
Non-Coding RNAs in the Regulation of Doxorubicin-Induced Cardiotoxicity

Abstract
1. Introduction
2. Mechanisms of Dox-Induced Cardiotoxicity
2.1. DNA Damage-Induced Apoptosis
2.2. Mitochondrial Dysfunction and ROS Production-Related Apoptosis
2.3. Ferroptosis
2.4. Pyroptosis
2.5. Autophagy
3. Non-Coding RNAs involved in the DIC
3.1. MicroRNAs and DOX-Induced Cardiotoxicity
3.1.1. Introduction to MicroRNAs
3.1.2. MiRNAs Directly Modulate p53 Signaling-Related Genes to Affect Cardiomyocyte Apoptosis in DIC
3.1.3. MiRNAs Directly Regulate Mitochondria-Dependent Apoptotic Genes in DIC
3.1.4. MiRNAs and DOX-Induced Oxidative Stress in Apoptosis
3.1.5. MiRNAs and Ferroptosis
3.1.6. MiRNAs and Endoplasmic Reticulum (ER) Stress-Related Apoptosis Induced by DOX
3.1.7. MiRNAs and Other DOX-Induced Mechanisms
3.2. Long Non-Coding RNAs (LncRNAs) and DOX-Induced Cardiotoxicity
3.2.1. Introduction of lncRNAs
3.2.2. LncRNAs Regulate DIC
3.3. CircRNAs and DOX-Induced Cardiotoxicity
3.3.1. Introduction to circRNAs
3.3.2. CircRNAs in Regulating DOX-Induced Cardiotoxicity
4. Discussion
4.1. Non-Coding RNAs Bear Differential Regulatory Mechanisms in DIC
4.2. Non-Coding RNAs’ Impacts on DOX-Induced Antitumor Effect and Cardiotoxicity
4.3. Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Octavia, Y.; Tocchetti, C.G.; Gabrielson, K.L.; Janssens, S.; Crijns, H.J.; Moens, A.L. Doxorubicin-induced cardiomyopathy: From molecular mechanisms to therapeutic strategies. J. Mol. Cell Cardiol. 2012, 52, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.P.; Soonpaa, M.H.; Field, D.; Simpson, E.; Lohe, M.R.-v.d.; Lee, H.K.; Sridhar, A.; Ware, S.M.; Green, N.; Li, X.; et al. Cardiac Troponin I–Interacting Kinase Affects Cardiomyocyte S-Phase Activity but Not Cardiomyocyte Proliferation. Circulation 2023, 147, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-induced cardiotoxicity: An update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed. Pharmacother. 2021, 139, 111708. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef]
- Davies, K.J.; Doroshow, J.H. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J. Biol. Chem. 1986, 261, 3060–3067. [Google Scholar] [CrossRef]
- Green, P.S.; Leeuwenburgh, C. Mitochondrial dysfunction is an early indicator of doxorubicin-induced apoptosis. Biochim. Biophys. Acta 2002, 1588, 94–101. [Google Scholar] [CrossRef]
- Arai, M.; Tomaru, K.; Takizawa, T.; Sekiguchi, K.; Yokoyama, T.; Suzuki, T.; Nagai, R. Sarcoplasmic reticulum genes are selectively down-regulated in cardiomyopathy produced by doxorubicin in rabbits. J. Mol. Cell Cardiol. 1998, 30, 243–254. [Google Scholar] [CrossRef]
- Arai, M.; Yoguchi, A.; Takizawa, T.; Yokoyama, T.; Kanda, T.; Kurabayashi, M.; Nagai, R. Mechanism of doxorubicin-induced inhibition of sarcoplasmic reticulum Ca(2+)-ATPase gene transcription. Circ. Res. 2000, 86, 8–14. [Google Scholar] [CrossRef]
- Li, Y.; Yan, J.; Yang, P. The mechanism and therapeutic strategies in doxorubicin-induced cardiotoxicity: Role of programmed cell death. Cell Stress. Chaperones 2024, 29, 666–680. [Google Scholar] [CrossRef]
- Fa, H.G.; Chang, W.G.; Zhang, X.J.; Xiao, D.D.; Wang, J.X. Noncoding RNAs in doxorubicin-induced cardiotoxicity and their potential as biomarkers and therapeutic targets. Acta Pharmacol. Sin. 2021, 42, 499–507. [Google Scholar] [CrossRef]
- Vedam, K.; Nishijima, Y.; Druhan, L.J.; Khan, M.; Moldovan, N.I.; Zweier, J.L.; Ilangovan, G. Role of heat shock factor-1 activation in the doxorubicin-induced heart failure in mice. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1832–H1841. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, T.; Shi, W.; Fang, J.; Deng, H.; Cui, G. Potential targets for intervention against doxorubicin-induced cardiotoxicity based on genetic studies: A systematic review of the literature. J. Mol. Cell Cardiol. 2020, 138, 88–98. [Google Scholar] [CrossRef]
- Ghigo, A.; Li, M.; Hirsch, E. New signal transduction paradigms in anthracycline-induced cardiotoxicity. Biochim. Biophys. Acta 2016, 1863, 1916–1925. [Google Scholar] [CrossRef]
- Parker, M.A.; King, V.; Howard, K.P. Nuclear magnetic resonance study of doxorubicin binding to cardiolipin containing magnetically oriented phospholipid bilayers. Biochim. Biophys. Acta 2001, 1514, 206–216. [Google Scholar] [CrossRef]
- Aryal, B.; Rao, V.A. Deficiency in Cardiolipin Reduces Doxorubicin-Induced Oxidative Stress and Mitochondrial Damage in Human B-Lymphocytes. PLoS ONE 2016, 11, e0158376. [Google Scholar] [CrossRef]
- Schlame, M.; Rua, D.; Greenberg, M.L. The biosynthesis and functional role of cardiolipin. Prog. Lipid Res. 2000, 39, 257–288. [Google Scholar] [CrossRef]
- Wu, B.B.; Leung, K.T.; Poon, E.N. Mitochondrial-Targeted Therapy for Doxorubicin-Induced Cardiotoxicity. Int. J. Mol. Sci. 2022, 23, 1912. [Google Scholar] [CrossRef]
- Carvalho, R.A.; Sousa, R.P.; Cadete, V.J.; Lopaschuk, G.D.; Palmeira, C.M.; Bjork, J.A.; Wallace, K.B. Metabolic remodeling associated with subchronic doxorubicin cardiomyopathy. Toxicology 2010, 270, 92–98. [Google Scholar] [CrossRef]
- Aries, A.; Paradis, P.; Lefebvre, C.; Schwartz, R.J.; Nemer, M. Essential role of GATA-4 in cell survival and drug-induced cardiotoxicity. Proc. Natl. Acad. Sci. USA 2004, 101, 6975–6980. [Google Scholar] [CrossRef]
- Suzuki, Y.J. Stress-induced activation of GATA-4 in cardiac muscle cells. Free Radic. Biol. Med. 2003, 34, 1589–1598. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, Y.; Wang, G.; Ren, J. Molecular Mechanisms and Therapeutic Targeting of Ferroptosis in Doxorubicin-Induced Cardiotoxicity. JACC Basic. Transl. Sci. 2024, 9, 811–826. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Yan, H.F.; Zou, T.; Tuo, Q.Z.; Xu, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: Mechanisms and links with diseases. Signal Transduct. Target. Ther. 2021, 6, 49. [Google Scholar] [CrossRef]
- Yi, X.; Wang, Q.; Zhang, M.; Shu, Q.; Zhu, J. Ferroptosis: A novel therapeutic target of natural products against doxorubicin-induced cardiotoxicity. Biomed. Pharmacother. 2024, 178, 117217. [Google Scholar] [CrossRef]
- Conrad, M.; Proneth, B. Broken hearts: Iron overload, ferroptosis and cardiomyopathy. Cell Res. 2019, 29, 263–264. [Google Scholar] [CrossRef]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef]
- Wang, Y.; Yan, S.; Liu, X.; Deng, F.; Wang, P.; Yang, L.; Hu, L.; Huang, K.; He, J. PRMT4 promotes ferroptosis to aggravate doxorubicin-induced cardiomyopathy via inhibition of the Nrf2/GPX4 pathway. Cell Death Differ. 2022, 29, 1982–1995. [Google Scholar] [CrossRef]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef]
- Meng, L.; Lin, H.; Zhang, J.; Lin, N.; Sun, Z.; Gao, F.; Luo, H.; Ni, T.; Luo, W.; Chi, J.; et al. Doxorubicin induces cardiomyocyte pyroptosis via the TINCR-mediated posttranscriptional stabilization of NLR family pyrin domain containing 3. J. Mol. Cell Cardiol. 2019, 136, 15–26. [Google Scholar] [CrossRef]
- Christidi, E.; Brunham, L.R. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis. 2021, 12, 339. [Google Scholar] [CrossRef]
- Ye, B.; Shi, X.; Xu, J.; Dai, S.; Xu, J.; Fan, X.; Han, B.; Han, J. Gasdermin D mediates doxorubicin-induced cardiomyocyte pyroptosis and cardiotoxicity via directly binding to doxorubicin and changes in mitochondrial damage. Transl. Res. 2022, 248, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Chen, Y.; Fan, X.; Han, J.; Zhong, L.; Zhang, Y.; Liu, Q.; Lin, J.; Huang, W.; Su, L.; et al. Emodin attenuates cardiomyocyte pyroptosis in doxorubicin-induced cardiotoxicity by directly binding to GSDMD. Phytomedicine 2023, 121, 155105. [Google Scholar] [CrossRef]
- Li, Z.; Song, Y.; Liu, L.; Hou, N.; An, X.; Zhan, D.; Li, Y.; Zhou, L.; Li, P.; Yu, L.; et al. miR-199a impairs autophagy and induces cardiac hypertrophy through mTOR activation. Cell Death Differ. 2017, 24, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Lekli, I.; Haines, D.D.; Balla, G.; Tosaki, A. Autophagy: An adaptive physiological countermeasure to cellular senescence and ischaemia/reperfusion-associated cardiac arrhythmias. J. Cell Mol. Med. 2017, 21, 1058–1072. [Google Scholar] [CrossRef]
- Kobayashi, S.; Volden, P.; Timm, D.; Mao, K.; Xu, X.; Liang, Q. Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyocyte death. J. Biol. Chem. 2010, 285, 793–804. [Google Scholar] [CrossRef]
- Velez, J.M.; Miriyala, S.; Nithipongvanitch, R.; Noel, T.; Plabplueng, C.D.; Oberley, T.; Jungsuwadee, P.; Van Remmen, H.; Vore, M.; St Clair, D.K. p53 Regulates oxidative stress-mediated retrograde signaling: A novel mechanism for chemotherapy-induced cardiac injury. PLoS ONE 2011, 6, e18005. [Google Scholar] [CrossRef]
- Tran, S.; Fairlie, W.D.; Lee, E.F. BECLIN1: Protein Structure, Function and Regulation. Cells 2021, 10, 1522. [Google Scholar] [CrossRef]
- He, H.; Luo, Y.; Qiao, Y.; Zhang, Z.; Yin, D.; Yao, J.; You, J.; He, M. Curcumin attenuates doxorubicin-induced cardiotoxicity via suppressing oxidative stress and preventing mitochondrial dysfunction mediated by 14-3-3gamma. Food Funct. 2018, 9, 4404–4418. [Google Scholar] [CrossRef]
- Bartlett, J.J.; Trivedi, P.C.; Yeung, P.; Kienesberger, P.C.; Pulinilkunnil, T. Doxorubicin impairs cardiomyocyte viability by suppressing transcription factor EB expression and disrupting autophagy. Biochem. J. 2016, 473, 3769–3789. [Google Scholar] [CrossRef]
- Takla, M.; Keshri, S.; Rubinsztein, D.C. The post-translational regulation of transcription factor EB (TFEB) in health and disease. EMBO Rep. 2023, 24, e57574. [Google Scholar] [CrossRef]
- Zhu, W.; Soonpaa, M.H.; Chen, H.; Shen, W.; Payne, R.M.; Liechty, E.A.; Caldwell, R.L.; Shou, W.; Field, L.J. Acute doxorubicin cardiotoxicity is associated with p53-induced inhibition of the mammalian target of rapamycin pathway. Circulation 2009, 119, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.B.; Wu, X.Y.; Gu, J.H.; Guo, Q.T.; Shen, W.X.; Lu, P.H. Activation of AMP-activated protein kinase contributes to doxorubicin-induced cell death and apoptosis in cultured myocardial H9c2 cells. Cell Biochem. Biophys. 2011, 60, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Koleini, N.; Kardami, E. Autophagy and mitophagy in the context of doxorubicin-induced cardiotoxicity. Oncotarget 2017, 8, 46663–46680. [Google Scholar] [CrossRef]
- Renu, K.; Abilash, V.G.; Tirupathi Pichiah, P.B.; Arunachalam, S. Molecular mechanism of doxorubicin-induced cardiomyopathy—An update. Eur. J. Pharmacol. 2018, 818, 241–253. [Google Scholar] [CrossRef]
- Ambros, V. microRNAs: Tiny regulators with great potential. Cell 2001, 107, 823–826. [Google Scholar] [CrossRef]
- Hutvagner, G.; Zamore, P.D. A microRNA in a multiple-turnover RNAi enzyme complex. Science 2002, 297, 2056–2060. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Wang, J.X.; Zhang, X.J.; Feng, C.; Sun, T.; Wang, K.; Wang, Y.; Zhou, L.Y.; Li, P.F. MicroRNA-532-3p regulates mitochondrial fission through targeting apoptosis repressor with caspase recruitment domain in doxorubicin cardiotoxicity. Cell Death Dis. 2015, 6, e1677. [Google Scholar] [CrossRef]
- Zhao, Y.; Ransom, J.F.; Li, A.; Vedantham, V.; von Drehle, M.; Muth, A.N.; Tsuchihashi, T.; McManus, M.T.; Schwartz, R.J.; Srivastava, D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell 2007, 129, 303–317. [Google Scholar] [CrossRef]
- van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef]
- Elia, L.; Contu, R.; Quintavalle, M.; Varrone, F.; Chimenti, C.; Russo, M.A.; Cimino, V.; De Marinis, L.; Frustaci, A.; Catalucci, D.; et al. Reciprocal regulation of microRNA-1 and insulin-like growth factor-1 signal transduction cascade in cardiac and skeletal muscle in physiological and pathological conditions. Circulation 2009, 120, 2377–2385. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Qi, Y.; Xu, L.; Tao, X.; Han, X.; Yin, L.; Peng, J. MicroRNA-140-5p aggravates doxorubicin-induced cardiotoxicity by promoting myocardial oxidative stress via targeting Nrf2 and Sirt2. Redox Biol. 2018, 15, 284–296. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Q.; Wei, C.; Zhao, L.; Guo, X.; Cui, X.; Shao, L.; Long, J.; Gu, J.; Zhao, M. MiR-378 modulates energy imbalance and apoptosis of mitochondria induced by doxorubicin. Am. J. Transl. Res. 2018, 10, 3600–3609. [Google Scholar]
- Wan, G.X.; Cheng, L.; Qin, H.L.; Zhang, Y.Z.; Wang, L.Y.; Zhang, Y.G. MiR-15b-5p is Involved in Doxorubicin-Induced Cardiotoxicity via Inhibiting Bmpr1a Signal in H9c2 Cardiomyocyte. Cardiovasc. Toxicol. 2019, 19, 264–275. [Google Scholar] [CrossRef]
- Tong, Z.; Jiang, B.; Wu, Y.; Liu, Y.; Li, Y.; Gao, M.; Jiang, Y.; Lv, Q.; Xiao, X. MiR-21 Protected Cardiomyocytes against Doxorubicin-Induced Apoptosis by Targeting BTG2. Int. J. Mol. Sci. 2015, 16, 14511–14525. [Google Scholar] [CrossRef]
- Xu, C.; Liu, C.H.; Zhang, D.L. MicroRNA-22 inhibition prevents doxorubicin-induced cardiotoxicity via upregulating SIRT1. Biochem. Biophys. Res. Commun. 2020, 521, 485–491. [Google Scholar] [CrossRef]
- Li, J.; Aung, L.H.; Long, B.; Qin, D.; An, S.; Li, P. miR-23a binds to p53 and enhances its association with miR-128 promoter. Sci. Rep. 2015, 5, 16422. [Google Scholar] [CrossRef]
- Du, J.; Hang, P.; Pan, Y.; Feng, B.; Zheng, Y.; Chen, T.; Zhao, L.; Du, Z. Inhibition of miR-23a attenuates doxorubicin-induced mitochondria-dependent cardiomyocyte apoptosis by targeting the PGC-1alpha/Drp1 pathway. Toxicol. Appl. Pharmacol. 2019, 369, 73–81. [Google Scholar] [CrossRef]
- Jing, X.; Yang, J.; Jiang, L.; Chen, J.; Wang, H. MicroRNA-29b Regulates the Mitochondria-Dependent Apoptotic Pathway by Targeting Bax in Doxorubicin Cardiotoxicity. Cell Physiol. Biochem. 2018, 48, 692–704. [Google Scholar] [CrossRef]
- Roca-Alonso, L.; Castellano, L.; Mills, A.; Dabrowska, A.F.; Sikkel, M.B.; Pellegrino, L.; Jacob, J.; Frampton, A.E.; Krell, J.; Coombes, R.C.; et al. Myocardial MiR-30 downregulation triggered by doxorubicin drives alterations in beta-adrenergic signaling and enhances apoptosis. Cell Death Dis. 2015, 6, e1754. [Google Scholar] [CrossRef]
- Piegari, E.; Russo, R.; Cappetta, D.; Esposito, G.; Urbanek, K.; Dell’Aversana, C.; Altucci, L.; Berrino, L.; Rossi, F.; De Angelis, A. MicroRNA-34a regulates doxorubicin-induced cardiotoxicity in rat. Oncotarget 2016, 7, 62312–62326. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.N.; Fu, Y.H.; Hu, Z.Q.; Li, W.Y.; Tang, C.M.; Fei, H.W.; Yang, H.; Lin, Q.X.; Gou, D.M.; Wu, S.L.; et al. Activation of miR-34a-5p/Sirt1/p66shc pathway contributes to doxorubicin-induced cardiotoxicity. Sci. Rep. 2017, 7, 11879. [Google Scholar] [CrossRef]
- Adlakha, Y.K.; Saini, N. miR-128 exerts pro-apoptotic effect in a p53 transcription-dependent and -independent manner via PUMA-Bak axis. Cell Death Dis. 2013, 4, e542. [Google Scholar] [CrossRef]
- Zhang, W.B.; Zheng, Y.F.; Wu, Y.G. Inhibition of miR-128-3p Attenuated Doxorubicin-Triggered Acute Cardiac Injury in Mice by the Regulation of PPAR-gamma. PPAR Res. 2021, 2021, 7595374. [Google Scholar] [CrossRef]
- Pakravan, G.; Foroughmand, A.M.; Peymani, M.; Ghaedi, K.; Hashemi, M.S.; Hajjari, M.; Nasr-Esfahani, M.H. Downregulation of miR-130a, antagonized doxorubicin-induced cardiotoxicity via increasing the PPARgamma expression in mESCs-derived cardiac cells. Cell Death Dis. 2018, 9, 758. [Google Scholar] [CrossRef]
- Horie, T.; Ono, K.; Nishi, H.; Nagao, K.; Kinoshita, M.; Watanabe, S.; Kuwabara, Y.; Nakashima, Y.; Takanabe-Mori, R.; Nishi, E.; et al. Acute doxorubicin cardiotoxicity is associated with miR-146a-induced inhibition of the neuregulin-ErbB pathway. Cardiovasc. Res. 2010, 87, 656–664. [Google Scholar] [CrossRef]
- Pan, J.A.; Tang, Y.; Yu, J.Y.; Zhang, H.; Zhang, J.F.; Wang, C.Q.; Gu, J. miR-146a attenuates apoptosis and modulates autophagy by targeting TAF9b/P53 pathway in doxorubicin-induced cardiotoxicity. Cell Death Dis. 2019, 10, 668. [Google Scholar] [CrossRef]
- Lin, Z.; Wang, J. Taxifolin protects against doxorubicin-induced cardiotoxicity and ferroptosis by adjusting microRNA-200a-mediated Nrf2 signaling pathway. Heliyon 2023, 9, e22011. [Google Scholar] [CrossRef]
- Hu, X.; Liu, H.; Wang, Z.; Hu, Z.; Li, L. miR-200a Attenuated Doxorubicin-Induced Cardiotoxicity through Upregulation of Nrf2 in Mice. Oxid. Med. Cell Longev. 2019, 2019, 1512326. [Google Scholar] [CrossRef]
- Tony, H.; Yu, K.; Qiutang, Z. MicroRNA-208a Silencing Attenuates Doxorubicin Induced Myocyte Apoptosis and Cardiac Dysfunction. Oxid. Med. Cell Longev. 2015, 2015, 597032. [Google Scholar] [CrossRef]
- Yin, Z.; Zhao, Y.; Li, H.; Yan, M.; Zhou, L.; Chen, C.; Wang, D.W. miR-320a mediates doxorubicin-induced cardiotoxicity by targeting VEGF signal pathway. Aging 2016, 8, 192–207. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wan, W.; Chen, T.; Tong, S.; Jiang, X.; Liu, W. miR-451 Silencing Inhibited Doxorubicin Exposure-Induced Cardiotoxicity in Mice. Biomed. Res. Int. 2019, 2019, 1528278. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Jiao, J.Q.; Li, Q.; Long, B.; Wang, K.; Liu, J.P.; Li, Y.R.; Li, P.F. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat. Med. 2011, 17, 71–78. [Google Scholar] [CrossRef]
- Wan, Q.; Xu, T.; Ding, W.; Zhang, X.; Ji, X.; Yu, T.; Yu, W.; Lin, Z.; Wang, J. miR-499-5p Attenuates Mitochondrial Fission and Cell Apoptosis via p21 in Doxorubicin Cardiotoxicity. Front. Genet. 2018, 9, 734. [Google Scholar] [CrossRef]
- Fu, J.; Peng, C.; Wang, W.; Jin, H.; Tang, Q.; Wei, X. Let-7 g is involved in doxorubicin induced myocardial injury. Env. Toxicol. Pharmacol. 2012, 33, 312–317. [Google Scholar] [CrossRef]
- Cai, H.; Tian, P.; Ju, J.; Wang, T.; Chen, X.; Wang, K.; Wang, F.; Yu, X.; Wang, S.; Wang, Y.; et al. Long noncoding RNA NONMMUT015745 inhibits doxorubicin-mediated cardiomyocyte apoptosis by regulating Rab2A-p53 axis. Cell Death Discov. 2022, 8, 364. [Google Scholar] [CrossRef]
- Guan, X.; Wang, Y.; Li, W.; Liu, X.; Jiang, J.; Bian, W.; Xu, C.; Sun, Y.; Zhang, C. The effects and mechanism of LncRNA NORAD on doxorubicin-induced cardiotoxicity. Toxicology 2023, 494, 153587. [Google Scholar] [CrossRef]
- Li, J.; Li, L.; Li, X.; Wu, S. Long noncoding RNA LINC00339 aggravates doxorubicin-induced cardiomyocyte apoptosis by targeting MiR-484. Biochem. Biophys. Res. Commun. 2018, 503, 3038–3043. [Google Scholar] [CrossRef]
- Zhuang, S.; Ma, Y.; Zeng, Y.; Lu, C.; Yang, F.; Jiang, N.; Ge, J.; Ju, H.; Zhong, C.; Wang, J.; et al. METTL14 promotes doxorubicin-induced cardiomyocyte ferroptosis by regulating the KCNQ1OT1-miR-7-5p-TFRC axis. Cell Biol. Toxicol. 2023, 39, 1015–1035. [Google Scholar] [CrossRef]
- Lu, Q.; Huo, J.; Liu, P.; Bai, L.; Ma, A. lncRNA HOXB-AS3 protects doxorubicin-induced cardiotoxicity by targeting miRNA-875-3p. Exp. Ther. Med. 2020, 19, 1388–1392. [Google Scholar] [CrossRef]
- Aung, L.H.H.; Chen, X.; Cueva Jumbo, J.C.; Li, Z.; Wang, S.Y.; Zhao, C.; Liu, Z.; Wang, Y.; Li, P. Cardiomyocyte mitochondrial dynamic-related lncRNA 1 (CMDL-1) may serve as a potential therapeutic target in doxorubicin cardiotoxicity. Mol. Ther. Nucleic Acids 2021, 25, 638–651. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Xia, W.; Chen, D.; Ye, Y.; Hu, T.; Li, S.; Hou, M. Exosomal LncRNA-NEAT1 derived from MIF-treated mesenchymal stem cells protected against doxorubicin-induced cardiac senescence through sponging miR-221-3p. J. Nanobiotechnology 2020, 18, 157. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lin, X.; Li, J.; Zeng, G.; Xu, T. Long Noncoding RNA SOX2-OT Aggravates Doxorubicin-Induced Apoptosis of Cardiomyocyte by Targeting miR-942-5p/DP5. Drug Des. Devel Ther. 2021, 15, 481–492. [Google Scholar] [CrossRef]
- Tian, C.; Yang, Y.; Li, B.; Liu, M.; He, X.; Zhao, L.; Song, X.; Yu, T.; Chu, X.M. Doxorubicin-Induced Cardiotoxicity May Be Alleviated by Bone Marrow Mesenchymal Stem Cell-Derived Exosomal lncRNA via Inhibiting Inflammation. J. Inflamm. Res. 2022, 15, 4467–4486. [Google Scholar] [CrossRef]
- Xia, W.; Chen, H.; Xie, C.; Hou, M. Long-noncoding RNA MALAT1 sponges microRNA-92a-3p to inhibit doxorubicin-induced cardiac senescence by targeting ATG4a. Aging 2020, 12, 8241–8260. [Google Scholar] [CrossRef]
- Li, J.; Zhou, L.; Jiang, Y.; Gao, H.; Maierhaba, T.; Gong, H. Long noncoding RNA RMRP ameliorates doxorubicin-induced apoptosis by interacting with PFN1 in a P53-Dependent manner. Mol. Cell Probes 2023, 72, 101937. [Google Scholar] [CrossRef]
- Xie, Z.; Xia, W.; Hou, M. Long intergenic non-coding RNA-p21 mediates cardiac senescence via the Wnt/beta-catenin signaling pathway in doxorubicin-induced cardiotoxicity. Mol. Med. Rep. 2018, 17, 2695–2704. [Google Scholar] [CrossRef]
- Chen, L.; Yan, K.P.; Liu, X.C.; Wang, W.; Li, C.; Li, M.; Qiu, C.G. Valsartan regulates TGF-beta/Smads and TGF-beta/p38 pathways through lncRNA CHRF to improve doxorubicin-induced heart failure. Arch. Pharm. Res. 2018, 41, 101–109. [Google Scholar] [CrossRef]
- Zhang, S.; Yuan, Y.; Zhang, Z.; Guo, J.; Li, J.; Zhao, K.; Qin, Y.; Qiu, C. LncRNA FOXC2-AS1 protects cardiomyocytes from doxorubicin-induced cardiotoxicity through activation of WNT1-inducible signaling pathway protein-1. Biosci. Biotechnol. Biochem. 2019, 83, 653–658. [Google Scholar] [CrossRef]
- Li, H.Q.; Wu, Y.B.; Yin, C.S.; Chen, L.; Zhang, Q.; Hu, L.Q. Obestatin attenuated doxorubicin-induced cardiomyopathy via enhancing long noncoding Mhrt RNA expression. Biomed. Pharmacother. 2016, 81, 474–481. [Google Scholar] [CrossRef]
- Lu, D.; Chatterjee, S.; Xiao, K.; Riedel, I.; Huang, C.K.; Costa, A.; Cushman, S.; Neufeldt, D.; Rode, L.; Schmidt, A.; et al. A circular RNA derived from the insulin receptor locus protects against doxorubicin-induced cardiotoxicity. Eur. Heart J. 2022, 43, 4496–4511. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Wang, Y.; Wang, Y.; Dai, X.; Zhou, T.; Chen, J.; Tao, B.; Zhang, J.; Cao, F. The Tumor-Suppressive Human Circular RNA CircITCH Sponges miR-330-5p to Ameliorate Doxorubicin-Induced Cardiotoxicity Through Upregulating SIRT6, Survivin, and SERCA2a. Circ. Res. 2020, 127, e108–e125. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Du, W.W.; Wu, N.; Li, F.; Li, X.; Xie, Y.; Wang, S.; Yang, B.B. The circular RNA circNlgnmediates doxorubicin-inducedcardiac remodeling and fibrosis. Mol. Ther. Nucleic Acids 2022, 28, 175–189. [Google Scholar] [CrossRef]
- Wang, X.; Cheng, Z.; Xu, J.; Feng, M.; Zhang, H.; Zhang, L.; Qian, L. Circular RNA Arhgap12 modulates doxorubicin-induced cardiotoxicity by sponging miR-135a-5p. Life Sci. 2021, 265, 118788. [Google Scholar] [CrossRef]
- Ji, X.; Ding, W.; Xu, T.; Zheng, X.; Zhang, J.; Liu, M.; Liu, G.; Wang, J. MicroRNA-31-5p attenuates doxorubicin-induced cardiotoxicity via quaking and circular RNA Pan3. J. Mol. Cell Cardiol. 2020, 140, 56–67. [Google Scholar] [CrossRef]
- Zeng, Y.; Du, W.W.; Wu, Y.; Yang, Z.; Awan, F.M.; Li, X.; Yang, W.; Zhang, C.; Yang, Q.; Yee, A.; et al. A Circular RNA Binds To and Activates AKT Phosphorylation and Nuclear Localization Reducing Apoptosis and Enhancing Cardiac Repair. Theranostics 2017, 7, 3842–3855. [Google Scholar] [CrossRef]
- Han, J.; Zhang, L.; Hu, L.; Yu, H.; Xu, F.; Yang, B.; Zhang, R.; Zhang, Y.; An, Y. Circular RNA-Expression Profiling Reveals a Potential Role of Hsa_circ_0097435 in Heart Failure via Sponging Multiple MicroRNAs. Front. Genet. 2020, 11, 212. [Google Scholar] [CrossRef]
- Li, C.; Zhang, L.; Bu, X.; Wang, J.; Li, L.; Yang, Z. Circ-LTBP1 is involved in doxorubicin-induced intracellular toxicity in cardiomyocytes via miR-107/ADCY1 signal. Mol. Cell Biochem. 2022, 477, 1127–1138. [Google Scholar] [CrossRef]
- Li, B.; Cai, X.; Wang, Y.; Zhu, H.; Zhang, P.; Jiang, P.; Yang, X.; Sun, J.; Hong, L.; Shao, L. Circ-SKA3 Enhances Doxorubicin Toxicity in AC16 Cells Through miR-1303/TLR4 Axis. Int. Heart J. 2021, 62, 1112–1123. [Google Scholar] [CrossRef]
- Arola, O.J.; Saraste, A.; Pulkki, K.; Kallajoki, M.; Parvinen, M.; Voipio-Pulkki, L.M. Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Res. 2000, 60, 1789–1792. [Google Scholar]
- Delpy, E.; Hatem, S.N.; Andrieu, N.; de Vaumas, C.; Henaff, M.; Rucker-Martin, C.; Jaffrezou, J.P.; Laurent, G.; Levade, T.; Mercadier, J.J. Doxorubicin induces slow ceramide accumulation and late apoptosis in cultured adult rat ventricular myocytes. Cardiovasc. Res. 1999, 43, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Kawano, I.; Adamcova, M. MicroRNAs in doxorubicin-induced cardiotoxicity: The DNA damage response. Front. Pharmacol. 2022, 13, 1055911. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, A.; Piegari, E.; Cappetta, D.; Russo, R.; Esposito, G.; Ciuffreda, L.P.; Ferraiolo, F.A.; Frati, C.; Fagnoni, F.; Berrino, L.; et al. SIRT1 activation rescues doxorubicin-induced loss of functional competence of human cardiac progenitor cells. Int. J. Cardiol. 2015, 189, 30–44. [Google Scholar] [CrossRef]
- Ren, Y.; Sun, C.; Sun, Y.; Tan, H.; Wu, Y.; Cui, B.; Wu, Z. PPAR gamma protects cardiomyocytes against oxidative stress and apoptosis via Bcl-2 upregulation. Vasc. Pharmacol. 2009, 51, 169–174. [Google Scholar] [CrossRef]
- Goormaghtigh, E.; Chatelain, P.; Caspers, J.; Ruysschaert, J.M. Evidence of a complex between adriamycin derivatives and cardiolipin: Possible role in cardiotoxicity. Biochem. Pharmacol. 1980, 29, 3003–3010. [Google Scholar] [CrossRef]
- Ashley, N.; Poulton, J. Mitochondrial DNA is a direct target of anti-cancer anthracycline drugs. Biochem. Biophys. Res. Commun. 2009, 378, 450–455. [Google Scholar] [CrossRef]
- Childs, A.C.; Phaneuf, S.L.; Dirks, A.J.; Phillips, T.; Leeuwenburgh, C. Doxorubicin treatment in vivo causes cytochrome C release and cardiomyocyte apoptosis, as well as increased mitochondrial efficiency, superoxide dismutase activity, and Bcl-2:Bax ratio. Cancer Res. 2002, 62, 4592–4598. [Google Scholar]
- Sprenger, H.G.; Langer, T. The Good and the Bad of Mitochondrial Breakups. Trends Cell Biol. 2019, 29, 888–900. [Google Scholar] [CrossRef]
- Quiles, J.M.; Gustafsson, A.B. The role of mitochondrial fission in cardiovascular health and disease. Nat. Rev. Cardiol. 2022, 19, 723–736. [Google Scholar] [CrossRef]
- Liang, X.; Wang, S.; Wang, L.; Ceylan, A.F.; Ren, J.; Zhang, Y. Mitophagy inhibitor liensinine suppresses doxorubicin-induced cardiotoxicity through inhibition of Drp1-mediated maladaptive mitochondrial fission. Pharmacol. Res. 2020, 157, 104846. [Google Scholar] [CrossRef]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Rajesh, M.; Batkai, S.; Kashiwaya, Y.; Hasko, G.; Liaudet, L.; Szabo, C.; Pacher, P. Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1466–H1483. [Google Scholar] [CrossRef]
- Lebrecht, D.; Setzer, B.; Ketelsen, U.P.; Haberstroh, J.; Walker, U.A. Time-dependent and tissue-specific accumulation of mtDNA and respiratory chain defects in chronic doxorubicin cardiomyopathy. Circulation 2003, 108, 2423–2429. [Google Scholar] [CrossRef]
- Wallace, K.B.; Sardao, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef]
- Regula, K.M.; Ens, K.; Kirshenbaum, L.A. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ. Res. 2002, 91, 226–231. [Google Scholar] [CrossRef]
- Siveski-Iliskovic, N.; Kaul, N.; Singal, P.K. Probucol promotes endogenous antioxidants and provides protection against adriamycin-induced cardiomyopathy in rats. Circulation 1994, 89, 2829–2835. [Google Scholar] [CrossRef]
- Yen, H.C.; Oberley, T.D.; Vichitbandha, S.; Ho, Y.S.; St Clair, D.K. The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. J. Clin. Investig. 1996, 98, 1253–1260. [Google Scholar] [CrossRef]
- Deniaud, A.; Sharaf el dein, O.; Maillier, E.; Poncet, D.; Kroemer, G.; Lemaire, C.; Brenner, C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 2008, 27, 285–299. [Google Scholar] [CrossRef]
- Dodd, D.A.; Atkinson, J.B.; Olson, R.D.; Buck, S.; Cusack, B.J.; Fleischer, S.; Boucek, R.J., Jr. Doxorubicin cardiomyopathy is associated with a decrease in calcium release channel of the sarcoplasmic reticulum in a chronic rabbit model. J. Clin. Investig. 1993, 91, 1697–1705. [Google Scholar] [CrossRef]
- Gupta, S.K.; Garg, A.; Avramopoulos, P.; Engelhardt, S.; Streckfuss-Bomeke, K.; Batkai, S.; Thum, T. miR-212/132 Cluster Modulation Prevents Doxorubicin-Mediated Atrophy and Cardiotoxicity. Mol. Ther. 2019, 27, 17–28. [Google Scholar] [CrossRef]
- Luu, A.Z.; Chowdhury, B.; Al-Omran, M.; Teoh, H.; Hess, D.A.; Verma, S. Role of Endothelium in Doxorubicin-Induced Cardiomyopathy. JACC Basic. Transl. Sci. 2018, 3, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Octavia, Y.; Kararigas, G.; de Boer, M.; Chrifi, I.; Kietadisorn, R.; Swinnen, M.; Duimel, H.; Verheyen, F.K.; Brandt, M.M.; Fliegner, D.; et al. Folic acid reduces doxorubicin-induced cardiomyopathy by modulating endothelial nitric oxide synthase. J. Cell Mol. Med. 2017, 21, 3277–3287. [Google Scholar] [CrossRef]
- Fernandez-Fernandez, A.; Carvajal, D.A.; Lei, T.; McGoron, A.J. Chemotherapy-induced changes in cardiac capillary permeability measured by fluorescent multiple indicator dilution. Ann. Biomed. Eng. 2014, 42, 2405–2415. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, E.L.; Sidaway, J.E.; Cross, M.J. Cardiotoxic drugs Herceptin and doxorubicin inhibit cardiac microvascular endothelial cell barrier formation resulting in increased drug permeability. Biol. Open 2016, 5, 1362–1370. [Google Scholar] [CrossRef]
- Mattick, J.S.; Amaral, P.P.; Carninci, P.; Carpenter, S.; Chang, H.Y.; Chen, L.L.; Chen, R.; Dean, C.; Dinger, M.E.; Fitzgerald, K.A.; et al. Long non-coding RNAs: Definitions, functions, challenges and recommendations. Nat. Rev. Mol. Cell Biol. 2023, 24, 430–447. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, W.; Zhu, W.; Dong, J.; Cheng, Y.; Yin, Z.; Shen, F. Mechanisms and Functions of Long Non-Coding RNAs at Multiple Regulatory Levels. Int. J. Mol. Sci. 2019, 20, 5573. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, H.; Zhang, Y.; Li, H.; An, M.; Zhao, B.; Ding, H.; Xu, J.; Shang, H.; Han, X. Integrated analysis of lncRNA, miRNA and mRNA profiles reveals potential lncRNA functions during early HIV infection. J. Transl. Med. 2021, 19, 135. [Google Scholar] [CrossRef]
- Wu, X.S.; Wang, F.; Li, H.F.; Hu, Y.P.; Jiang, L.; Zhang, F.; Li, M.L.; Wang, X.A.; Jin, Y.P.; Zhang, Y.J.; et al. Author Correction: LncRNA-PAGBC acts as a microRNA sponge and promotes gallbladder tumorigenesis. EMBO Rep. 2024, 25, 5216–5219. [Google Scholar] [CrossRef]
- Anderson, K.M.; Anderson, D.M. LncRNAs at the heart of development and disease. Mamm. Genome 2022, 33, 354–365. [Google Scholar] [CrossRef]
- Thum, T.; Condorelli, G. Long noncoding RNAs and microRNAs in cardiovascular pathophysiology. Circ. Res. 2015, 116, 751–762. [Google Scholar] [CrossRef]
- Schneider, T.; Bindereif, A. Circular RNAs: Coding or noncoding? Cell Res. 2017, 27, 724–725. [Google Scholar] [CrossRef] [PubMed]
- Xing, X.; Tan, Z.; Zhi, X.; Sun, H.; Yang, J.; Li, L.; Liu, Y.; Wang, L.; Dong, Z.; Guo, H. Integrating analysis of circular RNA and mRNA expression profiles in doxorubicin induced cardiotoxicity mice. J. Appl. Toxicol. 2022, 42, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Liu, Z.; Han, H.; Wang, B.; Li, W.; Mao, C.; Liu, S. Peptide SMIM30 promotes HCC development by inducing SRC/YES1 membrane anchoring and MAPK pathway activation. J. Hepatol. 2020, 73, 1155–1169. [Google Scholar] [CrossRef]
- Lu, P.; Fan, J.; Li, B.; Wang, X.; Song, M. A novel protein encoded by circLARP1B promotes the proliferation and migration of vascular smooth muscle cells by suppressing cAMP signaling. Atherosclerosis 2024, 395, 117575. [Google Scholar] [CrossRef]
- Surina, S.; Fontanella, R.A.; Scisciola, L.; Marfella, R.; Paolisso, G.; Barbieri, M. miR-21 in Human Cardiomyopathies. Front. Cardiovasc. Med. 2021, 8, 767064. [Google Scholar] [CrossRef]
- Han, L.; Huang, D.; Wu, S.; Liu, S.; Wang, C.; Sheng, Y.; Lu, X.; Broxmeyer, H.E.; Wan, J.; Yang, L. Lipid droplet-associated lncRNA LIPTER preserves cardiac lipid metabolism. Nat. Cell Biol. 2023, 25, 1033–1046. [Google Scholar] [CrossRef]
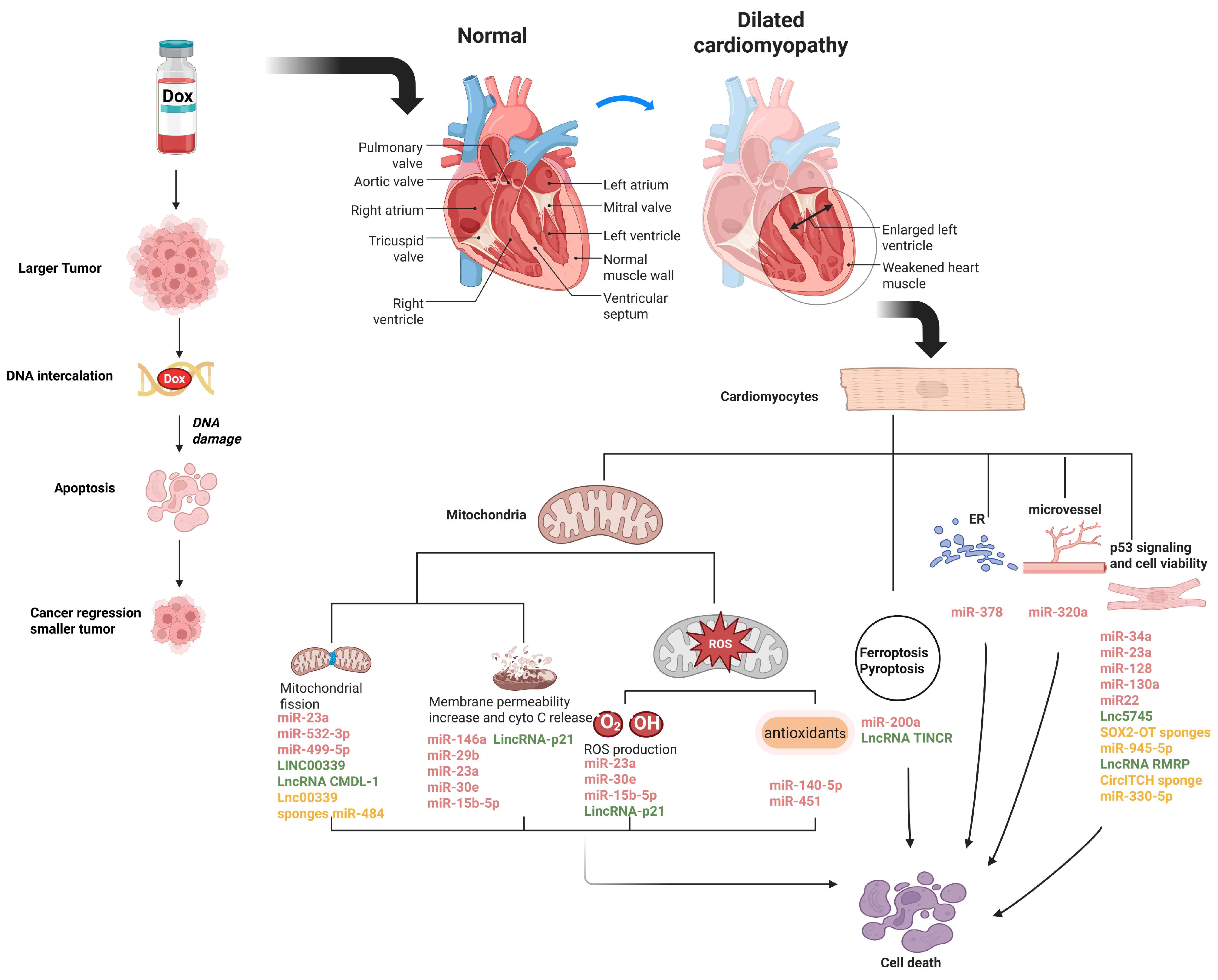


{kind=link}
{kind=link}
| NcRNA | Regulation | Targets | Biological Effects | Cell Type | Reference |
|---|---|---|---|---|---|
| miRNAs | |||||
| miR-15b-5p | Up | Bmpr1a | Mitochondrial dysfunction, ROS and apoptosis | Cardiomyocyte | [54] |
| miR-21-5p | Up | BTG2 | Apoptosis | Cardiomyocyte | [55] |
| miR-22 | Up | SIRT1 | apoptosis | Cardiomyocyte | [56] |
| miR-23a-3p | Up | P53 and PGC-1α, Drp1 | Mitochondrial dysfunction, ROS and apoptosis; fission | Cardiomyocyte | [57,58] |
| miR-29b | Down | Bax | Mitochondrial dysfunction and apoptosis | Cardiomyocyte | [59] |
| miR-30e | Down | β1AR, β2AR, Gia-2 and Bnip3L | ROS and apoptosis | Cardiomyocyte | [60] |
| miR-34a-5p | Up | SIRT1/P66shc | Apoptosis | Cardiomyocyte | [61,62] |
| miR-128 | Up | SIRT1, PPARγ | Apoptosis | Cardiomyocyte | [63,64] |
| miR-130a | Up | PPARγ | Apoptosis | mESC-derived cardiac cells | [65] |
| miR-140-5p | Up | Nrf2 and SIRT2 | Increase of ROS | Cardiomyocyte | [52] |
| miR-146a | Up/Down | ErbB4, TAF9b/P53 | Mitochondrial dysfunction and apoptosis | Cardiomyocyte | [66,67] |
| miR-200a | Down | Nrf2 | Ferroptosis | Cardiomyocyte | [68,69] |
| miR-208a-3p | Up | GATA4 | Apoptosis | Cardiomyocyte | [70] |
| miR-212-3p/ miR-132-3p | Down | Fitm2 | Apoptosis and atrophy | hiPSC-derived cardiomyocyte | [42] |
| miR-320a-3p | Up | VEGF-A | Reduced cardiac microvessel density and apoptosis | HUVEC | [71] |
| miR-378a-5p | Down | LDHA and PPIA | Energy metabolism disturbance and ER stress | Cardiomyocyte | [53] |
| miR-451-5p | Up | Cab39 | ROS and apoptosis | Cardiomyocyte | [72] |
| miR-499-5p | Down | p21 | Mitochondrial fission and apoptosis | Cardiomyocyte | [73,74] |
| miR-532-3p | Up | ARC | Mitochondrial fission and apoptosis | Cardiomyocyte | [48] |
| Let-7g-5p | Down | - | - | Cardiomyocyte | [75] |
| LncRNAs | |||||
| Lnc5745 | Down | Rab2A | p53-related apoptosis | Cardiomyocyte | [76] |
| LncRNA NORAD | Down | Apaf-1, p53 and fission | fission, ROS and apoptosis | Cardiomyocyte | [77] |
| LINC00339 | Up | miR-484 | Apoptosis | Cardiomyocyte | [78] |
| LncRNA KCNQ1OT1 | Modified | miR-7-5p | Ferroptosis | Cardiomyocyte | [79] |
| LncRNA HOXB-AS3 | Down | miRNA-875-3p | Protecting cardiomyocytes | Cardiomyocyte | [80] |
| LncRNA CDML-1 | Down | Drp1 | Apoptosis | Cardiomyocyte | [81] |
| LncRNA NEAT1 | - | miR-221-3p | Cardiac senescence | Cardiomyocyte | [82] |
| LncRNA SOX2-OT | Up | miR-942-5p/DP5 | Apoptosis | Cardiomyocyte | [83] |
| ElncRNA MSTRG 58791.2 | - | - | Inflammation | Cardiomyocyte | [84] |
| MALAT1 | - | miR-92a-3p/ATG4a | Cardiac senescence | Cardiomyocyte | [85] |
| LncRNA RMRP | Down | PFN1/p53 | Apoptosis | Cardiomyocyte | [86] |
| lincRNA-p21 | Up | Wnt/β-catenin | Oxidative stress and cardiac senescence | Cardiomyocyte | [87] |
| LncRNA CHRF | Up | TGF-β/Smads and TGF-β/p38 | Apoptosis | Cardiomyocyte | [88] |
| LncRNA FOXC2-AS1 | Down | WISP1 | cardiomyocyte viability | Cardiomyocyte | [89] |
| LncRNA Mhrt | Down | Nrf2 | Apoptosis | Cardiomyocyte | [90] |
| LncRNA TINCR | Up | IGF2BP1/NLRP3 | Pyroptosis | Cardiomyocyte | [29] |
| CircRNAs | |||||
| CircINSR | Down | SSBP1 | Apoptosis | Cardiomyocyte | [91] |
| CircITCH | Down | miR-330-5p | Apoptosis | Cardiomyocyte | [92] |
| CircNlgn | Up | Nlgn173 | Apoptosis and fibrosis | Cardiomyocyte | [93] |
| CircArhgap12 | Up | miR-135-5p | Apoptosis | Cardiomyocyte | [94] |
| CircPan3 | Down | - | Apoptosis | Cardiomyocyte | [95] |
| CircAmotl1 | - | Akt | Apoptosis | Cardiomyocyte | [96] |
| Has_circ_0097435 | Up | - | Apoptosis | Cardiomyocyte | [97] |
| Circ-LTBP1 | Up | miR-107/ADCY1 | Apoptosis and oxidative stress | Cardiomyocyte | [98] |
| Circ-SKA3 | Up | miR-1303/TLR4 | Apoptosis | Cardiomyocyte | [99] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, M.; Kim, I.-M.; Yang, L. Non-Coding RNAs in the Regulation of Doxorubicin-Induced Cardiotoxicity. Biomolecules 2025, 15, 800. https://doi.org/10.3390/biom15060800
Sun M, Kim I-M, Yang L. Non-Coding RNAs in the Regulation of Doxorubicin-Induced Cardiotoxicity. Biomolecules. 2025; 15(6):800. https://doi.org/10.3390/biom15060800
Chicago/Turabian StyleSun, Mengyao, Il-Man Kim, and Lei Yang. 2025. "Non-Coding RNAs in the Regulation of Doxorubicin-Induced Cardiotoxicity" Biomolecules 15, no. 6: 800. https://doi.org/10.3390/biom15060800
APA StyleSun, M., Kim, I.-M., & Yang, L. (2025). Non-Coding RNAs in the Regulation of Doxorubicin-Induced Cardiotoxicity. Biomolecules, 15(6), 800. https://doi.org/10.3390/biom15060800