

Role of Tumor Microenvironment in Prostate Cancer Immunometabolism

Abstract

1. Introduction
2. The Tumor Microenvironment in Prostate Cancer

{kind=link}
{kind=link}
{kind=link}
| Immune Cell Type | Function | Mechanisms of Immunosuppression |
|---|---|---|
| CD4+ T-cells | Coordinate immune response; differentiate into Th1, Th2, and Th17 cells; promote or inhibit tumor immunity. | Skewing towards Th2 and Th17 phenotypes; produce pro-inflammatory cytokines like IL-4 and IL-17 [85]. |
| CD8+ T-cells | Primary effector cells for killing cancer cells; recognize tumor antigens; induce apoptosis. | Suppressed by immunosuppressive cells and checkpoint molecules (e.g., PD-1/PD-L1) [86]. |
| Regulatory T-cells (Tregs) | Maintain immune homeostasis; suppress excessive immune responses; inhibit CD8+ T-cell activity. | Inhibit CD8+ cytotoxic T-cell function and promote immunosuppression. |
| Tumor-Associated Macrophages (TAMs) | Highly plastic; can be M1 (pro-inflammatory) or M2 (anti-inflammatory); promote tumor progression in M2 phenotype [61]. | Produce anti-inflammatory cytokines (IL-10, TGF-β) that suppress T-cells and promote angiogenesis [87]. |
| MDSCs | Heterogeneous population that inhibits T-cell and NK cell activation; promote expansion of Tregs. | Produce ROS, NO, and arginase to inhibit immune cell functions; associated with poor prognosis [64]. |
| Natural Killer (NK) cells | Recognize and eliminate tumor cells; impaired function due to immunosuppressive factors in the TME. | Impaired by factors like TGF-β and IL-10; less effective in controlling tumor growth [88,89]. |
3. Metabolic Reprogramming in Prostate Cancer
3.1. Overview of Metabolic Pathways
3.2. Metabolic Adaptations of Tumor Cells in Prostate Cancer
3.3. Impact of Tumor Metabolism on the Immune Microenvironment
4. Immunometabolic Changes in the Tumor Microenvironment
4.1. T-Cell Metabolism in the TME
4.2. Macrophage Polarization and Metabolism
4.3. MDSCs and Immunosuppression
4.4. Other Immune Cells Affected by TME Metabolism
5. Hypoxia and Immunometabolism in Prostate Cancer
5.1. Role of Hypoxia in Prostate Cancer Progression
5.2. Hypoxia-Induced Metabolic Shifts
5.3. Influence of Hypoxia on Immune Cells
6. Targeting Immunometabolism in Prostate Cancer Therapy
6.1. Current Therapeutic Approaches Targeting the TME
6.2. Metabolic Inhibitors in Cancer Therapy
6.3. Combination Therapies
6.4. Future Directions in Immunometabolism-Based Therapies
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PCa | Prostate cancer |
| ADT | Prostate-specific antigen |
| CRPC | Castration-resistant prostate cancer |
| TME | Tumor microenvironment |
| OXPHOS | Oxidative phosphorylation |
| HIFs | Hypoxia-inducible factors |
| CAFs | Cancer-associated fibroblasts |
| ECM | Extracellular matrix |
| MDSCs | Myeloid-derived suppressor cells |
| Tregs | Regulatory T-cells |
| PD-L1 | Programmed death-ligand 1 |
| VEGF | Vascular endothelial growth factor |
| ROS | Reactive oxygen species |
| NO | Nitric oxide |
| NK cells | Natural killer cells |
| MHC | Major histocompatibility complex |
| TGF-β | Transforming growth factor beta |
| IL-10 | Interleukin-10 |
| PGE2 | Prostaglandin E2 |
| FAO | Fatty acid oxidation |
| TCA | Tricarboxylic acid |
| HK2 | Hexokinase 2 |
| PKM2 | Pyruvate kinase M2 |
| FASN | Fatty acid synthase |
| GLUTs | Glucose transporters |
| LDH | Lactate dehydrogenase |
| MCTs | Monocarboxylate transporters |
| ASCT2 | Alanine-serine-cysteine transporter 2 |
| GLS | Glutaminase |
| SQLE | Squalene epoxidase |
| LDL | Low-density lipoprotein |
| CAR-T | Chimeric Antigen Receptor T-cell |
| PSMA | Prostate-specific membrane antigen |
| 2-DG | 2-Deoxyglucose |
References
- Schafer, E.J.; Laversanne, M.; Sung, H.; Soerjomataram, I.; Briganti, A.; Dahut, W.; Bray, F.; Jemal, A. Recent Patterns and Trends in Global Prostate Cancer Incidence and Mortality: An Update. Eur. Urol. 2025, 87, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Dee, E.C.; Iyengar, R.; Narayan, A.; Feliciano, E.J.G.; Wu, J.F.; Ho, F.D.V.; Ng, K.; Willmann, J.; Cabaero, M.L.L.; Tan, A.K.N.G.; et al. National Cancer System Characteristics and Prostate Cancer Outcomes: An Analysis of Global Data. Prostate 2025, 85, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Filho, A.M.; Laversanne, M.; Ferlay, J.; Colombet, M.; Piñeros, M.; Znaor, A.; Parkin, D.M.; Soerjomataram, I.; Bray, F. The GLOBOCAN 2022 cancer estimates: Data sources, methods, and a snapshot of the cancer burden worldwide. Int. J. Cancer 2025, 156, 1336–1346. [Google Scholar] [CrossRef]
- Elmadani, M.; Mokaya, P.O.; Omer, A.A.A.; Kiptulon, E.K.; Klara, S.; Orsolya, M. Cancer burden in Europe: A systematic analysis of the GLOBOCAN database (2022). BMC Cancer 2025, 25, 447. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.W.; Cheon, G.J. Prostate-Specific Membrane Antigen PET Imaging in Prostate Cancer: Opportunities and Challenges. Korean J. Radiol. 2018, 19, 819–831. [Google Scholar] [CrossRef]
- Ruckle, H.C.; Klee, G.G.; Oesterling, J.E. Prostate-specific antigen: Concepts for staging prostate cancer and monitoring response to therapy. Mayo Clin. Proc. 1994, 69, 69–79. [Google Scholar] [CrossRef]
- Caram, M.E.; Skolarus, T.A.; Cooney, K.A. Limitations of Prostate-specific Antigen Testing After a Prostate Cancer Diagnosis. Eur. Urol. 2016, 70, 209–210. [Google Scholar] [CrossRef]
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van der Kwast, T.; Bristow, R.G. Prostate cancer. Nat. Rev. Dis. Primers 2021, 7, 9. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Bi, J. Long non-coding RNAs correlate with genomic stability in prostate cancer: A clinical outcome and survival analysis. Genomics 2021, 113, 3141–3151. [Google Scholar] [CrossRef]
- Mehralivand, S.; Thomas, C.; Puhr, M.; Claessens, F.; van de Merbel, A.F.; Dubrovska, A.; Jenster, G.; Bernemann, C.; Sommer, U.; Erb, H.H.H. New advances of the androgen receptor in prostate cancer: Report from the 1st International Androgen Receptor Symposium. J. Transl. Med. 2024, 22, 71. [Google Scholar] [CrossRef]
- Ziglioli, F.; Patera, A.; Isgrò, G.; Campobasso, D.; Guarino, G.; Maestroni, U. Impact of modifiable lifestyle risk factors for prostate cancer prevention: A review of the literature. Front. Oncol. 2023, 13, 1203791. [Google Scholar] [CrossRef] [PubMed]
- Desai, K.; McManus, J.M.; Sharifi, N. Hormonal Therapy for Prostate Cancer. Endocr. Rev. 2021, 42, 354–373. [Google Scholar] [CrossRef]
- Hawley, J.E.; Obradovic, A.Z.; Dallos, M.C.; Lim, E.A.; Runcie, K.; Ager, C.R.; McKiernan, J.; Anderson, C.B.; Decastro, G.J.; Weintraub, J.; et al. Anti-PD-1 immunotherapy with androgen deprivation therapy induces robust immune infiltration in metastatic castration-sensitive prostate cancer. Cancer Cell 2023, 41, 1972–1988.e1975. [Google Scholar] [CrossRef]
- Houben, L.H.P.; Overkamp, M.; van Kraaij, P.; Trommelen, J.; van Roermund, J.G.H.; de Vries, P.; de Laet, K.; van der Meer, S.; Mikkelsen, U.R.; Verdijk, L.B.; et al. Resistance Exercise Training Increases Muscle Mass and Strength in Prostate Cancer Patients on Androgen Deprivation Therapy. Med. Sci. Sports Exerc. 2023, 55, 614–624. [Google Scholar] [CrossRef]
- Yanagisawa, T.; Rajwa, P.; Thibault, C.; Gandaglia, G.; Mori, K.; Kawada, T.; Fukuokaya, W.; Shim, S.R.; Mostafaei, H.; Motlagh, R.S.; et al. Androgen Receptor Signaling Inhibitors in Addition to Docetaxel with Androgen Deprivation Therapy for Metastatic Hormone-sensitive Prostate Cancer: A Systematic Review and Meta-analysis. Eur. Urol. 2022, 82, 584–598. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Parker, C.; Eeles, R.A.; Schröder, F.; Tomlins, S.A.; Tannock, I.; Drake, C.G.; de Bono, J.S. Prostate cancer. Lancet 2016, 387, 70–82. [Google Scholar] [CrossRef]
- Ge, R.; Wang, Z.; Montironi, R.; Jiang, Z.; Cheng, M.; Santoni, M.; Huang, K.; Massari, F.; Lu, X.; Cimadamore, A.; et al. Epigenetic modulations and lineage plasticity in advanced prostate cancer. Ann. Oncol. 2020, 31, 470–479. [Google Scholar] [CrossRef]
- Kang, J.; La Manna, F.; Bonollo, F.; Sampson, N.; Alberts, I.L.; Mingels, C.; Afshar-Oromieh, A.; Thalmann, G.N.; Karkampouna, S. Tumor microenvironment mechanisms and bone metastatic disease progression of prostate cancer. Cancer Lett. 2022, 530, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Karthaus, W.R.; Lee, Y.S.; Gao, V.R.; Wu, C.; Russo, J.W.; Liu, M.; Mota, J.M.; Abida, W.; Linton, E.; et al. Tumor Microenvironment-Derived NRG1 Promotes Antiandrogen Resistance in Prostate Cancer. Cancer Cell 2020, 38, 279–296.e279. [Google Scholar] [CrossRef]
- Yu, G.; Bao, J.; Zhan, M.; Wang, J.; Li, X.; Gu, X.; Song, S.; Yang, Q.; Liu, Y.; Wang, Z.; et al. Comprehensive Analysis of m5C Methylation Regulatory Genes and Tumor Microenvironment in Prostate Cancer. Front. Immunol. 2022, 13, 914577. [Google Scholar] [CrossRef]
- Brady, L.; Nelson, P.S. RISING STARS: Heterogeneity and the tumor microenvironment in neuroendocrine prostate cancer. J. Endocrinol. 2023, 256, e220211. [Google Scholar] [CrossRef] [PubMed]
- Boibessot, C.; Toren, P. Sex steroids in the tumor microenvironment and prostate cancer progression. Endocr. Relat. Cancer 2018, 25, R179–R196. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Lin, L.; Li, Q.; Liu, K.; Huang, Y.; Wang, X.; Cao, K.; Chen, X.; Cao, W.; Li, F.; et al. IGF-2 Preprograms Maturing Macrophages to Acquire Oxidative Phosphorylation-Dependent Anti-inflammatory Properties. Cell Metab. 2019, 29, 1363–1375.e1368. [Google Scholar] [CrossRef]
- Erin, N.; Grahovac, J.; Brozovic, A.; Efferth, T. Tumor microenvironment and epithelial mesenchymal transition as targets to overcome tumor multidrug resistance. Drug Resist. Updat. 2020, 53, 100715. [Google Scholar] [CrossRef]
- Ackerman, D.; Simon, M.C. Hypoxia, lipids, and cancer: Surviving the harsh tumor microenvironment. Trends Cell Biol. 2014, 24, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Dehne, N.; Mora, J.; Namgaladze, D.; Weigert, A.; Brüne, B. Cancer cell and macrophage cross-talk in the tumor microenvironment. Curr. Opin. Pharmacol. 2017, 35, 12–19. [Google Scholar] [CrossRef]
- Jiang, X.; Guo, S.; Wang, S.; Zhang, Y.; Chen, H.; Wang, Y.; Liu, R.; Niu, Y.; Xu, Y. EIF4A3-Induced circARHGAP29 Promotes Aerobic Glycolysis in Docetaxel-Resistant Prostate Cancer through IGF2BP2/c-Myc/LDHA Signaling. Cancer Res. 2022, 82, 831–845. [Google Scholar] [CrossRef]
- Uo, T.; Ojo, K.K.; Sprenger, C.C.T.; Epilepsia, K.S.; Perera, B.G.K.; Damodarasamy, M.; Sun, S.; Kim, S.; Hogan, H.H.; Hulverson, M.A.; et al. A Compound That Inhibits Glycolysis in Prostate Cancer Controls Growth of Advanced Prostate Cancer. Mol. Cancer Ther. 2024, 23, 973–994. [Google Scholar] [CrossRef] [PubMed]
- Ippolito, L.; Comito, G.; Parri, M.; Iozzo, M.; Duatti, A.; Virgilio, F.; Lorito, N.; Bacci, M.; Pardella, E.; Sandrini, G.; et al. Lactate Rewires Lipid Metabolism and Sustains a Metabolic-Epigenetic Axis in Prostate Cancer. Cancer Res. 2022, 82, 1267–1282. [Google Scholar] [CrossRef]
- Chaudagar, K.; Hieromnimon, H.M.; Khurana, R.; Labadie, B.; Hirz, T.; Mei, S.; Hasan, R.; Shafran, J.; Kelley, A.; Apostolov, E.; et al. Reversal of Lactate and PD-1-mediated Macrophage Immunosuppression Controls Growth of PTEN/p53-deficient Prostate Cancer. Clin. Cancer Res. 2023, 29, 1952–1968. [Google Scholar] [CrossRef]
- Liu, Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis. 2006, 9, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, R.K.; Nassar, Z.D.; Hanson, A.R.; Iggo, R.; Townley, S.L.; Dehairs, J.; Mah, C.Y.; Helm, M.; Alizadeh-Ghodsi, M.; Pickering, M.; et al. ACSM1 and ACSM3 Regulate Fatty Acid Metabolism to Support Prostate Cancer Growth and Constrain Ferroptosis. Cancer Res. 2024, 84, 2313–2332. [Google Scholar] [CrossRef]
- Mosquera, M.J.; Kim, S.; Bareja, R.; Fang, Z.; Cai, S.; Pan, H.; Asad, M.; Martin, M.L.; Sigouros, M.; Rowdo, F.M.; et al. Extracellular Matrix in Synthetic Hydrogel-Based Prostate Cancer Organoids Regulate Therapeutic Response to EZH2 and DRD2 Inhibitors. Adv. Mater. 2022, 34, e2100096. [Google Scholar] [CrossRef]
- Liu, B.; Li, X.; Wang, D.; Yu, Y.; Lu, D.; Chen, L.; Lv, F.; Li, Y.; Cheng, L.; Song, Y.; et al. CEMIP promotes extracellular matrix-detached prostate cancer cell survival by inhibiting ferroptosis. Cancer Sci. 2022, 113, 2056–2070. [Google Scholar] [CrossRef] [PubMed]
- Penet, M.F.; Kakkad, S.; Pathak, A.P.; Krishnamachary, B.; Mironchik, Y.; Raman, V.; Solaiyappan, M.; Bhujwalla, Z.M. Structure and Function of a Prostate Cancer Dissemination-Permissive Extracellular Matrix. Clin. Cancer Res. 2017, 23, 2245–2254. [Google Scholar] [CrossRef]
- Luo, Z.W.; Xia, K.; Liu, Y.W.; Liu, J.H.; Rao, S.S.; Hu, X.K.; Chen, C.Y.; Xu, R.; Wang, Z.X.; Xie, H. Extracellular Vesicles from Akkermansia muciniphila Elicit Antitumor Immunity Against Prostate Cancer via Modulation of CD8+ T Cells and Macrophages. Int. J. Nanomed. 2021, 16, 2949–2963. [Google Scholar] [CrossRef]
- Wang, S.; Huang, M.; Chen, M.; Sun, Z.; Jiao, Y.; Ye, G.; Pan, J.; Ye, W.; Zhao, J.; Zhang, D. Zoledronic acid and thymosin α1 elicit antitumor immunity against prostate cancer by enhancing tumor inflammation and cytotoxic T cells. J. Immunother. Cancer 2023, 11, e006381. [Google Scholar] [CrossRef] [PubMed]
- Bhinder, B.; Ferguson, A.; Sigouros, M.; Uppal, M.; Elsaeed, A.G.; Bareja, R.; Alnajar, H.; Eng, K.W.; Conteduca, V.; Sboner, A.; et al. Immunogenomic Landscape of Neuroendocrine Prostate Cancer. Clin. Cancer Res. 2023, 29, 2933–2943. [Google Scholar] [CrossRef]
- Terzic, J.; Abu El Maaty, M.A.; Lutzing, R.; Vincent, A.; El Bizri, R.; Jung, M.; Keime, C.; Metzger, D. Hypoxia-inducible factor 1A inhibition overcomes castration resistance of prostate tumors. EMBO Mol. Med. 2023, 15, e17209. [Google Scholar] [CrossRef]
- Kimbro, K.S.; Simons, J.W. Hypoxia-inducible factor-1 in human breast and prostate cancer. Endocr. Relat. Cancer 2006, 13, 739–749. [Google Scholar] [CrossRef]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef]
- Yenyuwadee, S.; Aliazis, K.; Wang, Q.; Christofides, A.; Shah, R.; Patsoukis, N.; Boussiotis, V.A. Immune cellular components and signaling pathways in the tumor microenvironment. Semin. Cancer Biol. 2022, 86, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xu, Y.-X.; Wang, Y.-S.; Ren, Y.-Y.; Dong, X.-M.; Wu, P.; Xie, T.; Zhang, Q.; Zhou, J.-L. Prostate cancer microenvironment: Multidimensional regulation of immune cells, vascular system, stromal cells, and microbiota. Mol. Cancer 2024, 23, 229. [Google Scholar] [CrossRef]
- Zhang, Y.; Fan, A.; Li, Y.; Liu, Z.; Yu, L.; Guo, J.; Hou, J.; Li, X.; Chen, W. Single-cell RNA sequencing reveals that HSD17B2 in cancer-associated fibroblasts promotes the development and progression of castration-resistant prostate cancer. Cancer Lett. 2023, 566, 216244. [Google Scholar] [CrossRef]
- Wang, H.; Li, N.; Liu, Q.; Guo, J.; Pan, Q.; Cheng, B.; Xu, J.; Dong, B.; Yang, G.; Yang, B.; et al. Antiandrogen treatment induces stromal cell reprogramming to promote castration resistance in prostate cancer. Cancer Cell 2023, 41, 1345–1362.e1349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Liu, M.; Mai, F.; Li, X.; Wang, W.; Huang, Q.; Du, X.; Ding, W.; Li, Y.; Barwick, B.G.; et al. Interruption of KLF5 acetylation promotes PTEN-deficient prostate cancer progression by reprogramming cancer-associated fibroblasts. J. Clin. Investig. 2024, 134, e175949. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, F. Cancer-associated fibroblast-derived gene signatures predict radiotherapeutic survival in prostate cancer patients. J. Transl. Med. 2022, 20, 453. [Google Scholar] [CrossRef]
- Cheteh, E.H.; Sarne, V.; Ceder, S.; Bianchi, J.; Augsten, M.; Rundqvist, H.; Egevad, L.; Östman, A.; Wiman, K.G. Interleukin-6 derived from cancer-associated fibroblasts attenuates the p53 response to doxorubicin in prostate cancer cells. Cell Death Discov. 2020, 6, 42. [Google Scholar] [CrossRef]
- Bonollo, F.; Thalmann, G.N.; Kruithof-de Julio, M.; Karkampouna, S. The Role of Cancer-Associated Fibroblasts in Prostate Cancer Tumorigenesis. Cancers 2020, 12, 1887. [Google Scholar] [CrossRef]
- Wu, F.; Yang, J.; Liu, J.; Wang, Y.; Mu, J.; Zeng, Q.; Deng, S.; Zhou, H. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct. Target. Ther. 2021, 6, 218. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, C.; D’Angiolo, R.; Gentile, G.; Giovannelli, P.; Perillo, B.; Migliaccio, A.; Castoria, G.; Di Donato, M. The Androgen Regulation of Matrix Metalloproteases in Prostate Cancer and Its Related Tumor Microenvironment. Endocrines 2023, 4, 350–365. [Google Scholar] [CrossRef]
- Gong, Y.; Chippada-Venkata, U.D.; Oh, W.K. Roles of matrix metalloproteinases and their natural inhibitors in prostate cancer progression. Cancers 2014, 6, 1298–1327. [Google Scholar] [CrossRef]
- Zhang, F.; Ma, Y.; Li, D.; Wei, J.; Chen, K.; Zhang, E.; Liu, G.; Chu, X.; Liu, X.; Liu, W.; et al. Cancer associated fibroblasts and metabolic reprogramming: Unraveling the intricate crosstalk in tumor evolution. J. Hematol. Oncol. 2024, 17, 80. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, X.; Wang, L.; Hong, X.; Yang, J. Metabolic reprogramming and crosstalk of cancer-related fibroblasts and immune cells in the tumor microenvironment. Front. Endocrinol. 2022, 13, 988295. [Google Scholar] [CrossRef]
- Koinis, F.; Xagara, A.; Chantzara, E.; Leontopoulou, V.; Aidarinis, C.; Kotsakis, A. Myeloid-Derived Suppressor Cells in Prostate Cancer: Present Knowledge and Future Perspectives. Cells 2021, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Bronte, G.; Conteduca, V.; Landriscina, M.; Procopio, A.D. Circulating myeloid-derived suppressor cells and survival in prostate cancer patients: Systematic review and meta-analysis. Prostate Cancer Prostatic Dis. 2023, 26, 41–46. [Google Scholar] [CrossRef]
- Siemińska, I.; Baran, J. Myeloid-Derived Suppressor Cells as Key Players and Promising Therapy Targets in Prostate Cancer. Front. Oncol. 2022, 12, 862416. [Google Scholar] [CrossRef]
- San-Jose Manso, L.; Alfranca, A.; Moreno-Pérez, I.; Ruiz-Vico, M.; Velasco, C.; Toquero, P.; Pacheco, M.; Zapatero, A.; Aldave, D.; Celada, G.; et al. Immunome profiling in prostate cancer: A guide for clinicians. Front. Immunol. 2024, 15, 1398109. [Google Scholar] [CrossRef]
- Xu, F.; Wang, X.; Huang, Y.; Zhang, X.; Sun, W.; Du, Y.; Xu, Z.; Kou, H.; Zhu, S.; Liu, C.; et al. Prostate cancer cell-derived exosomal IL-8 fosters immune evasion by disturbing glucolipid metabolism of CD8+ T cell. Cell Rep. 2023, 42, 113424. [Google Scholar] [CrossRef]
- Stultz, J.; Fong, L. How to turn up the heat on the cold immune microenvironment of metastatic prostate cancer. Prostate Cancer Prostatic Dis. 2021, 24, 697–717. [Google Scholar] [CrossRef] [PubMed]
- Vogelzang, N.J.; Beer, T.M.; Gerritsen, W.; Oudard, S.; Wiechno, P.; Kukielka-Budny, B.; Samal, V.; Hajek, J.; Feyerabend, S.; Khoo, V.; et al. Efficacy and Safety of Autologous Dendritic Cell-Based Immunotherapy, Docetaxel, and Prednisone vs Placebo in Patients With Metastatic Castration-Resistant Prostate Cancer: The VIABLE Phase 3 Randomized Clinical Trial. JAMA Oncol. 2022, 8, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Molina, O.E.; LaRue, H.; Simonyan, D.; Hovington, H.; Têtu, B.; Fradet, V.; Lacombe, L.; Toren, P.; Bergeron, A.; Fradet, Y. High infiltration of CD209+ dendritic cells and CD163+ macrophages in the peritumor area of prostate cancer is predictive of late adverse outcomes. Front. Immunol. 2023, 14, 1205266. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Wu, W.; Jiang, Q. Crosstalk between Endothelial Cells and Tumor Cells: A New Era in Prostate Cancer Progression. Int. J. Mol. Sci. 2023, 24, 16893. [Google Scholar] [CrossRef]
- Paiva, A.E.; Lousado, L.; Almeida, V.M.; Andreotti, J.P.; Santos, G.S.P.; Azevedo, P.O.; Sena, I.F.G.; Prazeres, P.; Borges, I.T.; Azevedo, V.; et al. Endothelial Cells as Precursors for Osteoblasts in the Metastatic Prostate Cancer Bone. Neoplasia 2017, 19, 928–931. [Google Scholar] [CrossRef]
- Bhowmick, S.; Bhowmick, N.A. RARγ: The Bone of Contention for Endothelial Cells in Prostate Cancer Metastasis. Cancer Res. 2022, 82, 2975–2976. [Google Scholar] [CrossRef]
- Zhao, R.; Bei, X.; Yang, B.; Wang, X.; Jiang, C.; Shi, F.; Wang, X.; Zhu, Y.; Jing, Y.; Han, B.; et al. Endothelial cells promote metastasis of prostate cancer by enhancing autophagy. J. Exp. Clin. Cancer Res. 2018, 37, 221. [Google Scholar] [CrossRef]
- Teng, M.W.L.; Ngiow, S.F.; Ribas, A.; Smyth, M.J. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015, 75, 2139–2145. [Google Scholar] [CrossRef]
- Leone, P.; Malerba, E.; Susca, N.; Favoino, E.; Perosa, F.; Brunori, G.; Prete, M.; Racanelli, V. Endothelial cells in tumor microenvironment: Insights and perspectives. Front. Immunol. 2024, 15, 1367875. [Google Scholar] [CrossRef]
- D’Antonio, K.B.; Toubaji, A.; Albadine, R.; Mondul, A.M.; Platz, E.A.; Netto, G.J.; Getzenberg, R.H. Extracellular matrix associated protein CYR61 is linked to prostate cancer development. J. Urol. 2010, 183, 1604–1610. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.A.; Cooper, C.R.; Sikes, R.A. Changes in extracellular matrix (ECM) and ECM-associated proteins in the metastatic progression of prostate cancer. Reprod. Biol. Endocrinol. 2004, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Li, Y.; Zhang, S.; Wang, X.; Dou, H.; Yu, X.; Zhang, Z.; Yang, S.; Xiao, M. Extracellular matrix remodeling in tumor progression and immune escape: From mechanisms to treatments. Mol. Cancer 2023, 22, 48. [Google Scholar] [CrossRef]
- Luthold, C.; Hallal, T.; Labbé, D.P.; Bordeleau, F. The Extracellular Matrix Stiffening: A Trigger of Prostate Cancer Progression and Castration Resistance? Cancers 2022, 14, 2887. [Google Scholar] [CrossRef]
- Jianfeng, W.; Yutao, W.; Jianbin, B. TACR2 is associated with the immune microenvironment and inhibits migration and proliferation via the Wnt/β-catenin signaling pathway in prostate cancer. Cancer Cell Int. 2021, 21, 415. [Google Scholar] [CrossRef]
- Davidsson, S.; Ohlson, A.L.; Andersson, S.O.; Fall, K.; Meisner, A.; Fiorentino, M.; Andrén, O.; Rider, J.R. CD4 helper T cells, CD8 cytotoxic T cells, and FOXP3+ regulatory T cells with respect to lethal prostate cancer. Mod. Pathol. 2013, 26, 448–455. [Google Scholar] [CrossRef]
- Radej, S.; Szewc, M.; Maciejewski, R. Prostate Infiltration by Treg and Th17 Cells as an Immune Response to Propionibacterium acnes Infection in the Course of Benign Prostatic Hyperplasia and Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 8849. [Google Scholar] [CrossRef] [PubMed]
- Bohner, P.; Chevalier, M.F.; Cesson, V.; Rodrigues-Dias, S.C.; Dartiguenave, F.; Burruni, R.; Tawadros, T.; Valerio, M.; Lucca, I.; Nardelli-Haefliger, D.; et al. Double Positive CD4+CD8+ T Cells Are Enriched in Urological Cancers and Favor T Helper-2 Polarization. Front. Immunol. 2019, 10, 622. [Google Scholar] [CrossRef]
- Comito, G.; Iscaro, A.; Bacci, M.; Morandi, A.; Ippolito, L.; Parri, M.; Montagnani, I.; Raspollini, M.R.; Serni, S.; Simeoni, L.; et al. Lactate modulates CD4+ T-cell polarization and induces an immunosuppressive environment, which sustains prostate carcinoma progression via TLR8/miR21 axis. Oncogene 2019, 38, 3681–3695. [Google Scholar] [CrossRef]
- Dahut, M.; Fousek, K.; Horn, L.A.; Angstadt, S.; Qin, H.; Hamilton, D.H.; Schlom, J.; Palena, C. Fulvestrant increases the susceptibility of enzalutamide-resistant prostate cancer cells to NK-mediated lysis. J. Immunother. Cancer 2023, 11, e007386. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Wang, Y.; Zhao, Y.; Kim, J.J.; Li, H.; Meng, C.; Chen, F.; Zhang, J.; Mak, D.H.; Van, V.; et al. Immune checkpoint B7-H3 is a therapeutic vulnerability in prostate cancer harboring PTEN and TP53 deficiencies. Sci. Transl. Med. 2023, 15, eadf6724. [Google Scholar] [CrossRef]
- Su, H.; Wang, Y.; Li, H. RNA m6A Methylation Regulators Multi-Omics Analysis in Prostate Cancer. Front. Genet. 2021, 12, 768041. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Chakraborty, G.; Raja, R.; Kale, S.; Kundu, G.C. Prostaglandin E2 regulates tumor angiogenesis in prostate cancer. Cancer Res. 2008, 68, 7750–7759. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Grebhardt, S.; Shi, J.; Peipe, I.; Zhang, J.; Mayer, D. Prostaglandin E2 stimulates S100A8 expression by activating protein kinase A and CCAAT/enhancer-binding-protein-beta in prostate cancer cells. Int. J. Biochem. Cell Biol. 2012, 44, 1919–1928. [Google Scholar] [CrossRef]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations. Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef]
- Lazarova, M.; Steinle, A. Impairment of NKG2D-Mediated Tumor Immunity by TGF-β. Front. Immunol. 2019, 10, 2689. [Google Scholar] [CrossRef]
- Masmoudi, D.; Villalba, M.; Alix-Panabières, C. Natural killer cells: The immune frontline against circulating tumor cells. J. Exp. Clin. Cancer Res. 2025, 44, 118. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- de Wet, L.; Williams, A.; Gillard, M.; Kregel, S.; Lamperis, S.; Gutgesell, L.C.; Vellky, J.E.; Brown, R.; Conger, K.; Paner, G.P.; et al. SOX2 mediates metabolic reprogramming of prostate cancer cells. Oncogene 2022, 41, 1190–1202. [Google Scholar] [CrossRef]
- Ma, S.; Chen, Y.; Quan, P.; Zhang, J.; Han, S.; Wang, G.; Qi, R.; Zhang, X.; Wang, F.; Yuan, J.; et al. NPAS2 promotes aerobic glycolysis and tumor growth in prostate cancer through HIF-1A signaling. BMC Cancer 2023, 23, 280. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Yang, Z.; Li, J.; Sun, Y.; Zhang, X.; Qu, Z.; Luo, Y.; Zhang, L. Effects of glutamate and aspartate on prostate cancer and breast cancer: A Mendelian randomization study. BMC Genom. 2022, 23, 213. [Google Scholar] [CrossRef]
- Chakraborty, G.; Nandakumar, S.; Hirani, R.; Nguyen, B.; Stopsack, K.H.; Kreitzer, C.; Rajanala, S.H.; Ghale, R.; Mazzu, Y.Z.; Pillarsetty, N.V.K.; et al. The Impact of PIK3R1 Mutations and Insulin-PI3K-Glycolytic Pathway Regulation in Prostate Cancer. Clin. Cancer Res. 2022, 28, 3603–3617. [Google Scholar] [CrossRef]
- Hu, C.; Xu, H.; Li, Z.; Liu, D.; Zhang, S.; Fang, F.; Wang, L. Juglone promotes antitumor activity against prostate cancer via suppressing glycolysis and oxidative phosphorylation. Phytother. Res. 2023, 37, 515–526. [Google Scholar] [CrossRef]
- Lv, C.; Fu, S.; Dong, Q.; Yu, Z.; Zhang, G.; Kong, C.; Fu, C.; Zeng, Y. PAGE4 promotes prostate cancer cells survive under oxidative stress through modulating MAPK/JNK/ERK pathway. J. Exp. Clin. Cancer Res. 2019, 38, 24. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, H.J.; Figueira, M.I.; Vaz, C.V.; Carvalho, T.M.A.; Brás, L.A.; Madureira, P.A.; Oliveira, P.J.; Sardão, V.A.; Socorro, S. Glutaminolysis is a metabolic route essential for survival and growth of prostate cancer cells and a target of 5α-dihydrotestosterone regulation. Cell. Oncol. 2021, 44, 385–403. [Google Scholar] [CrossRef] [PubMed]
- Morales, A.; Greenberg, M.; Nardi, F.; Gil, V.; Hayward, S.W.; Crawford, S.E.; Franco, O.E. Loss of ephrin B2 receptor (EPHB2) sets lipid rheostat by regulating proteins DGAT1 and ATGL inducing lipid droplet storage in prostate cancer cells. Lab. Investig. 2021, 101, 921–934. [Google Scholar] [CrossRef]
- Pujana-Vaquerizo, M.; Bozal-Basterra, L.; Carracedo, A. Metabolic adaptations in prostate cancer. Br. J. Cancer 2024, 131, 1250–1262. [Google Scholar] [CrossRef]
- Cutruzzolà, F.; Giardina, G.; Marani, M.; Macone, A.; Paiardini, A.; Rinaldo, S.; Paone, A. Glucose Metabolism in the Progression of Prostate Cancer. Front. Physiol. 2017, 8, 97. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Tomás, R.; Pérez-Guillén, I. Lactate in the Tumor Microenvironment: An Essential Molecule in Cancer Progression and Treatment. Cancers 2020, 12, 3244. [Google Scholar] [CrossRef]
- Ahmad, F.; Cherukuri, M.K.; Choyke, P.L. Metabolic reprogramming in prostate cancer. Br. J. Cancer 2021, 125, 1185–1196. [Google Scholar] [CrossRef]
- Beier, A.-M.K.; Puhr, M.; Stope, M.B.; Thomas, C.; Erb, H.H.H. Metabolic changes during prostate cancer development and progression. J. Cancer Res. Clin. Oncol. 2023, 149, 2259–2270. [Google Scholar] [CrossRef] [PubMed]
- Aurilio, G.; Cimadamore, A.; Mazzucchelli, R.; Lopez-Beltran, A.; Verri, E.; Scarpelli, M.; Massari, F.; Cheng, L.; Santoni, M.; Montironi, R. Androgen Receptor Signaling Pathway in Prostate Cancer: From Genetics to Clinical Applications. Cells 2020, 9, 2653. [Google Scholar] [CrossRef]
- Tan, M.H.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef]
- Shangguan, X.; He, J.; Ma, Z.; Zhang, W.; Ji, Y.; Shen, K.; Yue, Z.; Li, W.; Xin, Z.; Zheng, Q.; et al. SUMOylation controls the binding of hexokinase 2 to mitochondria and protects against prostate cancer tumorigenesis. Nat. Commun. 2021, 12, 1812. [Google Scholar] [CrossRef]
- Feng, T.; Wang, J.; Cheng, K.; Lu, Q.; Zhao, R.; Wang, S.; Zhang, Q.; Ge, L.; Pan, J.; Song, G.; et al. IL13Rα1 prevents a castration resistant phenotype of prostate cancer by targeting hexokinase 2 for ubiquitin-mediated degradation. Cancer Biol. Med. 2021, 19, 1008–1028. [Google Scholar] [CrossRef] [PubMed]
- Galbraith, L.; Leung, H.Y.; Ahmad, I. Lipid pathway deregulation in advanced prostate cancer. Pharmacol. Res. 2018, 131, 177–184. [Google Scholar] [CrossRef]
- Fridman, E.S.; Ginini, L.; Gil, Z. The Role of Extracellular Vesicles in Metabolic Reprogramming of the Tumor Microenvironment. Cells 2022, 11, 1433. [Google Scholar] [CrossRef]
- Xi, Y.; Shen, Y.; Chen, L.; Tan, L.; Shen, W.; Niu, X. Exosome-mediated metabolic reprogramming: Implications in esophageal carcinoma progression and tumor microenvironment remodeling. Cytokine Growth Factor Rev. 2023, 73, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wang, J.; Chen, L.; Wang, H.; Liang, C.-Z.; Huang, J.; Xu, L.-F. The role of glutamine metabolism in castration-resistant prostate cancer. Asian J. Androl. 2023, 25, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Wang, X.; Gong, Z.; Yu, M.; Wu, H.; Zhang, D. Exosome-mediated metabolic reprogramming: The emerging role in tumor microenvironment remodeling and its influence on cancer progression. Signal Transduct. Target. Ther. 2020, 5, 242. [Google Scholar] [CrossRef]
- Stelmach-Mardas, M.; Warchoł, W.; Garczyk, A.; Warchoł, E.; Korczak, J.; Litwiniuk, M.; Brajer-Luftmann, B.; Mardas, M. Influence of Androgen Deprivation Therapy on the Development of Sarcopenia in Patients with Prostate Cancer: A Systematic Review. Nutrients 2024, 16, 656. [Google Scholar] [CrossRef]
- Wright, H.H.; Walker, M.A.; Broadbent, S.; Linton, C.; Keech, J.J.; Rune, K.T.; Davis, C.L.; Morris, M.; Zhang, A.; Newton, R.U.; et al. The effect of dietary interventions or patterns on the cardiometabolic health of individuals treated with androgen deprivation therapy for prostate cancer: A systematic review. Maturitas 2024, 184, 107940. [Google Scholar] [CrossRef]
- Brett, S.I.; Kim, Y.; Biggs, C.N.; Chin, J.L.; Leong, H.S. Extracellular vesicles such as prostate cancer cell fragments as a fluid biopsy for prostate cancer. Prostate Cancer Prostatic Dis. 2015, 18, 213–220. [Google Scholar] [CrossRef]
- True, L.D.; Zhang, H.; Ye, M.; Huang, C.Y.; Nelson, P.S.; von Haller, P.D.; Tjoelker, L.W.; Kim, J.S.; Qian, W.J.; Smith, R.D.; et al. CD90/THY1 is overexpressed in prostate cancer-associated fibroblasts and could serve as a cancer biomarker. Mod. Pathol. 2010, 23, 1346–1356. [Google Scholar] [CrossRef] [PubMed]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- von Amsberg, G.; Alsdorf, W.; Karagiannis, P.; Coym, A.; Kaune, M.; Werner, S.; Graefen, M.; Bokemeyer, C.; Merkens, L.; Dyshlovoy, S.A. Immunotherapy in Advanced Prostate Cancer-Light at the End of the Tunnel? Int. J. Mol. Sci. 2022, 23, 2569. [Google Scholar] [CrossRef]
- Xia, C.; Yin, S.; To, K.K.W.; Fu, L. CD39/CD73/A2AR pathway and cancer immunotherapy. Mol. Cancer 2023, 22, 44. [Google Scholar] [CrossRef]
- Liou, G.-Y.; C’Lay-Pettis, R.; Kavuri, S. Involvement of Reactive Oxygen Species in Prostate Cancer and Its Disparity in African Descendants. Int. J. Mol. Sci. 2024, 25, 6665. [Google Scholar] [CrossRef] [PubMed]
- Zahm, C.D.; Johnson, L.E.; McNeel, D.G. Increased indoleamine 2,3-dioxygenase activity and expression in prostate cancer following targeted immunotherapy. Cancer Immunol. Immunother. 2019, 68, 1661–1669. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.-J.; Wang, L.; Li, Z.; Ku, C.-L.; Ho, P.-C. Metabolic challenges and interventions in CAR T cell therapy. Sci. Immunol. 2023, 8, eabq3016. [Google Scholar] [CrossRef]
- Jiang, Z.; He, J.; Zhang, B.; Wang, L.; Long, C.; Zhao, B.; Yang, Y.; Du, L.; Luo, W.; Hu, J.; et al. A Potential “Anti-Warburg Effect” in Circulating Tumor Cell-mediated Metastatic Progression? Aging Dis. 2024, 16, 269–282. [Google Scholar] [CrossRef]
- Song, C.; Zhang, J.; Liu, X.; Li, M.; Wang, D.; Kang, Z.; Yu, J.; Chen, J.; Pan, H.; Wang, H.; et al. PTEN loss promotes Warburg effect and prostate cancer cell growth by inducing FBP1 degradation. Front. Oncol. 2022, 12, 911466. [Google Scholar] [CrossRef]
- Zohar, Y.; Mabjeesh, N.J. Targeting HIF-1 for prostate cancer: A synthesis of preclinical evidence. Expert. Opin. Ther. Targets 2023, 27, 715–731. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; He, Z.; Yuan, Y.; Xie, J.; Zhou, Y.; Guo, B.; Guo, J. Docetaxel suppressed cell proliferation through Smad3/HIF-1α-mediated glycolysis in prostate cancer cells. Cell Commun. Signal 2022, 20, 194. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef]
- Koo, S.-J.; Garg, N.J. Metabolic programming of macrophage functions and pathogens control. Redox Biol. 2019, 24, 101198. [Google Scholar] [CrossRef]
- Gu, Q.; Qi, A.; Wang, N.; Zhou, Z.; Zhou, X. Macrophage dynamics in prostate cancer: Molecular to therapeutic insights. Biomed. Pharmacother. 2024, 177, 117002. [Google Scholar] [CrossRef]
- Setrerrahmane, S.; Xu, H. Tumor-related interleukins: Old validated targets for new anti-cancer drug development. Mol. Cancer 2017, 16, 153. [Google Scholar] [CrossRef]
- Chen, Z.; Han, F.; Du, Y.; Shi, H.; Zhou, W. Hypoxic microenvironment in cancer: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Basak, U.; Sarkar, T.; Mukherjee, S.; Chakraborty, S.; Dutta, A.; Dutta, S.; Nayak, D.; Kaushik, S.; Das, T.; Sa, G. Tumor-associated macrophages: An effective player of the tumor microenvironment. Front. Immunol. 2023, 14, 1295257. [Google Scholar] [CrossRef]
- Hofer, F.; Di Sario, G.; Musiu, C.; Sartoris, S.; De Sanctis, F.; Ugel, S. A Complex Metabolic Network Confers Immunosuppressive Functions to Myeloid-Derived Suppressor Cells (MDSCs) within the Tumour Microenvironment. Cells 2021, 10, 2700. [Google Scholar] [CrossRef]
- Gonzalez-Menendez, P.; Hevia, D.; Mayo, J.C.; Sainz, R.M. The dark side of glucose transporters in prostate cancer: Are they a new feature to characterize carcinomas? Int. J. Cancer 2018, 142, 2414–2424. [Google Scholar] [CrossRef] [PubMed]
- Mudhish, E.A.; Siddique, A.B.; Ebrahim, H.Y.; Abdelwahed, K.S.; King, J.A.; El Sayed, K.A. The Tobacco β-Cembrenediol: A Prostate Cancer Recurrence Suppressor Lead and Prospective Scaffold via Modulation of Indoleamine 2,3-Dioxygenase and Tryptophan Dioxygenase. Nutrients 2022, 14, 1505. [Google Scholar] [CrossRef]
- Zhu, T.; Li, Y.; Wang, Y.; Li, D. The Application of Dendritic Cells Vaccines in Tumor Therapy and Their Combination with Biomimetic Nanoparticles. Vaccines 2025, 13, 337. [Google Scholar] [CrossRef]
- Peng, X.; He, Y.; Huang, J.; Tao, Y.; Liu, S. Metabolism of Dendritic Cells in Tumor Microenvironment: For Immunotherapy. Front. Immunol. 2021, 12, 613492. [Google Scholar] [CrossRef]
- Cornel, A.M.; Mimpen, I.L.; Nierkens, S. MHC Class I Downregulation in Cancer: Underlying Mechanisms and Potential Targets for Cancer Immunotherapy. Cancers 2020, 12, 1760. [Google Scholar] [CrossRef]
- Slattery, K.; Gardiner, C.M. NK Cell Metabolism and TGFβ—Implications for Immunotherapy. Front. Immunol. 2019, 10, 2915. [Google Scholar] [CrossRef] [PubMed]
- Angelin, A.; Gil-de-Gómez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J.; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293.e7. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.A.; Vissers, J.P.C.; Nanda, J.; Stewart, G.D.; Husi, H.; Habib, F.K.; Hammond, D.E.; Gethings, L.A. The influence of hypoxia on the prostate cancer proteome. Clin. Chem. Lab. Med. 2020, 58, 980–993. [Google Scholar] [CrossRef]
- Marignol, L.; Coffey, M.; Lawler, M.; Hollywood, D. Hypoxia in prostate cancer: A powerful shield against tumour destruction? Cancer Treat. Rev. 2008, 34, 313–327. [Google Scholar] [CrossRef]
- Silva, V.L.; Ruiz, A.; Ali, A.; Pereira, S.; Seitsonen, J.; Ruokolainen, J.; Furlong, F.; Coulter, J.; Al-Jamal, W.T. Hypoxia-targeted cupric-tirapazamine liposomes potentiate radiotherapy in prostate cancer spheroids. Int. J. Pharm. 2021, 607, 121018. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, O.; Galvão, D.A.; Taaffe, D.R.; Chee, R.; Spry, N.; Newton, R.U. Exercise modulation of tumour perfusion and hypoxia to improve radiotherapy response in prostate cancer. Prostate Cancer Prostatic Dis. 2021, 24, 1–14. [Google Scholar] [CrossRef]
- Belisario, D.C.; Kopecka, J.; Pasino, M.; Akman, M.; De Smaele, E.; Donadelli, M.; Riganti, C. Hypoxia Dictates Metabolic Rewiring of Tumors: Implications for Chemoresistance. Cells 2020, 9, 2598. [Google Scholar] [CrossRef]
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer metabolism and the Warburg effect: The role of HIF-1 and PI3K. Mol. Biol. Rep. 2015, 42, 841–851. [Google Scholar] [CrossRef]
- Kukurugya, M.A.; Rosset, S.; Titov, D.V. The Warburg Effect is the result of faster ATP production by glycolysis than respiration. Proc. Natl. Acad. Sci. USA 2024, 121, e2409509121. [Google Scholar] [CrossRef]
- Gu, X.-Y.; Yang, J.-L.; Lai, R.; Zhou, Z.-J.; Tang, D.; Hu, L.; Zhao, L.-J. Impact of lactate on immune cell function in the tumor microenvironment: Mechanisms and therapeutic perspectives. Front. Immunol. 2025, 16, 1563303. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Q.; Huang, X.; Yang, M.; Zhou, S.; Li, Z.; Fang, Z.; Tang, Y.; Chen, Q.; Hou, H.; et al. Lactate in the tumor microenvironment: A rising star for targeted tumor therapy. Front. Nutr. 2023, 10, 1113739. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Li, T.; Li, X.; Zhang, L.; Sun, L.; He, X.; Zhong, X.; Jia, D.; Song, L.; Semenza, G.L.; et al. HIF-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep. 2014, 8, 1930–1942. [Google Scholar] [CrossRef]
- Sun, R.C.; Denko, N.C. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 2014, 19, 285–292. [Google Scholar] [CrossRef]
- Jianfeng, W.; Yutao, W.; Jianbin, B. Indolethylamine-N-Methyltransferase Inhibits Proliferation and Promotes Apoptosis of Human Prostate Cancer Cells: A Mechanistic Exploration. Front. Cell Dev. Biol. 2022, 10, 805402. [Google Scholar] [CrossRef]
- Sitkovsky, M.; Lukashev, D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat. Rev. Immunol. 2005, 5, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Wang, B.; Zhao, Q.; Zhang, Y.; Liu, Z.; Zheng, Z.; Liu, S.; Meng, L.; Xin, Y.; Jiang, X. Targeting hypoxia in the tumor microenvironment: A potential strategy to improve cancer immunotherapy. J. Exp. Clin. Cancer Res. 2021, 40, 24. [Google Scholar] [CrossRef] [PubMed]
- Claps, M.; Mennitto, A.; Guadalupi, V.; Sepe, P.; Stellato, M.; Zattarin, E.; Gillessen, S.S.; Sternberg, C.N.; Berruti, A.; De Braud, F.G.M.; et al. Immune-checkpoint inhibitors and metastatic prostate cancer therapy: Learning by making mistakes. Cancer Treat. Rev. 2020, 88, 102057. [Google Scholar] [CrossRef]
- Wu, Z.; Chen, Q.; Qu, L.; Li, M.; Wang, L.; Mir, M.C.; Carbonara, U.; Pandolfo, S.D.; Black, P.C.; Paul, A.K.; et al. Adverse Events of Immune Checkpoint Inhibitors Therapy for Urologic Cancer Patients in Clinical Trials: A Collaborative Systematic Review and Meta-analysis. Eur. Urol. 2022, 81, 414–425. [Google Scholar] [CrossRef]
- He, Y.; Xu, W.; Xiao, Y.-T.; Huang, H.; Gu, D.; Ren, S. Targeting signaling pathways in prostate cancer: Mechanisms and clinical trials. Signal Transduct. Target. Ther. 2022, 7, 198. [Google Scholar] [CrossRef]
- Ruiz de Porras, V.; Pardo, J.C.; Notario, L.; Etxaniz, O.; Font, A. Immune Checkpoint Inhibitors: A Promising Treatment Option for Metastatic Castration-Resistant Prostate Cancer? Int. J. Mol. Sci. 2021, 22, 4712. [Google Scholar] [CrossRef] [PubMed]
- Noori, M.; Azizi, S.; Mahjoubfar, A.; Abbasi Varaki, F.; Fayyaz, F.; Mousavian, A.H.; Bashash, D.; Kardoust Parizi, M.; Kasaeian, A. Efficacy and safety of immune checkpoint inhibitors for patients with prostate cancer: A systematic review and meta-analysis. Front. Immunol. 2023, 14, 1181051. [Google Scholar] [CrossRef]
- Zuccolotto, G.; Penna, A.; Fracasso, G.; Carpanese, D.; Montagner, I.M.; Dalla Santa, S.; Rosato, A. PSMA-Specific CAR-Engineered T Cells for Prostate Cancer: CD28 Outperforms Combined CD28-4-1BB “Super-Stimulation”. Front. Oncol. 2021, 11, 708073. [Google Scholar] [CrossRef]
- Liu, Z.-L.; Chen, H.-H.; Zheng, L.-L.; Sun, L.-P.; Shi, L. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduct. Target. Ther. 2023, 8, 198. [Google Scholar] [CrossRef] [PubMed]
- Niveau, C.; Cettour-Cave, M.; Mouret, S.; Sosa Cuevas, E.; Pezet, M.; Roubinet, B.; Gil, H.; De Fraipont, F.; Landemarre, L.; Charles, J.; et al. MCT1 lactate transporter blockade re-invigorates anti-tumor immunity through metabolic rewiring of dendritic cells in melanoma. Nat. Commun. 2025, 16, 1083. [Google Scholar] [CrossRef]
- Hossain, F.; Al-Khami, A.A.; Wyczechowska, D.; Hernandez, C.; Zheng, L.; Reiss, K.; Valle, L.D.; Trillo-Tinoco, J.; Maj, T.; Zou, W.; et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol. Res. 2015, 3, 1236–1247. [Google Scholar] [CrossRef]
- Luo, Y.; Yang, Z.; Yu, Y.; Zhang, P. HIF1α lactylation enhances KIAA1199 transcription to promote angiogenesis and vasculogenic mimicry in prostate cancer. Int. J. Biol. Macromol. 2022, 222, 2225–2243. [Google Scholar] [CrossRef] [PubMed]
- White, M.A.; Lin, C.; Rajapakshe, K.; Dong, J.; Shi, Y.; Tsouko, E.; Mukhopadhyay, R.; Jasso, D.; Dawood, W.; Coarfa, C.; et al. Glutamine Transporters Are Targets of Multiple Oncogenic Signaling Pathways in Prostate Cancer. Mol. Cancer Res. 2017, 15, 1017–1028. [Google Scholar] [CrossRef]
- Wang, J.-B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.B.; Dias, S.M.G.; Dang, C.V.; et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010, 18, 207–219. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, Y.; Wu, X.; Liu, Z.; Liu, X. A novel strategy to fuel cancer immunotherapy: Targeting glucose metabolism to remodel the tumor microenvironment. Front. Oncol. 2022, 12, 931104. [Google Scholar] [CrossRef]
- Divakaruni, A.S.; Hsieh, W.Y.; Minarrieta, L.; Duong, T.N.; Kim, K.K.O.; Desousa, B.R.; Andreyev, A.Y.; Bowman, C.E.; Caradonna, K.; Dranka, B.P.; et al. Etomoxir Inhibits Macrophage Polarization by Disrupting CoA Homeostasis. Cell Metab. 2018, 28, 490–503.e7. [Google Scholar] [CrossRef]
- Qiao, X.; Hu, Z.; Xiong, F.; Yang, Y.; Peng, C.; Wang, D.; Li, X. Lipid metabolism reprogramming in tumor-associated macrophages and implications for therapy. Lipids Health Dis. 2023, 22, 45. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, W.; Wang, W.; Ma, Y.; Wang, Y.; Drum, D.L.; Cai, J.; Blevins, H.; Lee, E.; Shah, S.; et al. CPT1A-mediated fatty acid oxidation confers cancer cell resistance to immune-mediated cytolytic killing. Proc. Natl. Acad. Sci. USA 2023, 120, e2302878120. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.-H.; Sun, I.-H.; Zhao, L.; Leone, R.D.; Sun, I.-M.; Xu, W.; Collins, S.L.; Tam, A.J.; Blosser, R.L.; Patel, C.H.; et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J. Clin. Investig. 2020, 130, 3865–3884. [Google Scholar] [CrossRef]
- Wang, J.-J.; Siu, M.K.-Y.; Jiang, Y.-X.; Leung, T.H.-Y.; Chan, D.W.; Wang, H.-G.; Ngan, H.Y.-S.; Chan, K.K.-L. A Combination of Glutaminase Inhibitor 968 and PD-L1 Blockade Boosts the Immune Response against Ovarian Cancer. Biomolecules 2021, 11, 1749. [Google Scholar] [CrossRef] [PubMed]
- Varghese, S.; Pramanik, S.; Williams, L.J.; Hodges, H.R.; Hudgens, C.W.; Fischer, G.M.; Luo, C.K.; Knighton, B.; Tan, L.; Lorenzi, P.L.; et al. The Glutaminase Inhibitor CB-839 (Telaglenastat) Enhances the Antimelanoma Activity of T-Cell-Mediated Immunotherapies. Mol. Cancer Ther. 2021, 20, 500–511. [Google Scholar] [CrossRef]
- Wang, J.; He, Y.; Hu, F.; Hu, C.; Sun, Y.; Yang, K.; Yang, S. Metabolic Reprogramming of Immune Cells in the Tumor Microenvironment. Int. J. Mol. Sci. 2024, 25, 12223. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, Y.; Song, H.; Li, Y.; Liu, Z.; Ye, Z.; Zhao, J.; Wu, Y.; Tang, J.; Yao, M. Metabolic reprogramming in tumor immune microenvironment: Impact on immune cell function and therapeutic implications. Cancer Lett. 2024, 597, 217076. [Google Scholar] [CrossRef]
- Yu, X.; Liu, R.; Gao, W.; Wang, X.; Zhang, Y. Single-cell omics traces the heterogeneity of prostate cancer cells and the tumor microenvironment. Cell Mol. Biol. Lett. 2023, 28, 38. [Google Scholar] [CrossRef]
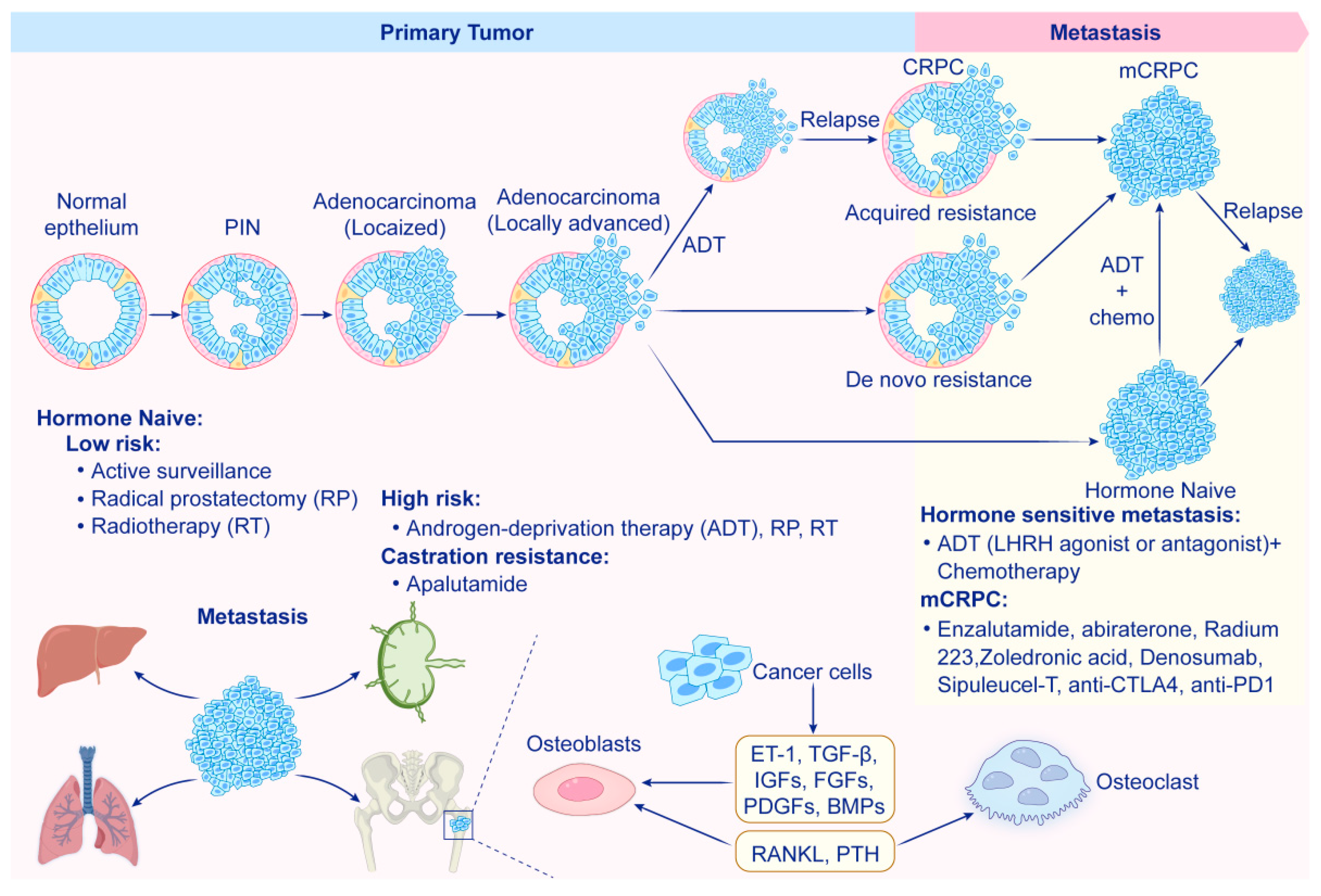
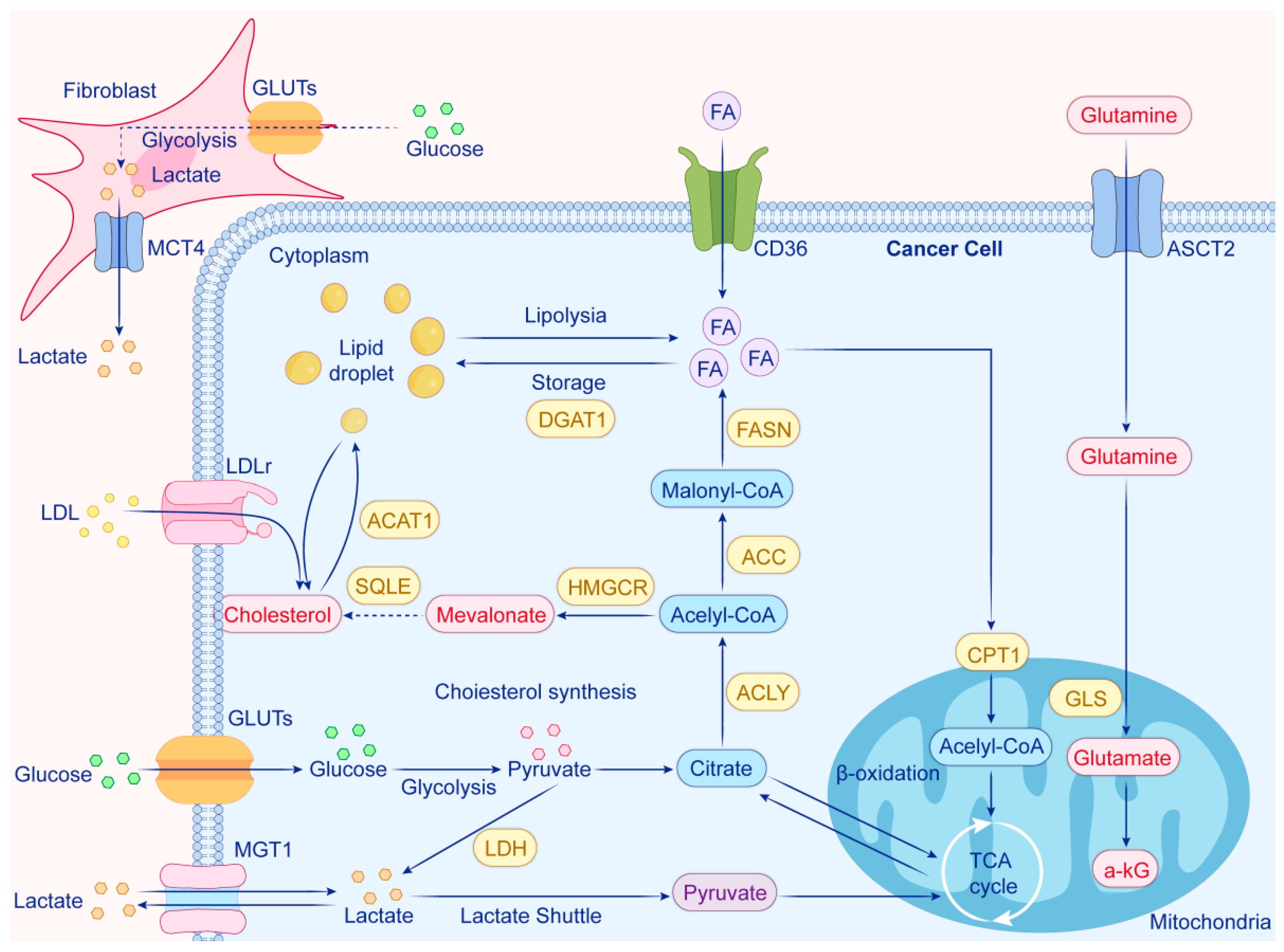
| Drug | Target/Mechanism | Condition | Status (2025) | Phase | NCT Identifier |
|---|---|---|---|---|---|
| Ipilimumab | CTLA4 | mCRPC | Completed | II | NCT02279862 |
| Ipilimumab | CTLA4 | mCRPC | Completed | III | NCT01057810 |
| Ipilimumab | CTLA4 | mCRPC | Completed | III | NCT00861614 |
| Atezolizumab | PD-L1 | mCRPC | Completed | I | NCT03024216 |
| Atezolizumab | PD-L1 | mCRPC | Active, not recruiting | III | NCT04446117 |
| Durvalumab | PD-L1 | mCRPC | Completed | II | NCT03204812 |
| Avelumab | PD-L1 | Neuroendocrine PCa | Completed | II | NCT03179410 |
| Pembrolizumab | PD1 | mCRPC | Recruiting | I/II | NCT02861573 |
| Pembrolizumab | PD1 | mCRPC | Completed | II | NCT03473925 |
| Pembrolizumab | PD1 | Hormone-sensitive PCa | Active, not recruiting | III | NCT04934722 |
| Pembrolizumab | PD1 | mCRPC | Active, not recruiting | III | NCT03834493 |
| Pembrolizumab | PD1 | mCRPC | Completed | III | NCT03834519 |
| Nivolumab | PD1 | mCRPC | Completed | II | NCT02601014 |
| Nivolumab | PD1 | mCRPC | Completed | III | NCT04100018 |
| Ipilimumab + Nivolumab | CTLA4&PD1 | mCRPC with CDK12 mutations | Completed | II | NCT03570619 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Chen, Y.; Wang, J. Role of Tumor Microenvironment in Prostate Cancer Immunometabolism. Biomolecules 2025, 15, 826. https://doi.org/10.3390/biom15060826
Wang Y, Chen Y, Wang J. Role of Tumor Microenvironment in Prostate Cancer Immunometabolism. Biomolecules. 2025; 15(6):826. https://doi.org/10.3390/biom15060826
Chicago/Turabian StyleWang, Yutao, Yiming Chen, and Jianfeng Wang. 2025. "Role of Tumor Microenvironment in Prostate Cancer Immunometabolism" Biomolecules 15, no. 6: 826. https://doi.org/10.3390/biom15060826
APA StyleWang, Y., Chen, Y., & Wang, J. (2025). Role of Tumor Microenvironment in Prostate Cancer Immunometabolism. Biomolecules, 15(6), 826. https://doi.org/10.3390/biom15060826