A.; Tomkinson, A.E. Direct interaction of DNA repair protein tyrosyl DNA phosphodiesterase 1 and the DNA ligase III catalytic domain is regulated by phosphorylation of its flexible N-terminus. J. Biol. Chem. 2021, 297, 100921. [Google Scholar] [CrossRef]
- Chiang, S.C.; Carroll, J.; El-Khamisy, S.F. TDP1 serine 81 promotes interaction with DNA ligase IIIalpha and facilitates cell survival following DNA damage. Cell Cycle 2010, 9, 588–595. [Google Scholar] [CrossRef]
- Moor, N.; Vasil’eva, I.; Lavrik, O. Functional Role of N-Terminal Extension of Human AP Endonuclease 1 In Coordination of Base Excision DNA Repair via Protein-Protein Interactions. Int. J. Mol. Sci. 2020, 21, 3122. [Google Scholar] [CrossRef]
- Nagelhus, T.A.; Haug, T.; Singh, K.K.; Keshav, K.F.; Skorpen, F.; Otterlei, M.; Bharati, S.; Lindmo, T.; Benichou, S.; Benarous, R.; et al. A sequence in the N-terminal region of human uracil-DNA glycosylase with homology to XPA interacts with the C-terminal part of the 34-kDa subunit of replication protein A. J. Biol. Chem. 1997, 272, 6561–6566. [Google Scholar] [CrossRef] [PubMed]
- Otterlei, M.; Warbrick, E.; Nagelhus, T.A.; Haug, T.; Slupphaug, G.; Akbari, M.; Aas, P.A.; Steinsbekk, K.; Bakke, O.; Krokan, H.E. Post-replicative base excision repair in replication foci. EMBO J. 1999, 18, 3834–3844. [Google Scholar] [CrossRef]
- Wen, X.; Casey Klockow, L.; Nekorchuk, M.; Sharifi, H.J.; de Noronha, C.M. The HIV1 protein Vpr acts to enhance constitutive DCAF1-dependent UNG2 turnover. PLoS ONE 2012, 7, e30939. [Google Scholar] [CrossRef]
- Schrofelbauer, B.; Yu, Q.; Zeitlin, S.G.; Landau, N.R. Human immunodeficiency virus type 1 Vpr induces the degradation of the UNG and SMUG uracil-DNA glycosylases. J. Virol. 2005, 79, 10978–10987. [Google Scholar] [CrossRef]
- Ahn, J.; Vu, T.; Novince, Z.; Guerrero-Santoro, J.; Rapic-Otrin, V.; Gronenborn, A.M. HIV-1 Vpr loads uracil DNA glycosylase-2 onto DCAF1, a substrate recognition subunit of a cullin 4A-ring E3 ubiquitin ligase for proteasome-dependent degradation. J. Biol. Chem. 2010, 285, 37333–37341. [Google Scholar] [CrossRef] [PubMed]
- Eldin, P.; Peron, S.; Galashevskaya, A.; Denis-Lagache, N.; Cogne, M.; Slupphaug, G.; Briant, L. Impact of HIV-1 Vpr manipulation of the DNA repair enzyme UNG2 on B lymphocyte class switch recombination. J. Transl. Med. 2020, 18, 310. [Google Scholar] [CrossRef]
- Screaton, R.A.; Kiessling, S.; Sansom, O.J.; Millar, C.B.; Maddison, K.; Bird, A.; Clarke, A.R.; Frisch, S.M. Fas-associated death domain protein interacts with methyl-CpG binding domain protein 4: A potential link between genome surveillance and apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 5211–5216. [Google Scholar] [CrossRef] [PubMed]
- Fukushige, S.; Kondo, E.; Gu, Z.; Suzuki, H.; Horii, A. RET finger protein enhances MBD2- and MBD4-dependent transcriptional repression. Biochem. Biophys. Res. Commun. 2006, 351, 85–92. [Google Scholar] [CrossRef]
- Kondo, E.; Gu, Z.; Horii, A.; Fukushige, S. The thymine DNA glycosylase MBD4 represses transcription and is associated with methylated p16(INK4a) and hMLH1 genes. Mol. Cell. Biol. 2005, 25, 4388–4396. [Google Scholar] [CrossRef]
- Meng, H.; Harrison, D.J.; Meehan, R.R. MBD4 interacts with and recruits USP7 to heterochromatic foci. J. Cell. Biochem. 2015, 116, 476–485. [Google Scholar] [CrossRef]
- Sannai, M.; Doneddu, V.; Giri, V.; Seeholzer, S.; Nicolas, E.; Yip, S.C.; Bassi, M.R.; Mancuso, P.; Cortellino, S.; Cigliano, A.; et al. Modification of the base excision repair enzyme MBD4 by the small ubiquitin-like molecule SUMO1. DNA Repair 2019, 82, 102687. [Google Scholar] [CrossRef]
- Tini, M.; Benecke, A.; Um, S.J.; Torchia, J.; Evans, R.M.; Chambon, P. Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Mol. Cell 2002, 9, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Iwai, S.; Hanaoka, F.; Sugasawa, K. Xeroderma pigmentosum group C protein interacts physically and functionally with thymine DNA glycosylase. EMBO J. 2003, 22, 164–173. [Google Scholar] [CrossRef]
- Boland, M.J.; Christman, J.K. Characterization of Dnmt3b:thymine-DNA glycosylase interaction and stimulation of thymine glycosylase-mediated repair by DNA methyltransferase(s) and RNA. J. Mol. Biol. 2008, 379, 492–504. [Google Scholar] [CrossRef]
- Miao, J.; Zhao, C.; Tang, K.; Xiong, X.; Wu, F.; Xue, W.; Duan, B.; Zhang, H.; Jing, X.; Li, W.; et al. TDG suppresses the migration and invasion of human colon cancer cells via the DNMT3A/TIMP2 axis. Int. J. Biol. Sci. 2022, 18, 2527–2539. [Google Scholar] [CrossRef]
- Hu, X.V.; Rodrigues, T.M.; Tao, H.; Baker, R.K.; Miraglia, L.; Orth, A.P.; Lyons, G.E.; Schultz, P.G.; Wu, X. Identification of RING finger protein 4 (RNF4) as a modulator of DNA demethylation through a functional genomics screen. Proc. Natl. Acad. Sci. USA 2010, 107, 15087–15092. [Google Scholar] [CrossRef] [PubMed]
- Shibata, E.; Dar, A.; Dutta, A. CRL4Cdt2 E3 ubiquitin ligase and proliferating cell nuclear antigen (PCNA) cooperate to degrade thymine DNA glycosylase in S phase. J. Biol. Chem. 2014, 289, 23056–23064. [Google Scholar] [CrossRef]
- Hardeland, U.; Steinacher, R.; Jiricny, J.; Schar, P. Modification of the human thymine-DNA glycosylase by ubiquitin-like proteins facilitates enzymatic turnover. EMBO J. 2002, 21, 1456–1464. [Google Scholar] [CrossRef] [PubMed]
- Minty, A.; Dumont, X.; Kaghad, M.; Caput, D. Covalent modification of p73alpha by SUMO-1. Two-hybrid screening with p73 identifies novel SUMO-1-interacting proteins and a SUMO-1 interaction motif. J. Biol. Chem. 2000, 275, 36316–36323. [Google Scholar] [CrossRef]
- Smet-Nocca, C.; Wieruszeski, J.M.; Leger, H.; Eilebrecht, S.; Benecke, A. SUMO-1 regulates the conformational dynamics of thymine-DNA Glycosylase regulatory domain and competes with its DNA binding activity. BMC Biochem. 2011, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Likhite, V.S.; Cass, E.I.; Anderson, S.D.; Yates, J.R.; Nardulli, A.M. Interaction of estrogen receptor alpha with 3-methyladenine DNA glycosylase modulates transcription and DNA repair. J. Biol. Chem. 2004, 279, 16875–16882. [Google Scholar] [CrossRef]
- Tang, S.; Stokasimov, E.; Cui, Y.; Pellman, D. Breakage of cytoplasmic chromosomes by pathological DNA base excision repair. Nature 2022, 606, 930–936. [Google Scholar] [CrossRef]
- Xia, L.; Zheng, L.; Lee, H.W.; Bates, S.E.; Federico, L.; Shen, B.; O’Connor, T.R. Human 3-methyladenine-DNA glycosylase: Effect of sequence context on excision, association with PCNA, and stimulation by AP endonuclease. J. Mol. Biol. 2005, 346, 1259–1274. [Google Scholar] [CrossRef]
- Miao, F.; Bouziane, M.; Dammann, R.; Masutani, C.; Hanaoka, F.; Pfeifer, G.; O’Connor, T.R. 3-Methyladenine-DNA glycosylase (MPG protein) interacts with human RAD23 proteins. J. Biol. Chem. 2000, 275, 28433–28438. [Google Scholar] [CrossRef]
- Jang, S.; Kumar, N.; Schaich, M.A.; Zhong, Z.; van Loon, B.; Watkins, S.C.; Van Houten, B. Cooperative interaction between AAG and UV-DDB in the removal of modified bases. Nucleic Acids Res. 2022, 50, 12856–12871. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Zhang, X.; Song, S.; Tian, C.; Yin, Y.; Xing, G.; He, F.; Zhang, L. Identification of UHRF1/2 as new N-methylpurine DNA glycosylase-interacting proteins. Biochem. Biophys. Res. Commun. 2013, 433, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.; Gu, Y.; Mahoney, W.; Lee, S.H.; Singh, K.K.; Lu, A.L. Human homolog of the MutY repair protein (hMYH) physically interacts with proteins involved in long patch DNA base excision repair. J. Biol. Chem. 2001, 276, 5547–5555. [Google Scholar] [CrossRef] [PubMed]
- Banda, D.M.; Nunez, N.N.; Burnside, M.A.; Bradshaw, K.M.; David, S.S. Repair of 8-oxoG:A mismatches by the MUTYH glycosylase: Mechanism, metals and medicine. Free Radic. Biol. Med. 2017, 107, 202–215. [Google Scholar] [CrossRef]
- Raetz, A.G.; David, S.S. When you’re strange: Unusual features of the MUTYH glycosylase and implications in cancer. DNA Repair 2019, 80, 16–25. [Google Scholar] [CrossRef]
- Gu, Y.; Parker, A.; Wilson, T.M.; Bai, H.; Chang, D.Y.; Lu, A.L. Human MutY homolog, a DNA glycosylase involved in base excision repair, physically and functionally interacts with mismatch repair proteins human MutS homolog 2/human MutS homolog 6. J. Biol. Chem. 2002, 277, 11135–11142. [Google Scholar] [CrossRef]
- Hwang, B.J.; Jin, J.; Gao, Y.; Shi, G.; Madabushi, A.; Yan, A.; Guan, X.; Zalzman, M.; Nakajima, S.; Lan, L.; et al. SIRT6 protein deacetylase interacts with MYH DNA glycosylase, APE1 endonuclease, and Rad9-Rad1-Hus1 checkpoint clamp. BMC Mol. Biol. 2015, 16, 12. [Google Scholar] [CrossRef]
- Dorn, J.; Ferrari, E.; Imhof, R.; Ziegler, N.; Hubscher, U. Regulation of human MutYH DNA glycosylase by the E3 ubiquitin ligase mule. J. Biol. Chem. 2014, 289, 7049–7058. [Google Scholar]
- Oyama, M.; Wakasugi, M.; Hama, T.; Hashidume, H.; Iwakami, Y.; Imai, R.; Hoshino, S.; Morioka, H.; Ishigaki, Y.; Nikaido, O.; et al. Human NTH1 physically interacts with p53 and proliferating cell nuclear antigen. Biochem. Biophys. Res. Commun. 2004, 321, 183–191. [Google Scholar] [CrossRef]
- Bessho, T. Nucleotide excision repair 3’ endonuclease XPG stimulates the activity of base excision repairenzyme thymine glycol DNA glycosylase. Nucleic Acids Res. 1999, 27, 979–983. [Google Scholar] [CrossRef]
- Klungland, A.; Hoss, M.; Gunz, D.; Constantinou, A.; Clarkson, S.G.; Doetsch, P.W.; Bolton, P.H.; Wood, R.D.; Lindahl, T. Base excision repair of oxidative DNA damage activated by XPG protein. Mol. Cell 1999, 3, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Limpose, K.L.; Trego, K.S.; Li, Z.; Leung, S.W.; Sarker, A.H.; Shah, J.A.; Ramalingam, S.S.; Werner, E.M.; Dynan, W.S.; Cooper, P.K.; et al. Overexpression of the base excision repair NTHL1 glycosylase causes genomic instability and early cellular hallmarks of cancer. Nucleic Acids Res. 2018, 46, 4515–4532. [Google Scholar] [CrossRef] [PubMed]
- Kar, A.; Degtyareva, N.P.; Doetsch, P.W. Human NTHL1 expression and subcellular distribution determines cisplatin sensitivity in human lung epithelial and non-small cell lung cancer cells. NAR Cancer 2024, 6, zcae006. [Google Scholar]
- Dantzer, F.; Luna, L.; Bjoras, M.; Seeberg, E. Human OGG1 undergoes serine phosphorylation and associates with the nuclear matrix and mitotic chromatin In Vivo. Nucleic Acids Res. 2002, 30, 2349–2357. [Google Scholar] [CrossRef]
- Park, M.J.; Park, J.H.; Hahm, S.H.; Ko, S.I.; Lee, Y.R.; Chung, J.H.; Sohn, S.Y.; Cho, Y.; Kang, L.W.; Han, Y.S. Repair activities of human 8-oxoguanine DNA glycosylase are stimulated by the interaction with human checkpoint sensor Rad9-Rad1-Hus1 complex. DNA Repair 2009, 8, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- Wysong, B.C.; Schuck, P.L.; Sridharan, M.; Carrison, S.; Murakami, Y.; Balakrishnan, L.; Stewart, J.A. Human CST Stimulates Base Excision Repair to Prevent the Accumulation of Oxidative DNA Damage. J. Mol. Biol. 2024, 436, 168672. [Google Scholar]
- Ramdzan, Z.M.; Pal, R.; Kaur, S.; Leduy, L.; Berube, G.; Davoudi, S.; Vadnais, C.; Nepveu, A. The function of CUX1 in oxidative DNA damage repair is needed to prevent premature senescence of mouse embryo fibroblasts. Oncotarget 2015, 6, 3613–3626. [Google Scholar] [CrossRef]
- Pal, R.; Ramdzan, Z.M.; Kaur, S.; Duquette, P.M.; Marcotte, R.; Leduy, L.; Davoudi, S.; Lamarche-Vane, N.; Iulianella, A.; Nepveu, A. CUX2 protein functions as an accessory factor in the repair of oxidative DNA damage. J. Biol. Chem. 2015, 290, 22520–22531. [Google Scholar] [PubMed]
- Ramdzan, Z.M.; Vadnais, C.; Pal, R.; Vandal, G.; Cadieux, C.; Leduy, L.; Davoudi, S.; Hulea, L.; Yao, L.; Karnezis, A.N.; et al. RAS transformation requires CUX1-dependent repair of oxidative DNA damage. PLoS Biol. 2014, 12, e1001807. [Google Scholar] [CrossRef]
- Xia, L.; Huang, W.; Bellani, M.; Seidman, M.M.; Wu, K.; Fan, D.; Nie, Y.; Cai, Y.; Zhang, Y.W.; Yu, L.R.; et al. CHD4 Has Oncogenic Functions in Initiating and Maintaining Epigenetic Suppression of Multiple Tumor Suppressor Genes. Cancer Cell 2017, 31, 653–668.e7. [Google Scholar] [CrossRef]
- Larsen, D.H.; Poinsignon, C.; Gudjonsson, T.; Dinant, C.; Payne, M.R.; Hari, F.J.; Rendtlew Danielsen, J.M.; Menard, P.; Sand, J.C.; Stucki, M.; et al. The chromatin-remodeling factor CHD4 coordinates signaling and repair after DNA damage. J. Cell Biol. 2010, 190, 731–740. [Google Scholar] [CrossRef]
- Smith, R.; Sellou, H.; Chapuis, C.; Huet, S.; Timinszky, G. CHD3 and CHD4 recruitment and chromatin remodeling activity at DNA breaks is promoted by early poly(ADP-ribose)-dependent chromatin relaxation. Nucleic Acids Res. 2018, 46, 6087–6098. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.R.; Parsons, J.L. The E3 Ubiquitin Ligase NEDD4L Targets OGG1 for Ubiquitylation and Modulates the Cellular DNA Damage Response. Front. Cell Dev. Biol. 2020, 8, 607060. [Google Scholar] [CrossRef]
- Das, A.; Boldogh, I.; Lee, J.W.; Harrigan, J.A.; Hegde, M.L.; Piotrowski, J.; de Souza Pinto, N.; Ramos, W.; Greenberg, M.M.; Hazra, T.K.; et al. The human Werner syndrome protein stimulates repair of oxidative DNA base damage by the DNA glycosylase NEIL1. J. Biol. Chem. 2007, 282, 26591–26602. [Google Scholar] [CrossRef]
- Dou, H.; Theriot, C.A.; Das, A.; Hegde, M.L.; Matsumoto, Y.; Boldogh, I.; Hazra, T.K.; Bhakat, K.K.; Mitra, S. Interaction of the human DNA glycosylase NEIL1 with proliferating cell nuclear antigen. The potential for replication-associated repair of oxidized bases in mammalian genomes. J. Biol. Chem. 2008, 283, 3130–3140. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.L.; Theriot, C.A.; Das, A.; Hegde, P.M.; Guo, Z.; Gary, R.K.; Hazra, T.K.; Shen, B.; Mitra, S. Physical and functional interaction between human oxidized base-specific DNA glycosylase NEIL1 and flap endonuclease 1. J. Biol. Chem. 2008, 283, 27028–27037. [Google Scholar] [CrossRef]
- Guan, X.; Bai, H.; Shi, G.; Theriot, C.A.; Hazra, T.K.; Mitra, S.; Lu, A.L. The human checkpoint sensor Rad9-Rad1-Hus1 interacts with and stimulates NEIL1 glycosylase. Nucleic Acids Res. 2007, 35, 2463–2472. [Google Scholar] [CrossRef]
- Rangaswamy, S.; Pandey, A.; Mitra, S.; Hegde, M.L. Pre-Replicative Repair of Oxidized Bases Maintains Fidelity in Mammalian Genomes: The Cowcatcher Role of NEIL1 DNA Glycosylase. Genes 2017, 8, 175. [Google Scholar] [CrossRef] [PubMed]
- Le Meur, R.A.; Pecen, T.J.; Le Meur, K.V.; Nagel, Z.D.; Chazin, W.J. Molecular basis and functional consequences of the interaction between the base excision repair DNA glycosylase NEIL1 and RPA. J. Biol. Chem. 2024, 300, 107579. [Google Scholar] [CrossRef]
- Sharma, N.; Chakravarthy, S.; Longley, M.J.; Copeland, W.C.; Prakash, A. The C-terminal tail of the NEIL1 DNA glycosylase interacts with the human mitochondrial single-stranded DNA binding protein. DNA Repair 2018, 65, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Bhakat, K.K.; Hazra, T.K.; Mitra, S. Acetylation of the human DNA glycosylase NEIL2 and inhibition of its activity. Nucleic Acids Res. 2004, 32, 3033–3039. [Google Scholar] [CrossRef]
- Myrup Holst, C.; Brondum Andersen, N.; Thinggaard, V.; Tilken, M.; Lautrup, S.; Tesauro, C.; Stevnsner, T. Phosphorylation of the Human DNA Glycosylase NEIL2 Is Affected by Oxidative Stress and Modulates Its Activity. Antioxidants 2023, 12, 355. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, J.; Wallace, S.S.; Chen, J.; Zhou, J.; D’Andrea, A.D. Cooperation of the NEIL3 and Fanconi anemia/BRCA pathways in interstrand crosslink repair. Nucleic Acids Res. 2020, 48, 3014–3028. [Google Scholar] [CrossRef]
- Wu, R.A.; Semlow, D.R.; Kamimae-Lanning, A.N.; Kochenova, O.V.; Chistol, G.; Hodskinson, M.R.; Amunugama, R.; Sparks, J.L.; Wang, M.; Deng, L.; et al. TRAIP is a master regulator of DNA interstrand crosslink repair. Nature 2019, 567, 267–272. [Google Scholar] [CrossRef]
- Chung, H.J.; Lee, J.R.; Kim, T.M.; Kim, S.; Park, K.; Kim, M.J.; Jung, E.; Kim, S.; Lee, E.A.; Ra, J.S.; et al. ZNF212 promotes genomic integrity through direct interaction with TRAIP. Nucleic Acids Res. 2023, 51, 631–649. [Google Scholar] [CrossRef]
- Chou, W.C.; Wang, H.C.; Wong, F.H.; Ding, S.L.; Wu, P.E.; Shieh, S.Y.; Shen, C.Y. Chk2-dependent phosphorylation of XRCC1 in the DNA damage response promotes base excision repair. EMBO J. 2008, 27, 3140–3150. [Google Scholar]
- Wang, S.; Gong, Z.; Chen, R.; Liu, Y.; Li, A.; Li, G.; Zhou, J. JWA regulates XRCC1 and functions as a novel base excision repair protein in oxidative-stress-induced DNA single-strand breaks. Nucleic Acids Res. 2009, 37, 1936–1950. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xu, B.; Yang, J.; He, L.; Zhang, Z.; Cheng, X.; Yu, H.; Liu, X.; Jin, T.; Peng, Y.; et al. UHRF2 commissions the completion of DNA demethylation through allosteric activation by 5hmC and K33-linked ubiquitination of XRCC1. Mol. Cell 2021, 81, 2960–2974.e7. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Ramdzan, Z.M.; Guiot, M.C.; Li, L.; Leduy, L.; Ramotar, D.; Sabri, S.; Abdulkarim, B.; Nepveu, A. CUX1 stimulates APE1 enzymatic activity and increases the resistance of glioblastoma cells to the mono-alkylating agent temozolomide. Neuro Oncol. 2018, 20, 484–493. [Google Scholar] [CrossRef]
- Dianova, I.I.; Bohr, V.A.; Dianov, G.L. Interaction of human AP endonuclease 1 with flap endonuclease 1 and proliferating cell nuclear antigen involved in long-patch base excision repair. Biochemistry 2001, 40, 12639–12644. [Google Scholar] [CrossRef]
- Bhakat, K.K.; Izumi, T.; Yang, S.H.; Hazra, T.K.; Mitra, S. Role of acetylated human AP-endonuclease (APE1/Ref-1) in regulation of the parathyroid hormone gene. EMBO J. 2003, 22, 6299–6309. [Google Scholar] [CrossRef]
- Busso, C.S.; Iwakuma, T.; Izumi, T. Ubiquitination of mammalian AP endonuclease (APE1) regulated by the p53-MDM2 signaling pathway. Oncogene 2009, 28, 1616–1625. [Google Scholar] [CrossRef]
- Zolner, A.E.; Abdou, I.; Ye, R.; Mani, R.S.; Fanta, M.; Yu, Y.; Douglas, P.; Tahbaz, N.; Fang, S.; Dobbs, T.; et al. Phosphorylation of polynucleotide kinase/phosphatase by DNA-dependent protein kinase and ataxia-telangiectasia mutated regulates its association with sites of DNA damage. Nucleic Acids Res. 2011, 39, 9224–9237. [Google Scholar] [CrossRef]
- Toueille, M.; El-Andaloussi, N.; Frouin, I.; Freire, R.; Funk, D.; Shevelev, I.; Friedrich-Heineken, E.; Villani, G.; Hottiger, M.O.; Hubscher, U. The human Rad9/Rad1/Hus1 damage sensor clamp interacts with DNA polymerase beta and increases its DNA substrate utilisation efficiency: Implications for DNA repair. Nucleic Acids Res. 2004, 32, 3316–3324. [Google Scholar] [CrossRef]
- Harrigan, J.A.; Opresko, P.L.; von Kobbe, C.; Kedar, P.S.; Prasad, R.; Wilson, S.H.; Bohr, V.A. The Werner syndrome protein stimulates DNA polymerase beta strand displacement synthesis via its helicase activity. J. Biol. Chem. 2003, 278, 22686–22695. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, J.A.; Wilson, D.M., 3rd; Prasad, R.; Opresko, P.L.; Beck, G.; May, A.; Wilson, S.H.; Bohr, V.A. The Werner syndrome protein operates in base excision repair and cooperates with DNA polymerase beta. Nucleic Acids Res. 2006, 34, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Ramdzan, Z.M.; Vickridge, E.; Li, L.; Faraco, C.C.F.; Djerir, B.; Leduy, L.; Marechal, A.; Nepveu, A. CUT Domains Stimulate Pol beta Enzymatic Activities to Accelerate Completion of Base Excision Repair. J. Mol. Biol. 2021, 433, 166806. [Google Scholar] [CrossRef]
- Hasan, S.; El-Andaloussi, N.; Hardeland, U.; Hassa, P.O.; Burki, C.; Imhof, R.; Schar, P.; Hottiger, M.O. Acetylation regulates the DNA end-trimming activity of DNA polymerase beta. Mol. Cell 2002, 10, 1213–1222. [Google Scholar] [CrossRef]
- Parsons, J.L.; Tait, P.S.; Finch, D.; Dianova, I.I.; Edelmann, M.J.; Khoronenkova, S.V.; Kessler, B.M.; Sharma, R.A.; McKenna, W.G.; Dianov, G.L. Ubiquitin ligase ARF-BP1/Mule modulates base excision repair. EMBO J. 2009, 28, 3207–3215. [Google Scholar] [CrossRef] [PubMed]
- Della-Maria, J.; Zhou, Y.; Tsai, M.S.; Kuhnlein, J.; Carney, J.P.; Paull, T.T.; Tomkinson, A.E. Human Mre11/human Rad50/Nbs1 and DNA ligase IIIalpha/XRCC1 protein complexes act together in an alternative nonhomologous end joining pathway. J. Biol. Chem. 2011, 286, 33845–33853. [Google Scholar] [CrossRef]
- De, A.; Campbell, C. A novel interaction between DNA ligase III and DNA polymerase gamma plays an essential role in mitochondrial DNA stability. Biochem. J. 2007, 402, 175–186. [Google Scholar] [CrossRef]
- Dianov, G.; Bischoff, C.; Piotrowski, J.; Bohr, V.A. Repair pathways for processing of 8-oxoguanine in DNA by mammalian cell extracts. J. Biol. Chem. 1998, 273, 33811–33816. [Google Scholar]
- Fortini, P.; Parlanti, E.; Sidorkina, O.M.; Laval, J.; Dogliotti, E. The type of DNA glycosylase determines the base excision repair pathway in mammalian cells. J. Biol. Chem. 1999, 274, 15230–15236. [Google Scholar] [CrossRef] [PubMed]
- Fortini, P.; Pascucci, B.; Parlanti, E.; D’Errico, M.; Simonelli, V.; Dogliotti, E. 8-Oxoguanine DNA damage: At the crossroad of alternative repair pathways. Mutat. Res. 2003, 531, 127–139. [Google Scholar] [CrossRef]
- Semlow, D.R.; Zhang, J.; Budzowska, M.; Drohat, A.C.; Walter, J.C. Replication-Dependent Unhooking of DNA Interstrand Cross-Links by the NEIL3 Glycosylase. Cell 2016, 167, 498–511.e414. [Google Scholar] [CrossRef]
- Hu, L.Y.; Chang, C.C.; Huang, Y.S.; Chou, W.C.; Lin, Y.M.; Ho, C.C.; Chen, W.T.; Shih, H.M.; Hsiung, C.N.; Wu, P.E.; et al. SUMOylation of XRCC1 activated by poly (ADP-ribosyl)ation regulates DNA repair. Hum. Mol. Genet. 2018, 27, 2306–2317. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.A.; Wilson, D.M., 3rd; Wong, D.; Demple, B. Interaction of human apurinic endonuclease and DNA polymerase beta in the base excision repair pathway. Proc. Natl. Acad. Sci. USA 1997, 94, 7166–7169. [Google Scholar] [CrossRef]
- Jiao, R.; Harrigan, J.A.; Shevelev, I.; Dietschy, T.; Selak, N.; Indig, F.E.; Piotrowski, J.; Janscak, P.; Bohr, V.A.; Stagljar, I. The Werner syndrome protein is required for recruitment of chromatin assembly factor 1 following DNA damage. Oncogene 2007, 26, 3811–3822. [Google Scholar] [CrossRef]
- Ramdzan, Z.M.; Vickridge, E.; Faraco, C.C.F.; Nepveu, A. CUT Domain Proteins in DNA Repair and Cancer. Cancers 2021, 13, 2953. [Google Scholar] [CrossRef]
- Janoshazi, A.K.; Horton, J.K.; Zhao, M.L.; Prasad, R.; Scappini, E.L.; Tucker, C.J.; Wilson, S.H. Shining light on the response to repair intermediates in DNA of living cells. DNA Repair 2020, 85, 102749. [Google Scholar] [CrossRef]
- El-Andaloussi, N.; Valovka, T.; Toueille, M.; Steinacher, R.; Focke, F.; Gehrig, P.; Covic, M.; Hassa, P.O.; Schar, P.; Hubscher, U.; et al. Arginine methylation regulates DNA polymerase beta. Mol. Cell 2006, 22, 51–62. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| BER Component | Proteins That Interact | Functional Role of Interactions | References |
|---|---|---|---|
| UNG2 | RPA2, PCNA | Recruits UNG2 to the replication fork to scan for uracil | [107,108] |
| CRL4 E3, Vpr | Regulates UNG2 protein levels | [109,110,111,112] | |
| SMUG1 | CRL4 E3, Vpr | Regulates SMUG1 protein levels | [109,110,111,112] |
| MBD4 | MLH1, FADD, HDAC, SIN3a, RPF | Regulates the apoptotic response to diverse DNA lesions | [23,113,114,115] |
| UHRF1 E3 ligase, SUMO1 | Regulates MBD4 protein levels | [116,117] | |
| TDG | CBP, p300, XPC-HR23B | Turnover of TDG | [118,119] |
| DNMT3a, DNMT3b, | Recruits TDG to the mismatch repair site | [120,121] | |
| RNF4 | Stimulates TDG activity | [122] | |
| CRL4 E3, PCNA, SUMO1 | Regulates TDG protein levels | [123,124,125,126] | |
| MPG/AAG | ERα, PCNA | Recruits MPG to the repair site | [127,128,129] |
| hHR23, UV-DDB | Stimulates MPG activity | [130,131] | |
| UHRF1 | [132] | ||
| MUTYH | PCNA, RPA | [133,134,135] | |
| hMSH6, SIRT6 | Stimulates MUTYH activity | [136,137] | |
| E3 ubiquitin ligase mule | Regulates MUTYH protein levels | [138] | |
| NTHL1 | PCNA, p53, XPG | Stimulates NTHL1 activity | [139,140,141,142,143] |
| OGG1 | PKC | Compartmentalization of OGG1 in the nucleus | [144] |
| 9-1-1 complex, CST, CUX1, CUX2 | Stimulates OGG1 activity | [145,146,147,148,149] | |
| CHD4 | Chromatin relaxation at the repair site | [150,151,152] | |
| E3 Ubiquitin ligase NEDD4L | Regulates OGG1 protein levels | [153] | |
| NEIL1 | WRN, PCNA, Fen-1, 9-1-1 complex | Stimulates NEIL1 activity | [154,155,156,157] |
| RPA, MtSSB | [158,159,160] | ||
| NEIL2 | p300, PKC | Regulates NEIL2 activity | [161,162] |
| NEIL3 | RUVBL1/2 complex, TRAIP, ZNF212 | Regulates NEIL3 recruitment to the ICL lesions | [163,164,165] |
| XRCC1 | CK2 | Regulates interaction between XRCC1 and APTX | [102,103] |
| Chk2 | Regulates interaction between XRCC1 and DNA glycosylase | [166] | |
| JWA, RAD23B E3 ubiquitin ligase UHRF2 | Regulates XRCC1 protein levels | [167,168] | |
| APE1 | SIRT1, SIRT6, RNF4, CST, CUX1 | Stimulates APE1 endonuclease activity | [68,122,137,146,169] |
| FEN1, PCNA, p300 | [170,171] | ||
| E3 ubiquitin ligase MDM2 | Regulates APE1 protein levels | [172] | |
| PNKP | DNA-PK, ATM | Regulates PNKP | [173] |
| Pol β | 9-1-1 complex, WRN, CST, CUX1, | Stimulate Pol β activity | [146,174,175,176,177] |
| p300, E3 ubiquitin ligase Mule & ARF | Regulates Pol β | [178,179] | |
| Ligase IIIα | Mre11:Rad50:Nbs1, Pol γ | [180,181] | |
| APTX | p53, nucleolin | [102] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rathnaiah, G.; Sweasy, J.B. Protein–Protein Interactions in Base Excision Repair. Biomolecules 2025, 15, 890. https://doi.org/10.3390/biom15060890
Rathnaiah G, Sweasy JB. Protein–Protein Interactions in Base Excision Repair. Biomolecules. 2025; 15(6):890. https://doi.org/10.3390/biom15060890
Chicago/Turabian StyleRathnaiah, Govardhan, and Joann B. Sweasy. 2025. "Protein–Protein Interactions in Base Excision Repair" Biomolecules 15, no. 6: 890. https://doi.org/10.3390/biom15060890
APA StyleRathnaiah, G., & Sweasy, J. B. (2025). Protein–Protein Interactions in Base Excision Repair. Biomolecules, 15(6), 890. https://doi.org/10.3390/biom15060890