
Tuning Autophagy for Improved Liver Transplant Outcomes: Insights from Experimental Models
Abstract

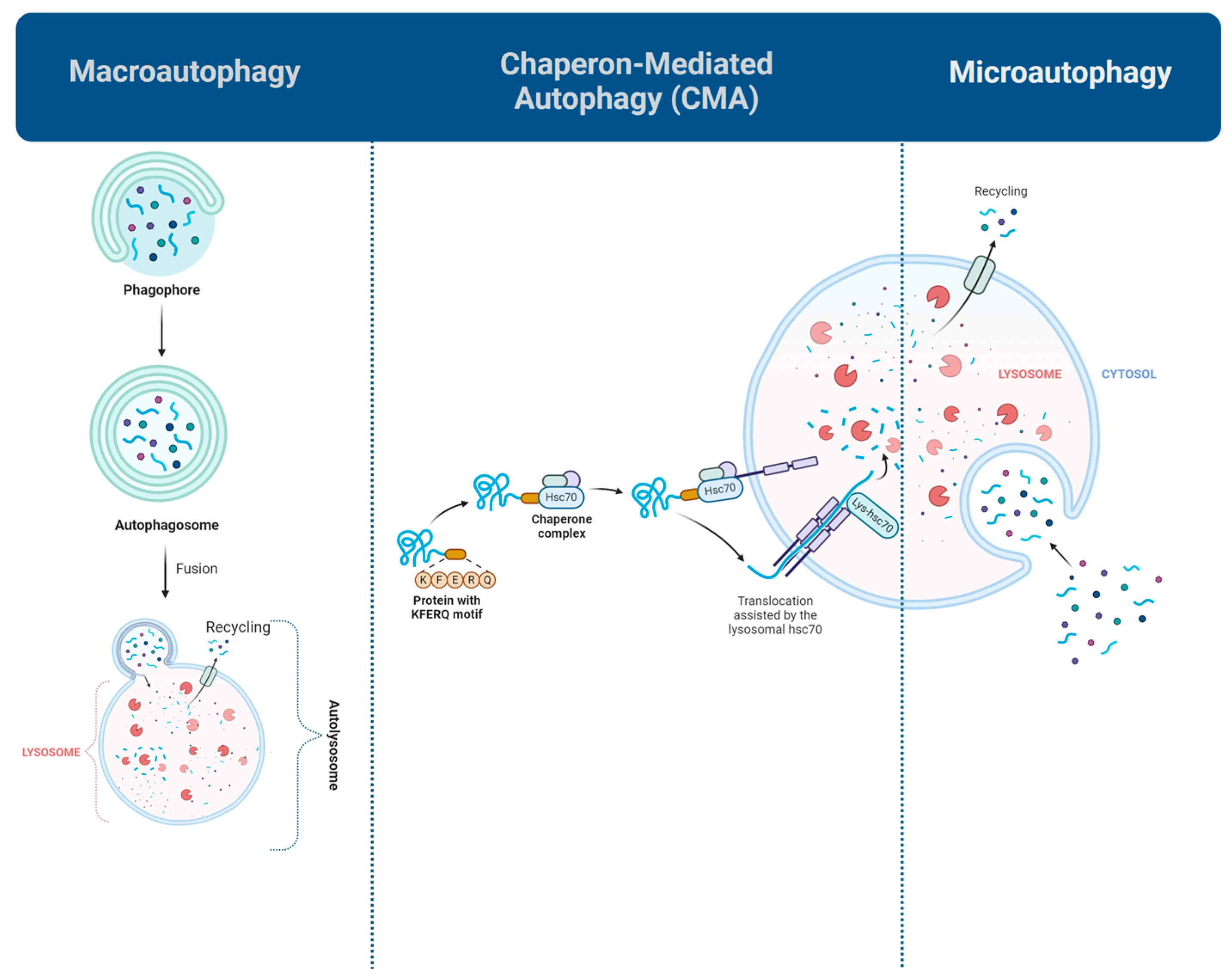
1. Introduction
2. Optimizing Autophagy Regulation Prior to Liver Transplantation
3. Regulating Autophagy Dynamics for Enhanced Protection Post Liver Transplantation
3.1. The Dual Role of Autophagy in I/RI
3.2. Therapeutic Modulation of Autophagy
3.3. Autophagy and Immune Responses
3.4. Modulating Autophagy in Kupffer Cells and T Cells
3.5. Fine-Tuning Autophagy: Balancing Activation and Inhibition
3.6. Mitophagy in Liver Transplantation
4. Autophagy and Xenotransplantation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 25HC | 25-Hydroxycholesterol |
| AdHO-1 | Ad encoding HO-1 |
| 3-MA | 3-methyladenine |
| AALD | Alcohol-associated liver disease |
| AMPK | AMP-activated protein kinase |
| AHXR | Acute humoral xenograft rejection |
| Bax | Bcl-2-associated X protein |
| Bcl-2 | B-cell lymphoma 2 |
| BMM | Bone marrow-derived macrophages |
| CDDO-Im | 2-Cyano-3,12-Dioxooleana-1,9-Dien-28-Imidazolide |
| CMA | Chaperone-mediated autophagy |
| CMC | Carboxymethylcellulose |
| CS | Cold storage |
| DCD | Donation after circulatory death |
| DMSO | Dimethyl sulfoxide |
| ER | Endoplasmic reticulum |
| Eva1a | Eva-1 homologous gene A |
| GSK3β | Glycogen synthase kinase 3 beta |
| HAR | Hyperacute rejection |
| HCC | Hepatocellular carcinoma |
| HMGB1 | High mobility group box 1 |
| HMP | Hypothermic machine perfusion |
| HNPE | Hypothermic deoxygenated (nitrogenated) perfusion |
| HO-1 | Heme Oxygenase-1 |
| HOPE | Hypothermic oxygenated perfusion |
| H/R | Hypoxia/Reoxygenation |
| Hsc70 | Heat shock cognate 71 kDa protein |
| HSPA8 | Heat shock protein family A member 8 |
| I/RI | Ischemia/reperfusion injury |
| IGL-1 | Institut Georges Lopez-1 |
| IL-1β | Interleukin-1 beta |
| IPC | Ischemic preconditioning |
| JNK | c-Jun N-terminal kinase |
| LAMP2A | Lysosomal-associated membrane protein 2 |
| LDH | Lactate Dehydrogenase |
| LPS | lipopolysaccharide |
| L-THP | Levo-tetrahydropalmatine |
| MASH | Metabolic dysfunction-associated steatohepatitis |
| mTOR | Mammalian target of rapamycin |
| NF-kappaB | Nuclear factor-kappa B |
| NK | Natural killer |
| NLRP3 | NOD-like receptor family pyrin domain-containing 3 |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| OA | Oleanolic Acid |
| OE | Overexpression |
| OLT | Orthotopic liver transplantation |
| PACAP | Pituitary adenylate cyclase-activating polypeptide |
| P38 MAPK | P38 mitogen-activated protein kinase |
| PEG35 | Polyethylene glycol 35 kDa |
| PI3K | Phosphatidylinositol-3-kinase |
| PPAR-γ | Peroxisome proliferators-activated receptor γ |
| PRRs | Pattern recognition receptors |
| ROS | Reactive oxygen species |
| SAHA | Suberoylanilide hydroxamic acid |
| SIRT1 | Sirtuin 1 |
| TanIIA | Tanshinone IIA |
| TLR4 | Toll-like receptor 4 |
| TMZ | Trimetazidine |
| TNF-α | Tumor necrosis factor-α |
| ULK1 | Unc-51-Like Autophagy-Activating Kinase 1 |
| UPR | Unfolded protein response |
| UW | University of Wisconsin |
| WI | Warm Ischemia |
References
- Liu, A.; Fang, H. Ischemic Preconditioning on Liver Ischemia Reperfusion Injury: How Far is the Bedside from the Bench? J. Investig. Surg. 2020, 33, 884–885. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.Y. Diverse functions of autophagy in liver physiology and liver diseases. Int. J. Mol. Sci. 2019, 20, 300. [Google Scholar] [CrossRef] [PubMed]
- Zaouali, M.A.; Panisello, A.; Lopez, A.; Folch, E.; Castro-Benítez, C.; Adam, R.; Roselló-Catafau, J. Cross-Talk Between Sirtuin 1 and High-Mobility Box 1 in Steatotic Liver Graft Preservation. Transplant. Proc. 2017, 49, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Wang, S.; Li, S.; Yang, Y.; Fang, Z.; Huang, H.; Wang, Y.; Fan, X.; Ye, Q. Hypothermic oxygenated machine perfusion alleviates liver injury in donation after circulatory death through activating autophagy in mice. Artif. Organs 2019, 43, E320–E332. [Google Scholar] [CrossRef]
- Rampes, S.; Ma, D. Hepatic ischemia-reperfusion injury in liver transplant setting: Mechanisms and protective strategies. J. Biomed. Res. 2019, 33, 221–234. [Google Scholar] [CrossRef]
- Lu, T.-Y.; Xu, X.-L.; Du, X.-G.; Wei, J.-H.; Yu, J.-N.; Deng, S.-L.; Qin, C. Advances in Innate Immunity to Overcome Immune Rejection during Xenotransplantation. Cells 2022, 11, 3865. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Huang, Q.; Peng, L.; Lin, S.; Liu, J.; Zhang, J.; Li, C.; Zhai, S.; Xu, Z.; Wang, S. The multifunctional roles of autophagy in the innate immune response: Implications for regulation of transplantation rejection. Front. Cell Dev. Biol. 2022, 10, 1007559. [Google Scholar] [CrossRef]
- Xiang, S.; Chen, K.; Xu, L.; Wang, T.; Guo, C. Bergenin exerts hepatoprotective effects by inhibiting the release of inflammatory factors, apoptosis and autophagy via the PPAR-γ pathway. Drug Des. Devel. Ther. 2020, 14, 129–143. [Google Scholar] [CrossRef]
- Wang, W.; Wu, L.; Li, J.; Ji, J.; Chen, K.; Yu, Q.; Li, S.; Feng, J.; Liu, T.; Zhang, J.; et al. Alleviation of hepatic ischemia reperfusion injury by oleanolic acid pretreatment via reducing HMGB1 release and inhibiting apoptosis and autophagy. Mediat. Inflamm. 2019, 2019, 3240713. [Google Scholar] [CrossRef]
- Su, S.; Wu, J.; Gong, T.; He, K.; Feng, C.; Zhang, M.; Li, B.; Xia, X. Inhibition of high mobility group box 1-toll-like receptor-4 signaling by glycyrrhizin contributes to the attenuation of cold ischemic injury of liver in a rat model. Transplant. Proc. 2016, 48, 191–198. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, Q.; Dai, W.; Li, S.; Feng, J.; Li, J.; Liu, T.; Xu, S.; Wang, W.; Lu, X.; et al. Quercetin pretreatment attenuates hepatic ischemia reperfusion-induced apoptosis and autophagy by inhibiting ERK/NF-κB pathway. Gastroenterol. Res. Pract. 2017, 2017, 9724217. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, L.; Deng, Y.; Li, X.; Li, G.; Zhou, J.; Cheng, D.; Yang, Y.; Yang, Q.; Chen, G.; et al. Inhibition of autophagy prolongs recipient survival through promoting CD8+ T cell apoptosis in a rat liver transplantation model. Front. Immunol. 2019, 10, 1356. [Google Scholar] [CrossRef] [PubMed]
- Kouroumalis, E.; Voumvouraki, A.; Augoustaki, A.; Samonakis, D.N. Autophagy in liver diseases. World J. Hepatol. 2021, 13, 6–65. [Google Scholar] [CrossRef]
- Tang, C.; Livingston, M.J.; Liu, Z.; Dong, Z. Autophagy in kidney homeostasis and disease. Nat. Rev. Nephrol. 2020, 16, 489–508. [Google Scholar] [CrossRef]
- Bizargity, P.; Schröppel, B. Autophagy: Basic Principles and Relevance to Transplant Immunity. Am. J. Transplant. 2014, 14, 1731–1739. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, J.; Yu, B.; Huang, L.; Dai, B.; Liu, J.; Tang, J. The role of autophagy in lung ischemia/reperfusion injury after lung transplantation in rats. Am. J. Transl. Res. 2016, 8, 3593–3602. [Google Scholar] [PubMed]
- Yin, X.-M.; Ding, W.-X.; Gao, W. Autophagy in the liver. Hepatology 2008, 47, 1773–1785. [Google Scholar] [CrossRef]
- Álvarez-Mercado, A.I.; Rojano-Alfonso, C.; Micó-Carnero, M.; Caballeria-Casals, A.; Peralta, C.; Casillas-Ramírez, A. New Insights Into the Role of Autophagy in Liver Surgery in the Setting of Metabolic Syndrome and Related Diseases. Front. Cell Dev. Biol. 2021, 9, 670273. [Google Scholar] [CrossRef]
- Williams, J.A.; Manley, S.; Ding, W.-X. New advances in molecular mechanisms and emerging therapeutic targets in alcoholic liver diseases. World J. Gastroenterol. 2014, 20, 12908–12933. [Google Scholar] [CrossRef]
- González-Rodríguez, Á.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; Miquilena-Colina, M.E.; Vargas-Castrillón, J.; Iacono, O.L.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef]
- Wang, L.; Ou, J.-H.J. Hepatitis C virus and autophagy. Biol. Chem. 2015, 396, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Liang, C.; Chen, W.-L.; Jung, J.U.; Ou, J.-H.J. Perturbation of autophagic pathway by hepatitis C virus. Autophagy 2008, 4, 830–831. [Google Scholar] [CrossRef] [PubMed]
- Chao, X.; Qian, H.; Wang, S.; Fulte, S.; Ding, W.-X. Autophagy and liver cancer. Clin. Mol. Hepatol. 2020, 26, 606–617. [Google Scholar] [CrossRef]
- Kolahdouzmohammadi, M.; Kolahdouz-Mohammadi, R.; Tabatabaei, S.A.; Franco, B.; Totonchi, M. Revisiting the Role of Autophagy in Cardiac Differentiation: A Comprehensive Review of Interplay with Other Signaling Pathways. Genes 2023, 14, 1328. [Google Scholar] [CrossRef]
- Kolahdouzmohammadi, M.; Totonchi, M.; Pahlavan, S. The role of iPSC modeling toward projection of autophagy pathway in disease pathogenesis: Leader or follower. Stem Cell Rev. Rep. 2021, 17, 539–561. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Zhao, L.; Zhang, F.; Li, L. Regulation of autophagy protects against liver injury in liver surgery-induced ischaemia/reperfusion. J. Cell Mol. Med. 2021, 25, 9905–9917. [Google Scholar] [CrossRef]
- Menzies, R.A.; Gold, P.H. The turnover of mitochondria in a variety of tissues of young adult and aged rats. J. Biol. Chem. 1971, 246, 2425–2429. [Google Scholar] [CrossRef]
- Burra, P.; Zanetto, A.; Russo, F.; Germani, G. Organ Preservation in Liver Transplantation. Semin Liver Dis. 2018, 38, 260–269. [Google Scholar]
- Bejaoui, M. Emerging concepts in liver graft preservation. World J. Gastroenterol. 2015, 21, 396. [Google Scholar] [CrossRef]
- Carini, R.; Castino, R.; De Cesaris, M.G.; Splendore, R.; Démoz, M.; Albano, E.; Isidoro, C. Preconditioning-induced cytoprotection in hepatocytes requires Ca2+-dependent exocytosis of lysosomes. J. Cell Sci. 2004, 117, 1065–1077. [Google Scholar] [CrossRef]
- Wang, Q.; Zuurbier, C.J.; Huhn, R.; Torregroza, C.; Hollmann, M.W.; Preckel, B.; van Den Brom, C.E.; Weber, N.C. Pharmacological cardioprotection against ischemia reperfusion injury—The search for a clinically effective therapy. Cells 2023, 12, 1432. [Google Scholar] [CrossRef] [PubMed]
- Cowled, P.; Fitridge, R. Pathophysiology of reperfusion injury. In Mechanisms of Vascular Disease: A Reference Book for Vascular Specialists; University of Adelaide Press: Adelaide, Australia, 2011; Chapter 18. [Google Scholar]
- Panisello-Roselló, A.; Verde, E.; Lopez, A.; Flores, M.; Folch-Puy, E.; Rolo, A.; Palmeira, C.; Hotter, G.; Carbonell, T.; Adam, R.; et al. Cytoprotective mechanisms in fatty liver preservation against cold ischemia injury: A comparison between IGL-1 and HTK. Int. J. Mol. Sci. 2018, 19, 348. [Google Scholar] [CrossRef]
- Nakamura, K.; Kageyama, S.; Yue, S.; Huang, J.; Fujii, T.; Ke, B.; Sosa, R.A.; Reed, E.F.; Datta, N.; Zarrinpar, A.; et al. Heme oxygenase-1 regulates sirtuin-1–autophagy pathway in liver transplantation: From mouse to human. Am. J. Transplant. 2018, 18, 1110–1121. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhu, J.; Yue, S.; Lu, L.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Wang, X.; Zhai, Y. The Dichotomy of Endoplasmic Reticulum Stress Response in Liver Ischemia-Reperfusion Injury. Transplantation 2016, 100, 365–372. [Google Scholar] [CrossRef]
- Saidi, R.F.; Kenari, S.K.H. Liver Ischemia/Reperfusion Injury: An Overview. J. Investig. Surg. 2014, 27, 366–379. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Han, S.; Chen, X.; Li, X.; Xia, N.; Pu, L. Eva1a inhibits NLRP3 activation to reduce liver ischemia-reperfusion injury via inducing autophagy in kupffer cells. Mol. Immunol. 2021, 132, 82–92. [Google Scholar] [CrossRef]
- Wang, J.; Deng, M.; Wu, H.; Wang, M.; Gong, J.; Bai, H.; Wu, Y.; Pan, J.; Chen, Y.; Li, S. Suberoylanilide hydroxamic acid alleviates orthotopic liver transplantation-induced hepatic ischemia-reperfusion injury by regulating the AKT/GSK3β/NF-κB and AKT/mTOR pathways in rat Kupffer cells. Int. J. Mol. Med. 2020, 45, 1875–1887. [Google Scholar] [CrossRef]
- Liu, H.; Dong, J.; Song, S.; Zhao, Y.; Wang, J.; Fu, Z.; Yang, J. Spermidine ameliorates liver ischaemia-reperfusion injury through the regulation of autophagy by the AMPK-mTOR-ULK1 signalling pathway. Biochem. Biophys. Res. Commun. 2019, 519, 227–233. [Google Scholar] [CrossRef]
- Kong, D.; Hua, X.; Qin, T.; Zhang, J.; He, K.; Xia, Q. Inhibition of glycogen synthase kinase 3β protects liver against ischemia/reperfusion injury by activating 5′ adenosine monophosphate-activated protein kinase-mediated autophagy. Hepatol. Res. 2019, 49, 462–472. [Google Scholar] [CrossRef]
- Xu, D.; Chen, L.; Chen, X.; Wen, Y.; Yu, C.; Yao, J.; Wu, H.; Wang, X.; Xia, Q.; Kong, X. The triterpenoid cddo-imidazolide ameliorates mouse liver ischemia-reperfusion injury through activating the nrf2/ho-1 pathway enhanced autophagy. Cell Death Dis. 2017, 8, e2983. [Google Scholar] [CrossRef]
- Xue, Z.; Zhang, Y.; Liu, Y.; Zhang, C.; Shen, X.-D.; Gao, F.; Busuttil, R.W.; Zheng, S.; Kupiec-Weglinski, J.W.; Ji, H. PACAP neuropeptide promotes Hepatocellular Protection via CREB-KLF4 dependent autophagy in mouse liver Ischemia Reperfusion Injury. Theranostics 2020, 10, 4453–4465. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2014, 22, 377–388. [Google Scholar] [CrossRef]
- Puleston, D.J.; Zhang, H.; Powell, T.J.; Lipina, E.; Sims, S.; Panse, I.; Watson, A.S.; Cerundolo, V.; Townsend, A.R.; Klenerman, P. Autophagy is a critical regulator of memory CD8+ T cell formation. Elife 2014, 3, e03706. [Google Scholar] [CrossRef] [PubMed]
- Germic, N.; Frangez, Z.; Yousefi, S.; Simon, H.U. Regulation of the innate immune system by autophagy: Monocytes, macrophages, dendritic cells and antigen presentation. Cell Death Differ. 2019, 26, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, Q.; Mo, W.; Yu, Q.; Xu, S.; Li, J.; Li, S.; Feng, J.; Wu, L.; Lu, X. The protective effects of shikonin on hepatic ischemia/reperfusion injury are mediated by the activation of the PI3K/Akt pathway. Sci. Rep. 2017, 7, 44785. [Google Scholar] [CrossRef]
- Xu, Y.; Tang, Y.; Lu, J.; Zhang, W.; Zhu, Y.; Zhang, S.; Ma, G.; Jiang, P.; Zhang, W. PINK1-mediated mitophagy protects against hepatic ischemia/reperfusion injury by restraining NLRP3 inflammasome activation. Free Radic. Biol. Med. 2020, 160, 871–886. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Luo, J.; Xiong, Y.; Liu, Z.; Ye, Q. 25-Hydroxycholesterol mitigates hepatic ischemia reperfusion injury via mediating mitophagy. Int. Immunopharmacol. 2021, 96, 107643. [Google Scholar] [CrossRef]
- Teixeira da Silva, R.; Machado, I.F.; Teodoro, J.S.; Panisello-Roselló, A.; Roselló-Catafau, J.; Rolo, A.P.; Palmeira, C.M. PEG35 as a Preconditioning Agent against Hypoxia/Reoxygenation Injury. Int. J. Mol. Sci. 2022, 23, 1156. [Google Scholar] [CrossRef]
- Lee, S.C.; Kim, K.H.; Kim, O.H.; Lee, S.K.; Kim, S.J. Activation of Autophagy by Everolimus Confers Hepatoprotection Against Ischemia–Reperfusion Injury. Am. J. Transplant. 2016, 16, 2042–2054. [Google Scholar] [CrossRef]
- Lin, Y.; Sheng, M.; Weng, Y.; Xu, R.; Lu, N.; Du, H.; Yu, W. Berberine protects against ischemia/reperfusion injury after orthotopic liver transplantation via activating Sirt1/FoxO3α induced autophagy. Biochem. Biophys. Res. Commun. 2017, 483, 885–891. [Google Scholar] [CrossRef]
- Jiang, T.; Zhan, F.; Rao, Z.; Pan, X.; Zhong, W.; Sun, Y.; Wang, P.; Lu, L.; Zhou, H.; Wang, X. Combined ischemic and rapamycin preconditioning alleviated liver ischemia and reperfusion injury by restoring autophagy in aged mice. Int. Immunopharmacol. 2019, 74, 105711. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Huang, L.; Guo, E.; Li, R.; Yang, J.; Li, A.; Yang, Y.; Liu, S.; Hu, J.; Jiang, X.; et al. Baicalein pretreatment reduces liver ischemia/reperfusion injury via induction of autophagy in rats. Sci. Rep. 2016, 6, 25042. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Zhou, J.; Dai, X.; Zhou, H.; Pan, X.; Wang, X.; Zhang, F.; Rao, J.; Lu, L. Short-term starvation attenuates liver ischemia-reperfusion injury (IRI) by Sirt1-autophagy signaling in mice. Am. J. Transl. Res. 2016, 8, 3364–3375. [Google Scholar] [PubMed]
- Yu, Q.; Wu, L.; Liu, T.; Li, S.; Feng, J.; Mao, Y.; Fan, X.; Guo, C.; Wu, J. Protective effects of levo-tetrahydropalmatine on hepatic ischemia/reperfusion injury are mediated by inhibition of the ERK/NF-κB pathway. Int. Immunopharmacol. 2019, 70, 435–445. [Google Scholar] [CrossRef]
- Wang, Y.; Ni, Q.; Ye, Q.; Liu, F.; Fu, Z.; Wang, Q. Tanshinone IIA activates autophagy to reduce liver ischemia-reperfusion injury by MEK/ERK/mTOR pathway. Pharmazie 2018, 73, 396–401. [Google Scholar]
- Deng, S.; Pan, D.; Dai, Y.; Wu, J. Editorial: Solutions for organ transplantation: Xenotransplantation and interspecies chimaeras. Front. Cell Dev. Biol. 2023, 11, 1194654. [Google Scholar] [CrossRef]
- Pallet, N.; Livingston, M.; Dong, Z. Emerging functions of autophagy in kidney transplantation. Am. J. Transplant. 2014, 14, 13–20. [Google Scholar] [CrossRef]
- Kuai, Z.; Chao, X.; He, Y.; Ren, W. Metformin attenuates inflammation and boosts autophagy in the liver and intestine of chronologically aged rats. Exp. Gerontol. 2023, 184, 112331. [Google Scholar] [CrossRef]
- Xia, Y.; He, F.; Moukeila Yacouba, M.B.; Zhou, H.; Li, J.; Xiong, Y.; Zhang, J.; Li, H.; Wang, Y.; Ke, J. Adenosine A2A receptor regulates autophagy flux and apoptosis to alleviate ischemia-reperfusion injury via the cAMP/PKA signaling pathway. Front. Cardiovasc. Med. 2022, 9, 755619. [Google Scholar] [CrossRef]
- Totonchi, H.; Mokarram, P.; Karima, S.; Rezaei, R.; Dastghaib, S.; Koohpeyma, F.; Noori, S.; Azarpira, N. Resveratrol promotes liver cell survival in mice liver-induced ischemia-reperfusion through unfolded protein response: A possible approach in liver transplantation. BMC Pharmacol. Toxicol. 2022, 23, 74. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study (Author, Year) | Experimental Model | Groups & Sample Size | Preservation Mode | Intervention (Autophagy Modulator) | Modulator Role (Inducer/Inhibitor) | Main Results (Quantitative) |
|---|---|---|---|---|---|---|
| Zaouali et al., 2017 [3] | Isolated perfused rat liver model (Steatotic and Non-Steatotic) | Three Groups (16 Zücker rats per group, 8 Obese and 8 Lean): Control, IGL-1, IGL-1 + TMZ | CS | TMZ (10−6 M) in IGL-1 solution | Inducer. (Induces autophagy via SIRT1) | IGL-1 + TMZ showed significant reductions in liver injury markers (AST, ALT, and GLDH) in Non-Steatotic and Steatotic Livers. |
| Zeng et al., 2019 [4] | Mouse (C57BL/6) | Eight Groups (six mice per group): Control, CS, HNPE, HOPE, CS-Reperfusion, HNPE-Reperfusion, HOPE-Reperfusion, 3-MA | CS | 3-MA (30 mg/kg 1 h before WI) | Inhibitor. (Attenuates protective effect of HOPE) | HOPE showed significant reduction in liver injury markers (AST, ALT) and increased in autophagy markers (LC3B-II, Atg5, ULK1) vs. CS and HNPE. |
| Nakamura et al., 2017 [34] | Mouse (C57BL/6) | Three Groups (4–6 mice per group): Control, AdHO-1, Adβ-gal | CS | HO-1 overexpression (via genetically modified macrophages) | Inducer. (Stimulates SIRT1) | AdHO-1-treated OLT in mouse and human models showed reduced liver injury marker (ALT) and enhanced autophagy markers (SIRT1 and LC3B) compared to controls with low HO-1 expression. |
| Human (OLT model) | Two Groups: Low-HO-1, High-HO-1 (51 OLT patients) | EX527 (Administered 30 min before OLT surgery) | Inhibitor. (Blocks SIRT1, reduces autophagy, diminishes HO-1-mediated hepatoprotection) |
| Study (Author, Year) | Experimental Model | Groups & Sample Size | I/RI Induction Method | Intervention (Autophagy Modulator) | Type of Autophagy | Modulator Role (Inducer/Inhibitor) | Main Results (Quantitative) |
|---|---|---|---|---|---|---|---|
| Lee et al., 2016 [50] | In vitro: (AML12 cells) | Three Groups: Control, IRI, and IRI + Everolimus | Following one hour in Krebs–Henseleit buffer containing antimycin A and 2-deoxyglucose, the cells were reperfused with complete media. | Everolimus (100 nM) | Macroautophagy | Inducer. (promotes autophagy by inhibiting mTOR) | IRI + Everolimus showed enhanced autophagy markers, and decreased apoptosis compared to IRI alone. |
| In vivo: (BALB/c mice) | Four Groups (15 mice per group): Sham + Saline, Sham + Everolimus, IRI + Saline, and IRI + Everolimus | 45 min of hepatic ischemia followed by reperfusion. | Everolimus (1 mg/kg, 24 h before and immediately after IRI induction) | IRI + Everolimus showed significant IL-6 and TNF-α reduction, lower liver enzyme levels (AST, ALT, and ammonia), and improved liver histopathology compared to IRI + Saline. | |||
| Wang et al., 2020 [38] | In vitro: (Kupffer Cells) | Six Groups: Normal, lipopolysaccharide (LPS), LPS + DMSO, LPS + SAHA, LPS + Scramble-siRNA, and LPS + SAHA + ATG5-siRNA | LPS stimulation to induce inflammation. | SAHA (3 μM) | Macroautophagy | Inducer. (promotes autophagy through inhibition of the AKT/mTOR pathway) | LPS + SAHA inhibited M1 polarization of Kupffer cells and showed increased autophagy markers compared to LPS alone. |
| In vivo: (Sprague-Dawley rats) | Eight Groups (five rats per group): Sham, IRI, IRI + DMSO, IRI + SAHA, IRI + SAHA + Clodronate liposomes, IRI + SAHA + Chloroquine, IRI + Scramble-shRNA, and IRI + SAHA + ATG5-shRNA | Modified Kamada’s two-cuff technique. | SAHA (50 mg/kg, 12 h prior to OLT and continued q12 h after reperfusion) | IRI + SAHA showed decreased levels of IL-1β, TNF-α, AST, and ALT, along with enhanced histopathology, and reduced M1 polarization of Kupffer cells compared to IRI alone. | |||
| Liu et al., 2019 [39] | In vivo: (C57BL/6 mice) | Four Groups (six mice per group): Sham, Sham + Spermidine, IRI, and IRI + Spermidine | 60 min of partial hepatic ischemia (70%) followed by reperfusion. | Spermidine (3 mM, 2 mL/day for four weeks before IRI induction) | Macroautophagy | Inducer. (stimulates autophagy through the AMPK-mTOR-ULK1 pathway) | IRI + Spermidine showed reduced IL-6, TNF-α, AST, ALT, and improved histopathology compared to IRI alone. |
| Lin et al., 2017 [51] | In vivo: (Wistar rats) | Four Groups (six rats per group): Sham, OLT, OLT + Berberine, and OLT + Berberine + EX527 | OLT using the Kamada’s two-cuff technique followed by reperfusion. | Berberine (100 mg/kg/day, for two weeks before OLT) | Sirt1/FoxO3α Induced Autophagy | Inducer. (activates autophagy via the Sirt1/FoxO3α pathway) | OLT + Berberine showed reduced ALT, AST, improved histopathology, and decreased oxidative stress compared to OLT alone. |
| Jiang et al., 2019 [52] | In vivo: (C57BL/6 mice) | Six Groups (six mice per group): Sham, IRI, Ischemic preconditioning (IPC), Rapamycin, IPC + Rapamycin, and IPC + Rapamycin + 3-MA | 90 min of segmental (70%) hepatic ischemia followed by reperfusion. | Rapamycin (1 mg/kg, 1 h before ischemia) | Macroautophagy | Inducer. (promotes autophagy by inhibiting mTOR) | IPC + Rapamycin showed reduced ALT, and improved histopathology, compared to IPC alone. |
| Nakamura et al., 2017 [34] | In vivo: (C57BL/6 mice) | Three Groups (six mice per group): BMMs, BMM (ad encoding HO-1), and BMM (Adβ-gal) | 20 h of CS in UW solution followed by OLT and reperfusion. | HO-1 (2.5 × 109 pfu) via adoptive transfer of Ad encoding HO-1-transfected BMMs | Macroautophagy | Inducer. (activates autophagy through the SIRT1) | BMM (Ad encoding HO-1) showed reduced ALT levels, and improved histopathology (lower Suzuki score) compared to BMM (Adβ-gal) |
| Kong et al., 2019 [40] | In vitro: (primary hepatocytes) | Three Groups: H/R + DMSO, H/R + SB216763, and H/R + SB216763 + 3-MA | Cells underwent 4 h of hypoxia (1% O2) followed by 2 h of reoxygenation. | SB216763 | Macroautophagy | Inducer. (activates autophagy through the inhibition of GSK3β) | H/R + SB216763 showed reduced LDH release (lower cell death), and increased autophagy compared to control group. |
| In vivo: (C57BL/6 mice) | Four Groups (4–6 mice per group): Sham, DMSO + IRI, IRI + SB216763, and IRI + SB216763 + 3-MA | 90 min of partial hepatic ischemia (70%) followed by reperfusion. | SB216763 (20 mg/kg, 1 h before ischemia) | IRI + SB216763 showed reduced AST, ALT, improved histopathology, compared to DMSO + IRI. | |||
| Xu et al., 2017 [41] | In vitro: (primary hepatocytes) | Two Groups: DMSO + H/R and CDDO-Im + H/R | Cells underwent 4 h of hypoxia (1% O2) followed by 2 h of reoxygenation. | CDDO-Im (200 μM) | Macroautophagy | Inducer. (activates autophagy through the Nrf2/HO-1 pathway) | CDDO-Im + H/R showed reduced LDH release (less cell death), and enhanced mitochondrial function (lower mtDNA release, improved ROS clearance) compared to DMSO + H/R. |
| In vivo: (C57BL/6 mice) | Three Groups (4–6 mice per group): Sham, IRI+DMSO, and IRI + CDDO-Im. | 90 min of partial hepatic ischemia followed by reperfusion. | CDDO-Im (2 mg/kg, 3 h before ischemia) | IRI + CDDO-Im. showed reduced AST, ALT, and improved histopathology, compared to IRI + DMSO. | |||
| Xue et al., 2020 [42] | In vitro: (primary hepatocytes) | Four Groups: PBS, H2O2, H2O2 + PACAP+DMSO, and H2O2 + PACAP + 3-MA | Hydrogen peroxide (H2O2) 0.4 mM. | PACAP38 (10 nM) | Macroautophagy | Inducer. (activates autophagy through the CREB-KLF4 pathway) | H2O2 + PACAP showed reduced LDH release |
| In vivo: (C57BL/6 mice) | Four Groups (12 mice per group): Sham, PBS, PACAP + DMSO, and PACAP + 3-MA. | 20 h of storage at 4 °C in UW solution followed by syngeneic OLT and reperfusion. | PACAP38 (1 mg/kg at the time of liver graft procurement and before reperfusion) | PACAP + DMSO showed reduced ALT, improved histopathology, and decreased necrosis compared to PBS group. | |||
| Wu et al., 2017 [11] | In vitro: (primary hepatocytes) | Seven Groups: Control, QE, DMSO, H/R, H/R + QE 5 μM, H/R + QE 10 μM, and H/R + QE 20 μM | Cells underwent 24 h of hypoxia (3% O2) followed by 2 h of reoxygenation. | Quercetin (QE) (20 μM) | Macroautophagy | Inhibitor. (inhibits autophagy through ERK/NF-κB pathway) | H/R + QE showed decreased apoptosis, reduced autophagy markers, and enhanced cell viability compared to H/R alone. |
| In vivo: (Balb/c mice) | Eight Groups (5 and 24 mice per group): Control, Vehicle, Low-QE, High-QE, Sham, IRI, QE100 + IRI, and QE200 + IRI | 45 min of 70% hepatic WI followed by reperfusion. | Quercetin (100 mg/kg or 200 mg/kg, before IRI) | IRI + QE showed reduced ALT, AST, improved histopathology, reduced pro-inflammatory cytokines (TNF-α, IL-6), and decreased apoptosis compared to IRI alone. | |||
| Wang et al., 2019 [9] | In vivo: (Balb/c mice) | Five Groups (18 mice per group): Sham, CMC, IRI, Low-OA (30 mg/kg), and High-OA (60 mg/kg) | 60 min of partial (70%) hepatic ischemia followed by reperfusion. | Oleanolic Acid (OA) (30 mg/kg or 60 mg/kg, for seven days before surgery) | Macroautophagy | Inhibitor. (inhibits autophagy through JNK phosphorylation and HMGB1 suppression) | Low-OA and High-OA groups showed reduced ALT, AST, and improved histopathology, compared to IRI group. |
| Liu et al., 2016 [53] | In vitro: (primary rat hepatocytes) | Four Groups: Control, H/R, H/R + Baicalein, and H/R + Baicalein + 3-MA | Cells underwent 6 h of hypoxia (1% O2) followed by 2 h of reoxygenation. | Baicalein (5 μM) | Macroautophagy | Inducer. (activates autophagy via HO-1) | H/R + Baicalein showed improved cell viability, and reduced LDH release compared to H/R alone. |
| In vivo: (Sprague-Dawley rats) | Three Groups (six rats per group): Sham, IRI, and Baicalein + IRI | 60 min of partial hepatic WI followed by reperfusion. | Baicalein (100 mg/kg, 1 h before ischemia) | Baicalein + IRI showed reduced ALT, AST, improved histopathology, and reduced necrosis, compared to IRI alone. | |||
| Qin et al., 2016 [54] | In Vivo: (C57BL/6 mice) | Two Main Groups: Sham (control, one day-STS, two day-STS, and three day-STS), IRI (control, one day-STS, two day-STS, and three day-STS). | 90 min of partial hepatic ischemia followed by reperfusion. | Short-term starvation (STS) for one, two, or three days before IRI | Macroautophagy | Inducer. (enhances autophagy via Sirt1 activation) | IRI + STS showed reduced ALT, AST, improved histopathology, decreased IL-1β, TNF-α, and apoptosis markers compared to IRI alone. |
| Xiang et al., 2020 [8] | In vitro: (LO2 hepatocytes) | Four Groups: Control, H/R, H/R + Bergenin, and H/R + Bergenin + GW9662 | Cells underwent 24 h of hypoxia (3% O2) followed by 2 h of reoxygenation. | Bergenin (3 mM) | Peroxisome proliferators activated receptor γ (PPAR-γ) pathway mediated autophagy | Inhibitor. (inhibits autophagy through the PPAR-γ Pathway) | H/R + Bergenin showed enhanced cell viability, reduced apoptosis, and decreased autophagy markers compared to H/R. |
| In vivo: (Balb/c mice) | Five Groups (24 mice per group): Sham, IRI, IRI + Bergenin (10 mg/kg), IRI + Bergenin (20 mg/kg), and IRI + Bergenin (40 mg/kg). | 60 min of partial hepatic ischemia (70%) followed by reperfusion. | Bergenin (10, 20, 40 mg/kg, for three days) (once before the operation, once per day) | IRI + Bergenin showed reduced ALT, AST, improved histopathology, and decreased inflammation, compared to IRI alone. | |||
| Chen et al., 2019 [12] | In vivo: (Lewis and Brown Norway rats) | Three Groups (five rats per group): Syngeneic control, Allogeneic, and Allogeneic + 3-MA | OLT using the Two-Cuff Method. | 3-MA (24 mg/kg, every three days, starting one day before transplantation, dosage reduced by half every nine days) | Macroautophagy | Inhibitor. | Allogeneic + 3-MA group showed prolonged survival time, reduced ALT, AST, and improved histopathology compared to Allogeneic group without 3-MA. |
| Wang et al., 2021 [37] | In vitro: (Kupffer cells) | Mixed Groups: Control, IRI + Eva1a OE, and IRI + Eva1a KD | - | Eva1a Overexpression (OE) and Knockdown (KD). | Macroautophagy | Inducer. (Eva1a OE Promotes autophagy by interacting with ATG16L1). | IRI + Eva1a KD showed increased TNF-α, increased ALT, AST, compared to IRI alone. |
| In vivo: (C57BL/6 mice) | Four Groups (4–6 mice per group): Sham, IRI, IRI + Eva1a OE, and IRI + Eva1a KD. | 90 min of partial hepatic ischemia (70%) followed by reperfusion. | Eva1a-OE and Knockdown (KD). | ||||
| Yu et al., 2019 [55] | In Vivo: (BALB/c mice) | Six Groups (six mice per group): Control, Sham, L-THP (40 mg/kg), IRI, L-THP (20 mg/kg) + IRI, and L-THP (40 mg/kg) + IRI | 45 min of partial hepatic ischemia (70%) followed by reperfusion. | Levo-tetrahydropalmatine (L-THP) (20 mg/kg and 40 mg/kg, for five days before IRI). | Macroautophagy | Inhibitor. (suppresses autophagy via inhibition of ERK/NF-κB signaling pathway) | L-THP + IRI showed reduced AST and ALT levels, decreased TNF-α and IL-6, and improved liver architecture compared to IRI alone. |
| Liu et al., 2017 [46] | In vivo: (Balb/c mice) | Four Groups (18 mice per group): Control, IRI, IRI + Shikonin 7.5 mg/kg, and IRI + Shikonin 12.5 mg/kg | 45 min of segmental (70%) hepatic ischemia followed by reperfusion. | Shikonin (7.5 mg/kg or 12.5 mg/kg, 2 h before ischemia). | Macroautophagy | Inhibitor. (inhibits autophagy via the PI3K/Akt pathway) | IRI + Shikonin showed reduced ALT, AST, improved histopathology, and decreased IL-1β, TNF-α, IL-6, compared to IRI alone. |
| Wang et al., 2018 [56] | In vivo: (C57BL/6 mice) | Four groups (six mice per group): Vehicle, TanIIA, IRI + Vehicle, and IRI + TanIIA | 60 min of partial hepatic ischemia (70%) followed by reperfusion | Tanshinone IIA (TanIIA) (20 mg/kg/day, for three days before IRI). | Macroautophagy | Inducer. (activates autophagy through the MEK/ERK/mTOR pathway) | IRI + TanIIA showed reduced ALT, AST, improved histopathology, and reduced ROS production, compared to IRI+Vehicle group. |
| Cao et al., 2021 [48] | In vivo: (Sprague-Dawley rats) | Six Groups (six rats per group): Sham, 25HC, IRI (3) + Vehicle, IRI (3) + 25HC, IRI (24) + Vehicle, and IRI (24) + 25HC. | 60 min of partial hepatic ischemia (70%) followed by reperfusion. | 25-Hydroxycholesterol (25HC) (30 mg/kg 4 h before ischemia). | Mitophagy | Inducer. (activates PINK1/Parkin-dependent mitophagy) | IRI + 25HC showed reduced ALT, AST, improved histopathology, decreased NLRP3 activation, and reduced IL-1β, compared to IRI + Vehicle. |
| Teixeira da Silva et al., 2022 [49] | In vitro: (HepG2 cells) | Six groups: Control, Control + 1% PEG35, Control + 5% PEG35, H/R, 1% PEG35 + H/R, and 5% PEG35 + H/R | H/R: 2 h of hypoxia (hypoxia mimic solution) followed by 2 h of reoxygenation (complete medium) | PEG35 (1% or 5%, preconditioning for 1 h before H/R) | Mitophagy | Inducer. (enhances mitochondrial biogenesis and recovery of membrane potential). | 5% PEG35 + H/R showed increased cell viability, preserved mitochondrial membrane potential, reduced ROS, maintained ATP levels, and improved mitochondrial dynamics by enhancing fusion and reducing fission compared to H/R alone. |
| Xu et al., 2020 [47] | In vitro: (Kupffer cells) | Two Groups: Control (Vector and PINK1 OE), H/R (Vector and PINK1 OE) | Cells were exposed to 4 h of hypoxia (1% O2) followed by 4 h of reoxygenation. | Mitophagy | Inducer. (promotes PINK1/Parkin-dependent mitophagy) | H/R + PINK1 OE showed enhanced autophagy, decreased IL-1β, and TNF-α compared to H/R alone. | |
| In vivo: (C57BL/6 mice) | Two Groups (4–6 mice per group): Vector (Control), PINK1 OE | 60 min of partial hepatic ischemia followed by reperfusion. | PINK1-OE (50 μg pcDNA3.1-mPINK1 plasmid 72 h before IRI). | PINK1 OE showed reduced ALT, AST, and improved histopathology compared to control group. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolahdouzmohammadi, M.; Oldani, G. Tuning Autophagy for Improved Liver Transplant Outcomes: Insights from Experimental Models. Biomolecules 2025, 15, 797. https://doi.org/10.3390/biom15060797
Kolahdouzmohammadi M, Oldani G. Tuning Autophagy for Improved Liver Transplant Outcomes: Insights from Experimental Models. Biomolecules. 2025; 15(6):797. https://doi.org/10.3390/biom15060797
Chicago/Turabian StyleKolahdouzmohammadi, Mina, and Graziano Oldani. 2025. "Tuning Autophagy for Improved Liver Transplant Outcomes: Insights from Experimental Models" Biomolecules 15, no. 6: 797. https://doi.org/10.3390/biom15060797
APA StyleKolahdouzmohammadi, M., & Oldani, G. (2025). Tuning Autophagy for Improved Liver Transplant Outcomes: Insights from Experimental Models. Biomolecules, 15(6), 797. https://doi.org/10.3390/biom15060797