
Pre-Amplification of Cell-Free DNA: Balancing Amplification Errors with Enhanced Sensitivity

 , , ,
, , ,  , ,
, ,  , and
, and 
Abstract
1. Introduction
2. Methods and Materials
2.1. Sample Collection and cfDNA Extraction and Quantification
2.2. Pre-Amplification Using T-Oligo Primed PCR
2.3. Droplet Digital PCR
2.4. Statistics
3. Results
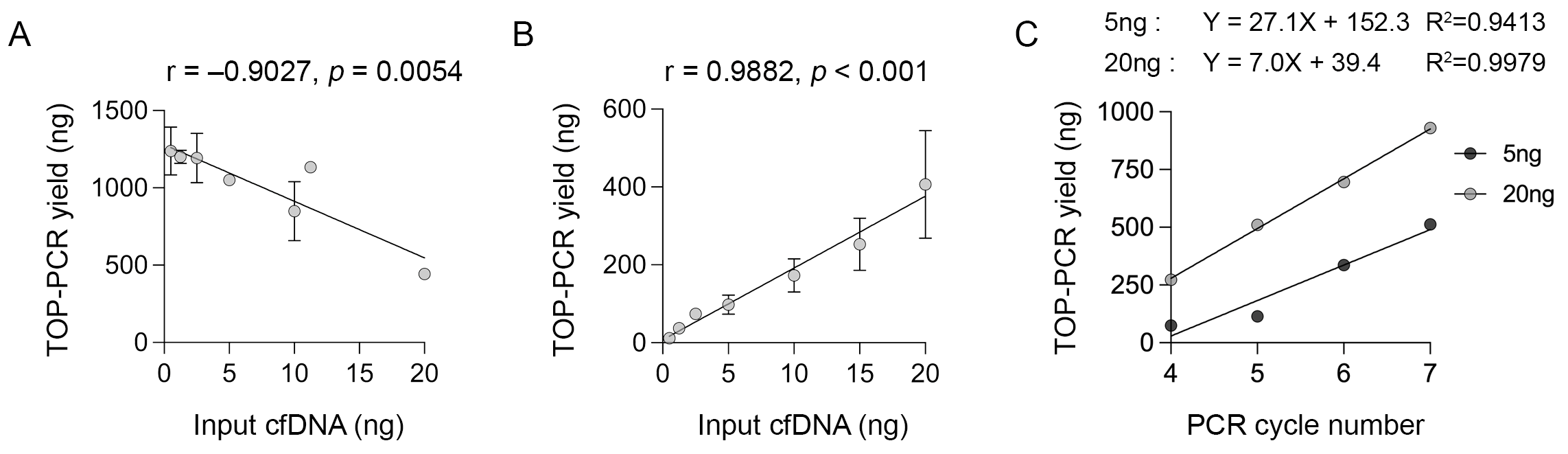
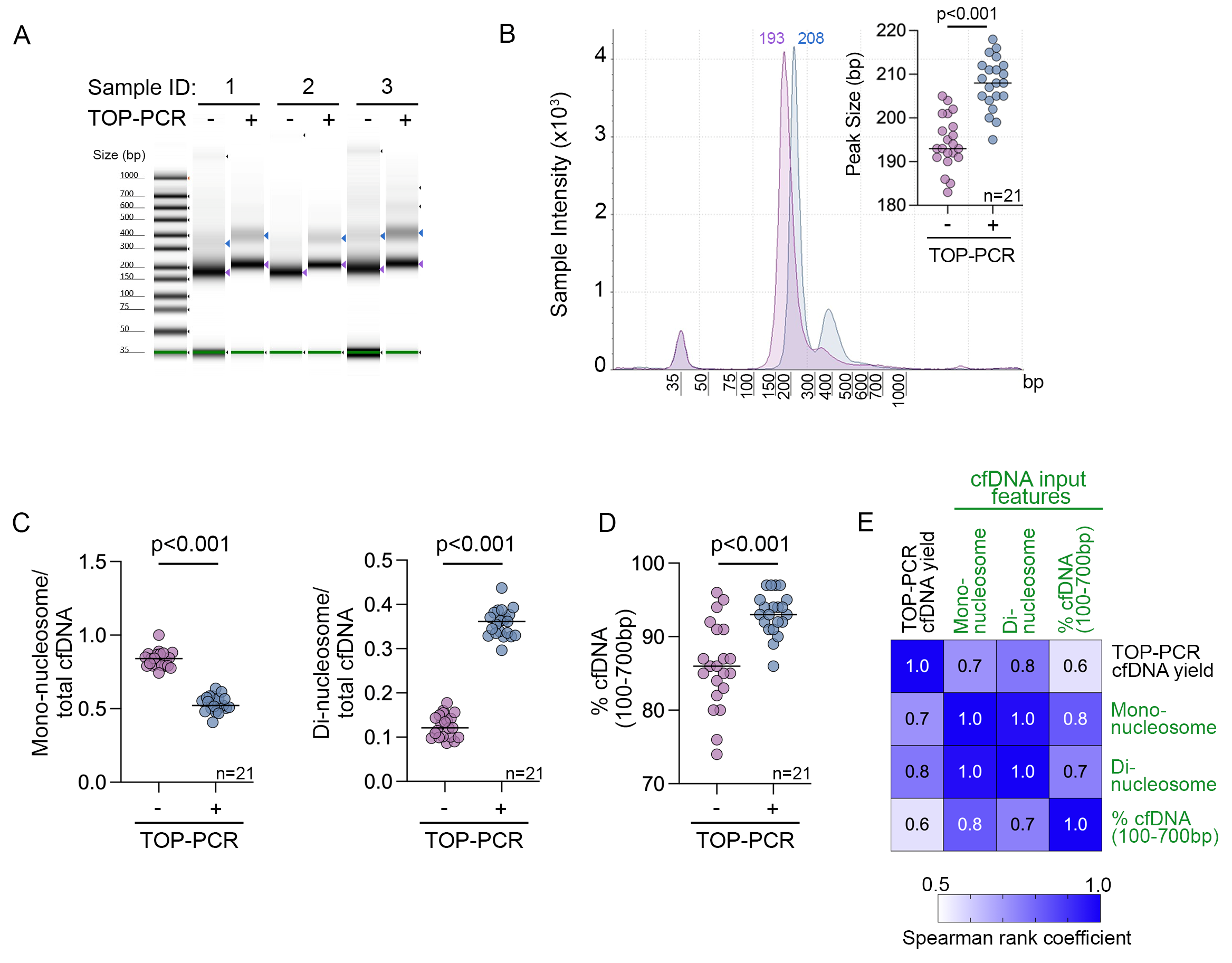
3.1. Performance and Optimisation of TOP-PCR cfDNA Pre-Amplification
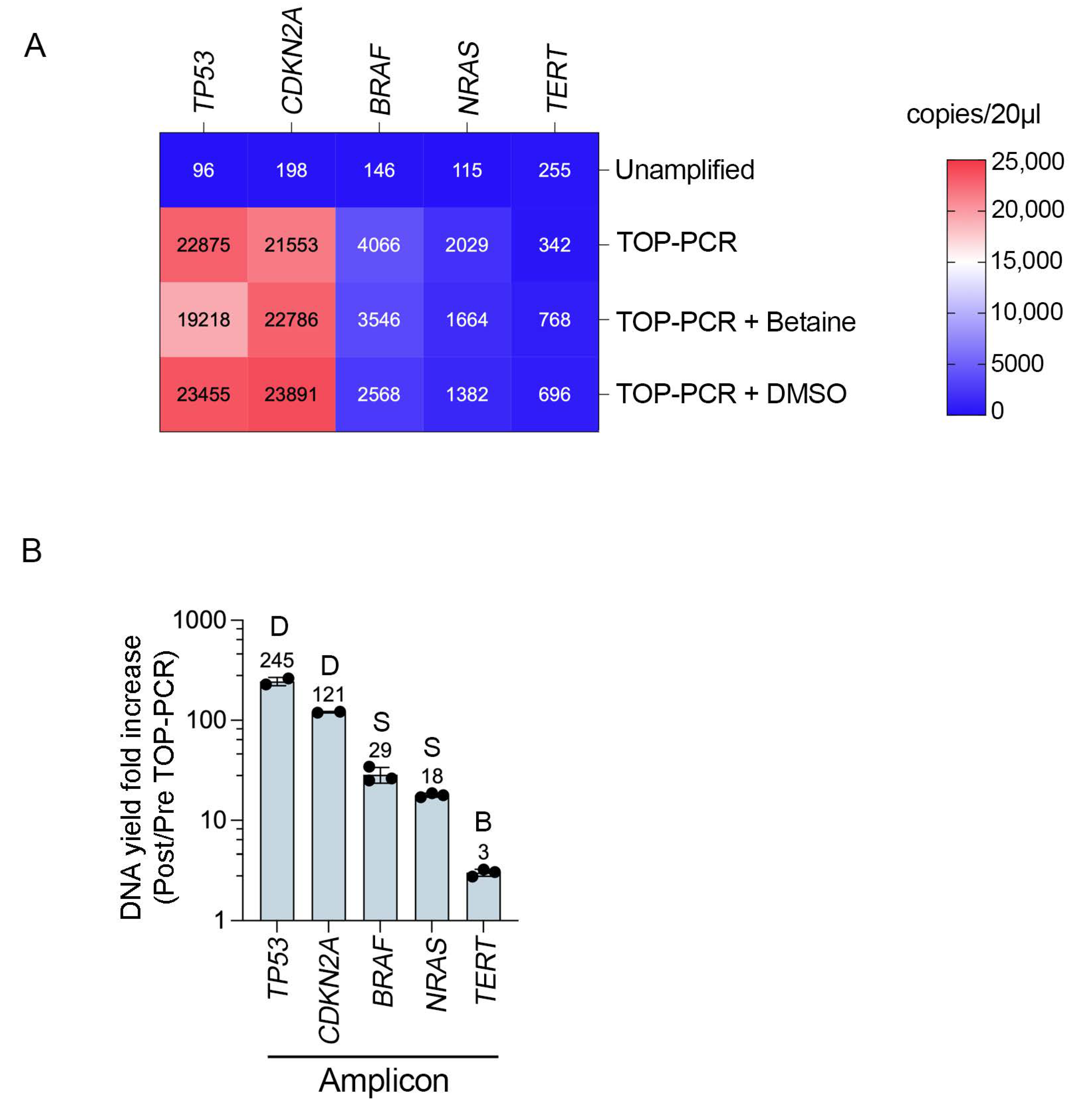
3.2. TOP-PCR Pre-Amplification Efficiency Varies Across Cancer Gene Targets
3.3. TOP-PCR Pre-Amplification Increases ctDNA Testing and Detection Sensitivity
3.4. TOP-PCR-Errors and Impact on Rare Variant Detection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peng, Y.; Mei, W.; Ma, K.; Zeng, C. Circulating Tumor DNA and Minimal Residual Disease (MRD) in Solid Tumors: Current Horizons and Future Perspectives. Front. Oncol. 2021, 11, 763790. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Long, G.V.; Boyd, S.; Lo, S.; Menzies, A.M.; Tembe, V.; Guminski, A.; Jakrot, V.; Scolyer, R.A.; Mann, G.J.; et al. Circulating tumour DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann. Oncol. 2017, 28, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Saw, R.P.; Thompson, J.F.; Lo, S.; Spillane, A.J.; Shannon, K.F.; Stretch, J.R.; Howle, J.; Menzies, A.M.; Carlino, M.S.; et al. Pre-operative ctDNA predicts survival in high-risk stage III cutaneous melanoma patients. Ann. Oncol. 2019, 30, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Ignatiadis, M.; Sledge, G.W.; Jeffrey, S.S. Liquid biopsy enters the clinic-implementation issues and future challenges. Nat. Rev. Clin. Oncol. 2021, 18, 297–312. [Google Scholar] [CrossRef]
- Chin, R.I.; Chen, K.; Usmani, A.; Chua, C.; Harris, P.K.; Binkley, M.S.; Azad, T.D.; Dudley, J.C.; Chaudhuri, A.A. Detection of Solid Tumor Molecular Residual Disease (MRD) Using Circulating Tumor DNA (ctDNA). Mol. Diagn. Ther. 2019, 23, 311–331. [Google Scholar] [CrossRef]
- Silva, T.F.d.; Azevedo, J.C.d.; Teixeira, E.B.; Casseb, S.M.M.; Moreira, F.C.; Assumpção, P.P.d.; Santos, S.E.B.d.; Calcagno, D.Q. From haystack to high precision: Advanced sequencing methods to unraveling circulating tumor DNA mutations. Front. Mol. Biosci. 2024, 11, 1423470. [Google Scholar] [CrossRef]
- Chen, M.; Zhao, H. Next-generation sequencing in liquid biopsy: Cancer screening and early detection. Human. Genom. 2019, 13, 34. [Google Scholar] [CrossRef]
- Semenkovich, N.P.; Szymanski, J.J.; Earland, N.; Chauhan, P.S.; Pellini, B.; Chaudhuri, A.A. Genomic approaches to cancer and minimal residual disease detection using circulating tumor DNA. J. Immunother. Cancer 2023, 11, e006284. [Google Scholar] [CrossRef]
- Tébar-Martínez, R.; Martín-Arana, J.; Gimeno-Valiente, F.; Tarazona, N.; Rentero-Garrido, P.; Cervantes, A. Strategies for improving detection of circulating tumor DNA using next generation sequencing. Cancer Treat. Rev. 2023, 119, 102595. [Google Scholar] [CrossRef]
- Cheng, A.P.; Widman, A.J.; Arora, A.; Rusinek, I.; Sossin, A.; Rajagopalan, S.; Midler, N.; Hooper, W.F.; Murray, R.M.; Halmos, D.; et al. Error-corrected flow-based sequencing at whole-genome scale and its application to circulating cell-free DNA profiling. Nat. Methods 2025, 22, 973–981. [Google Scholar] [CrossRef]
- Nai, Y.S.; Chen, T.H.; Huang, Y.F.; Midha, M.K.; Shiau, H.C.; Shen, C.Y.; Chen, C.J.; Yu, A.L.; Chiu, K.P. T Oligo-Primed Polymerase Chain Reaction (TOP-PCR): A Robust Method for the Amplification of Minute DNA Fragments in Body Fluids. Sci. Rep. 2017, 7, 40767. [Google Scholar] [CrossRef] [PubMed]
- Bronkhorst, A.J.; Ungerer, V.; Holdenrieder, S. Comparison of methods for the isolation of cell-free DNA from cell culture supernatant. Tumour Biol. 2020, 42, 1010428320916314. [Google Scholar] [CrossRef] [PubMed]
- Ungerer, V.; Bronkhorst, A.J.; Van den Ackerveken, P.; Herzog, M.; Holdenrieder, S. Serial profiling of cell-free DNA and nucleosome histone modifications in cell cultures. Sci. Rep. 2021, 11, 9460. [Google Scholar] [CrossRef] [PubMed]
- Bronkhorst, A.J.; Wentzel, J.F.; Aucamp, J.; van Dyk, E.; du Plessis, L.; Pretorius, P.J. Characterization of the cell-free DNA released by cultured cancer cells. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 157–165. [Google Scholar] [CrossRef]
- Cha, R.S.; Thilly, W.G. Specificity, efficiency, and fidelity of PCR. PCR Methods Appl. 1993, 3, S18–S29. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, H.; Shang, S.; Meng, S.; Lin, T.; Zhang, Y.; Liu, H. Evaluation validation of a qPCR curve analysis method and conventional approaches. BMC Genom. 2021, 22, 680. [Google Scholar] [CrossRef]
- McDonald, B.R.; Contente-Cuomo, T.; Sammut, S.J.; Odenheimer-Bergman, A.; Ernst, B.; Perdigones, N.; Chin, S.F.; Farooq, M.; Mejia, R.; Cronin, P.A.; et al. Personalized circulating tumor DNA analysis to detect residual disease after neoadjuvant therapy in breast cancer. Sci. Transl. Med. 2019, 11, eaax7392. [Google Scholar] [CrossRef]
- Diefenbach, R.J.; Lee, J.H.; Menzies, A.M.; Carlino, M.S.; Long, G.V.; Saw, R.P.M.; Howle, J.R.; Spillane, A.J.; Scolyer, R.A.; Kefford, R.F.; et al. Design and Testing of a Custom Melanoma Next Generation Sequencing Panel for Analysis of Circulating Tumor DNA. Cancers 2020, 12, 2228. [Google Scholar] [CrossRef]
- Chabon, J.J.; Hamilton, E.G.; Kurtz, D.M.; Esfahani, M.S.; Moding, E.J.; Stehr, H.; Schroers-Martin, J.; Nabet, B.Y.; Chen, B.; Chaudhuri, A.A.; et al. Integrating genomic features for non-invasive early lung cancer detection. Nature 2020, 580, 245–251. [Google Scholar] [CrossRef]
- Sanz-Garcia, E.; Zhao, E.; Bratman, S.V.; Siu, L.L. Monitoring and adapting cancer treatment using circulating tumor DNA kinetics: Current research, opportunities, and challenges. Sci. Adv. 2022, 8, eabi8618. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra224. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Menzies, A.; Carlino, M.; Kefford, R.; Scolyer, R.; Long, G.; Rizos, H. Circulating tumor DNA (ctDNA) in metastatic melanoma (MM) patients (pts) with brain metastases (mets). J. Clin. Oncol. 2019, 37, 9581. [Google Scholar] [CrossRef]
- Husain, H.; Pavlick, D.C.; Fendler, B.J.; Madison, R.W.; Decker, B.; Gjoerup, O.; Parachoniak, C.A.; McLaughlin-Drubin, M.; Erlich, R.L.; Schrock, A.B.; et al. Tumor Fraction Correlates With Detection of Actionable Variants Across > 23,000 Circulating Tumor DNA Samples. JCO Precis. Oncol. 2022, 6, e2200261. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef]
- Costello, M.; Pugh, T.J.; Fennell, T.J.; Stewart, C.; Lichtenstein, L.; Meldrim, J.C.; Fostel, J.L.; Friedrich, D.C.; Perrin, D.; Dionne, D.; et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 2013, 41, e67. [Google Scholar] [CrossRef]
- Sanchez, C.; Roch, B.; Mazard, T.; Blache, P.; Dache, Z.A.A.; Pastor, B.; Pisareva, E.; Tanos, R.; Thierry, A.R. Circulating nuclear DNA structural features, origins, and complete size profile revealed by fragmentomics. JCI Insight 2021, 6, e144561. [Google Scholar] [CrossRef]
- Underhill, H.R.; Kitzman, J.O.; Hellwig, S.; Welker, N.C.; Daza, R.; Baker, D.N.; Gligorich, K.M.; Rostomily, R.C.; Bronner, M.P.; Shendure, J. Fragment Length of Circulating Tumor DNA. PLoS Genet. 2016, 12, e1006162. [Google Scholar] [CrossRef]
- Henriksen, T.V.; Drue, S.O.; Frydendahl, A.; Demuth, C.; Rasmussen, M.H.; Reinert, T.; Pedersen, J.S.; Andersen, C.L. Error Characterization and Statistical Modeling Improves Circulating Tumor DNA Detection by Droplet Digital PCR. Clin. Chem. 2022, 68, 657–667. [Google Scholar] [CrossRef]
- Karunanathie, H.; Kee, P.S.; Ng, S.F.; Kennedy, M.A.; Chua, E.W. PCR enhancers: Types, mechanisms, and applications in long-range PCR. Biochimie 2022, 197, 130–143. [Google Scholar] [CrossRef]
- Colebatch, A.J.; Witkowski, T.; Waring, P.M.; McArthur, G.A.; Wong, S.Q.; Dobrovic, A. Optimizing Amplification of the GC-Rich TERT Promoter Region Using 7-Deaza-dGTP for Droplet Digital PCR Quantification of TERT Promoter Mutations. Clin. Chem. 2018, 64, 745–747. [Google Scholar] [CrossRef] [PubMed]
- McEvoy, A.C.; Calapre, L.; Pereira, M.R.; Giardina, T.; Robinson, C.; Khattak, M.A.; Meniawy, T.M.; Pritchard, A.L.; Hayward, N.K.; Amanuel, B.; et al. Sensitive droplet digital PCR method for detection of TERT promoter mutations in cell free DNA from patients with metastatic melanoma. Oncotarget 2017, 8, 78890–78900. [Google Scholar] [CrossRef] [PubMed]
- Aird, D.; Ross, M.G.; Chen, W.S.; Danielsson, M.; Fennell, T.; Russ, C.; Jaffe, D.B.; Nusbaum, C.; Gnirke, A. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011, 12, R18. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.A.; Liu, X.S. Identifying and mitigating bias in next-generation sequencing methods for chromatin biology. Nat. Rev. Genet. 2014, 15, 709–721. [Google Scholar] [CrossRef]
- Chan, W.Y.; Lee, J.H.; Stewart, A.; Diefenbach, R.J.; Gonzalez, M.; Menzies, A.M.; Blank, C.; Scolyer, R.A.; Long, G.V.; Rizos, H. Circulating tumour DNA dynamics predict recurrence in stage III melanoma patients receiving neoadjuvant immunotherapy. J. Exp. Clin. Cancer Res. 2024, 43, 238. [Google Scholar] [CrossRef]
- Chiu, K.-P. Introduction to One Drop of Body Fluid-PCR (1DF-PCR). J. Biomed. Res. Environ. Sci. 2022, 3, 1565–1566. [Google Scholar] [CrossRef]
- Jackson, J.B.; Choi, D.S.; Luketich, J.D.; Pennathur, A.; Ståhlberg, A.; Godfrey, T.E. Multiplex Preamplification of Serum DNA to Facilitate Reliable Detection of Extremely Rare Cancer Mutations in Circulating DNA by Digital PCR. J. Mol. Diagn. 2016, 18, 235–243. [Google Scholar] [CrossRef]
- Rowlands, V.; Rutkowski, A.J.; Meuser, E.; Carr, T.H.; Harrington, E.A.; Barrett, J.C. Optimisation of robust singleplex and multiplex droplet digital PCR assays for high confidence mutation detection in circulating tumour DNA. Sci. Rep. 2019, 9, 12620. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Patient | Progressed | Unamplified cfDNA Yield (ng) | Amplified cfDNA Yield (ng) a | Mutation 1 Droplets b | Mutation 2 Droplets b |
|---|---|---|---|---|---|
| 42193 | Yes | 68.7 | 501.6 | 0 | 7 |
| 52318 | Yes | 34.9 | 415.2 | 0 | 5 |
| 58647 | Yes | 54.5 | 423.6 | 0 | 8 |
| 01622 | Yes | 48.0 | 310.8 | 440 | 20 |
| 38965 | Yes | 135.3 | 612.0 | 16 | 30 |
| 44691 | Yes | 125.6 | 536.4 | 30 | 11 |
| 50487 | Yes | 65.4 | 436.8 | 110 | 48 |
| 01523 | No | 24.0 | 369.6 | 0 | 0 |
| 01509 | No | 21.0 | 418.8 | 0 | 0 |
| 44894 | No | 42.0 | 381.6 | 0 | 0 |
| 28124 | No | 61.1 | 308.4 | 0 | 0 |
| 39346 | No | 45.5 | 292.8 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, W.Y.; Stewart, A.; Diefenbach, R.J.; Gray, E.S.; Lee, J.H.; Scolyer, R.A.; Long, G.V.; Rizos, H. Pre-Amplification of Cell-Free DNA: Balancing Amplification Errors with Enhanced Sensitivity. Biomolecules 2025, 15, 883. https://doi.org/10.3390/biom15060883
Chan WY, Stewart A, Diefenbach RJ, Gray ES, Lee JH, Scolyer RA, Long GV, Rizos H. Pre-Amplification of Cell-Free DNA: Balancing Amplification Errors with Enhanced Sensitivity. Biomolecules. 2025; 15(6):883. https://doi.org/10.3390/biom15060883
Chicago/Turabian StyleChan, Wei Yen, Ashleigh Stewart, Russell J. Diefenbach, Elin S. Gray, Jenny H. Lee, Richard A. Scolyer, Georgina V. Long, and Helen Rizos. 2025. "Pre-Amplification of Cell-Free DNA: Balancing Amplification Errors with Enhanced Sensitivity" Biomolecules 15, no. 6: 883. https://doi.org/10.3390/biom15060883
APA StyleChan, W. Y., Stewart, A., Diefenbach, R. J., Gray, E. S., Lee, J. H., Scolyer, R. A., Long, G. V., & Rizos, H. (2025). Pre-Amplification of Cell-Free DNA: Balancing Amplification Errors with Enhanced Sensitivity. Biomolecules, 15(6), 883. https://doi.org/10.3390/biom15060883