

Alternative Splicing in Tumorigenesis and Cancer Therapy

Abstract
1. Introduction
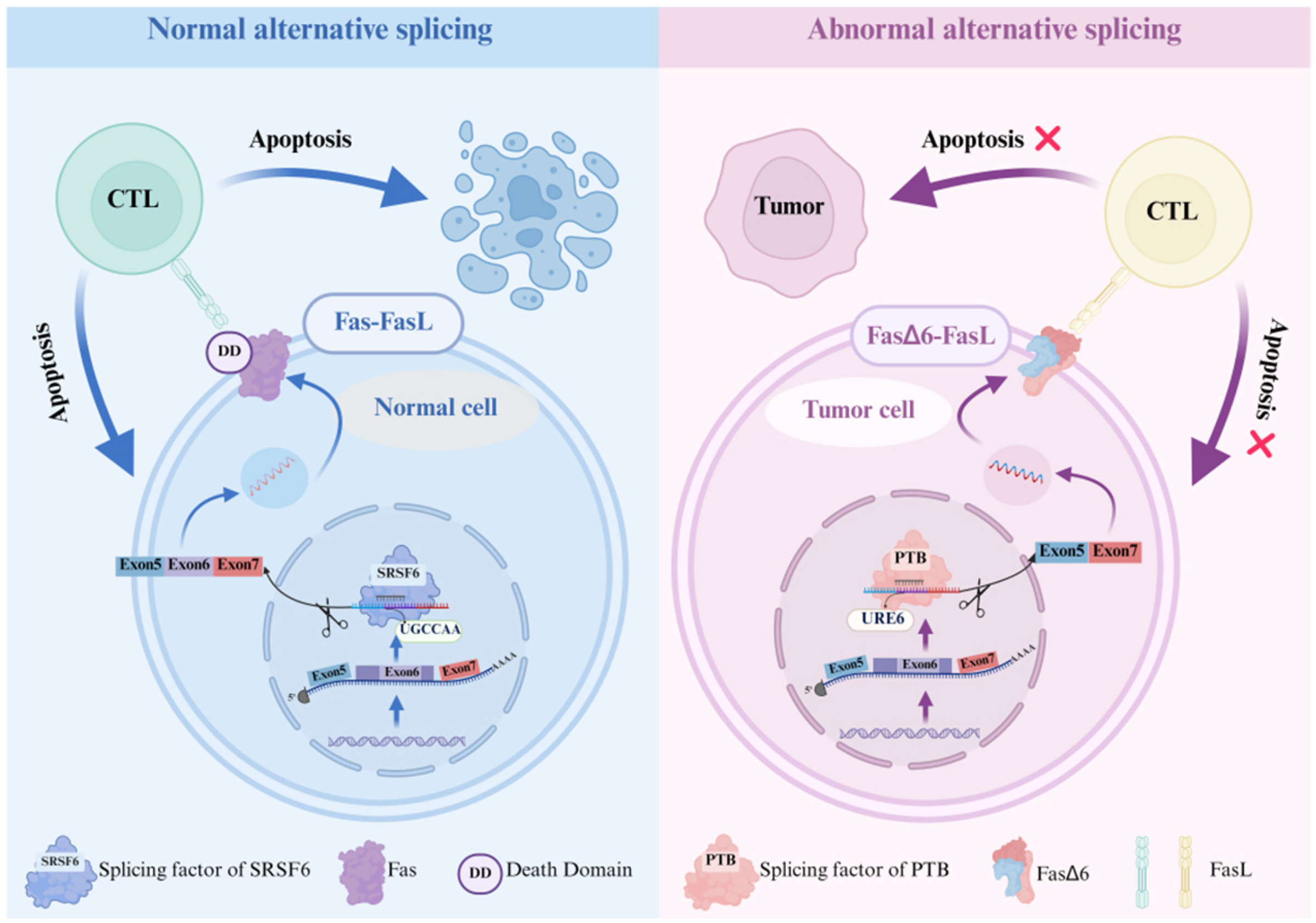
2. Normal and Abnormal Alternative Splicing
3. Abnormal Alternative Splicing and Cancer
3.1. Cis-Acting Splicing Mutations
3.2. Chromatin State
3.3. RNA Structure
3.4. Trans-Acting Splicing Mutations
3.5. Abnormal AS and NMD Regulation
3.6. Abberant Splicing in Breast Cancer
3.7. Abberant Splicing in Colorectal Cancer
3.8. Abberant Splicing in Lung Cancer
3.9. Other Malignancies
3.10. AS and Cancer Immunotherapy
3.10.1. AS and Immune Activation
3.10.2. AS in Immune Regulatory Molecules
3.11. AS and Cancer Therapy
3.11.1. Cancer Immunotherapy
3.11.2. SSOs and Cancer Therapy
3.11.3. Targeting Novel Splice Variants
4. Innovative Technologies in the Study of AS
4.1. Splicing Analysis Tools and Databases
4.2. Neoantigen Prediction Innovations
4.3. Multi-Omics Integration Strategy
5. Challenges and Difficulties
6. Conclusions and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACT | Adoptive Cell Therapy. |
| AML | Acute Myeloid Leukemia. |
| AS | Alternative Splicing. |
| AS Cancer Atlas | Alternative Splicing Cancer Atlas. |
| BC | Breast Cancer. |
| BCR | B-cell Receptor. |
| CAR-T | Chimeric Antigen Receptor T-cell. |
| CCS | Circular Consensus Sequencing. |
| CRC | Colorectal Cancer. |
| CHOP | Children’s Hospital of Philadelphia. |
| EMT | Epithelial-to-Mesenchymal Transition. |
| ESEs | Exonic Splicing Enhancers. |
| ESSs | Exonic Splicing Silencers. |
| ERs | Estrogen Receptors. |
| ER | Endoplasmic Reticulum. |
| FDSIs | Functionally Distinct Splice Isoforms. |
| FLT3-ITDs | FLT3 Internal Tandem Duplications. |
| GBM | Glioblastoma. |
| GBC | Gallbladder Cancer. |
| HCC | Hepatocellular Carcinoma. |
| HGSC | High-grade serous ovarian cancer. |
| HNSCC | Head and neck squamous cell carcinoma. |
| hnRNPs | Heterogeneous Nuclear Ribonucleoproteins. |
| ISEs | Intronic Splicing Enhancers. |
| ISSs | Intronic Splicing Silencers. |
| ICB | Immune Checkpoint Blockade. |
| ICI | Immune Checkpoint Inhibitor. |
| Ig | Immunoglobulin. |
| IRIS | Isoform Peptides from RNA Splicing for Immunotherapy Target Screening. |
| ISR | Integrated Stress Response. |
| IR | Insulin Receptor. |
| LUAD | Lung Adenocarcinoma. |
| ML | Machine Learning. |
| MHC | Major Histocompatibility Complex. |
| m6A | N6-methyladenosine. |
| MDS | Myelodysplastic syndromes. |
| MS | Mass Spectrometry. |
| NJs | neojunctions. |
| NMD | Nonsense—mediated Decay. |
| NSCLC | Non-small cell lung cancer. |
| NGS | Next-Generation Sequencing. |
| NEPC | Neuroendocrine prostate cancer. |
| NCI | National Cancer Institute. |
| ORFs | Open Reading Frames. |
| PBMCs | Peripheral Blood Mononuclear Cells. |
| PDAC | Pancreatic ductal adenocarcinoma. |
| PTCs | Premature Termination Codons. |
| RBPs | RNA-binding proteins. |
| RBM | RNA-binding Motif RNA. |
| ROS | Reactive Oxygen Species. |
| rMATS | replicate Multivariate Analysis of Transcript Splicing. |
| snRNAs | small nuclear RNAs. |
| snRNPs | small nuclear ribonucleoproteins. |
| SAVs | Splice-altering Variants. |
| SNAF | Spliced Neo Antigen Finder. |
| SNV | Single-Nucleotide Variant. |
| SMRT | Single Molecule, Real-Time. |
| SRs | Serine/Arginine-rich Proteins. |
| SSOs | Splice-switching Oligonucleotides. |
| TAMs | Tumor Associated Macrophages. |
| TCR | T-cell Receptor. |
| TCR-T | T-cell Receptor Engineered T-cell. |
| Tregs | Regulatory T Cells. |
| TME | Tumor Microenvironment. |
| TMB | Tumor Mutational Burden. |
| TKIs | Tyrosine Kinase Inhibitors. |
| TSAs | Tumor-Specific Antigens. |
| T-ALL | T-cell acute lymphoblastic leukemia. |
| TESLA | Tumor Neoantigen Selection Alliance. |
| URE6 | Uridine-rich sequence located in exon 6. |
| UCLA | University of California, Los Angeles. |
| WES | Whole-exome sequencing. |
References
- Huang, P.; Wen, F.; Tuerhong, N.; Yang, Y.; Li, Q. Neoantigens in cancer immunotherapy: Focusing on alternative splicing. Front. Immunol. 2024, 15, 1437774. [Google Scholar] [CrossRef]
- Blackwell, D.L.; Fraser, S.D.; Caluseriu, O.; Vivori, C.; Tyndall, A.V.; Lamont, R.E.; Parboosingh, J.S.; Innes, A.M.; Bernier, F.P.; Childs, S.J. Hnrnpul1 controls transcription, splicing, and modulates skeletal and limb development in vivo. G3 2022, 12, jkac067. [Google Scholar] [CrossRef]
- Sciarrillo, R.; Wojtuszkiewicz, A.; Assaraf, Y.G.; Jansen, G.; Kaspers, G.J.L.; Giovannetti, E.; Cloos, J. The role of alternative splicing in cancer: From oncogenesis to drug resistance. Drug Resist. Updat. 2020, 53, 100728. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Jia, Z.; Meng, X.; Chen, Y.; Wang, S.; Fu, C.; Yang, L.; Zhou, R.; Wang, B.; Cao, Y. Combined Metabolomic and Transcriptomic Analysis Reveals Allantoin Enhances Drought Tolerance in Rice. Int. J. Mol. Sci. 2022, 23, 14172. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yu, W.; Lai, M. Towards understandings of serine/arginine-rich splicing factors. Acta Pharm. Sin. B 2023, 13, 3181–3207. [Google Scholar] [CrossRef] [PubMed]
- Mehta, Z.; Touma, M. Post-Transcriptional Modification by Alternative Splicing and Pathogenic Splicing Variants in Cardiovascular Development and Congenital Heart Defects. Int. J. Mol. Sci. 2023, 24, 1555. [Google Scholar] [CrossRef]
- Schindler, N.R.; Braun, D.A. Antigenic targets in clear cell renal cell carcinoma. Kidney Cancer 2023, 7, 81–91. [Google Scholar] [CrossRef]
- Ouedraogo, W.; Ouangraoua, A. SimSpliceEvol2: Alternative splicing-aware simulation of biological sequence evolution and transcript phylogenies. BMC Bioinform. 2024, 25, 235. [Google Scholar] [CrossRef]
- Shender, V.O.; Anufrieva, K.S.; Shnaider, P.V.; Arapidi, G.P.; Pavlyukov, M.S.; Ivanova, O.M.; Malyants, I.K.; Stepanov, G.A.; Zhuravlev, E.; Ziganshin, R.H.; et al. Therapy-induced secretion of spliceosomal components mediates pro-survival crosstalk between ovarian cancer cells. Nat. Commun. 2024, 15, 5237. [Google Scholar] [CrossRef]
- Donnellan, L.; Young, C.; Simpson, B.S.; Acland, M.; Dhillon, V.S.; Costabile, M.; Fenech, M.; Hoffmann, P.; Deo, P. Proteomic Analysis of Methylglyoxal Modifications Reveals Susceptibility of Glycolytic Enzymes to Dicarbonyl Stress. Int. J. Mol. Sci. 2022, 23, 3689. [Google Scholar] [CrossRef]
- Freytag, M.; Kluth, M.; Bady, E.; Hube-Magg, C.; Makrypidi-Fraune, G.; Heinzer, H.; Höflmayer, D.; Weidemann, S.; Uhlig, R.; Huland, H.; et al. Epithelial splicing regulatory protein 1 and 2 (ESRP1 and ESRP2) upregulation predicts poor prognosis in prostate cancer. BMC Cancer 2020, 20, 1220. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, S.; Li, Y.; Liu, J.; Li, Q.; Zang, W.; Pan, Y. The Regulatory Network of hnRNPs Underlying Regulating PKM Alternative Splicing in Tumor Progression. Biomolecules 2024, 14, 566. [Google Scholar] [CrossRef] [PubMed]
- Bei, M.; Xu, J. SR proteins in cancer: Function, regulation, and small inhibitor. Cell Mol. Biol. Lett. 2024, 29, 78. [Google Scholar] [CrossRef]
- Pan, D.; Long, L.; Li, C.; Zhou, Y.; Liu, Q.; Zhao, Z.; Zhao, H.; Lin, W.; Zheng, Z.; Peng, L.; et al. Splicing factor hnRNPA1 regulates alternative splicing of LOXL2 to enhance the production of LOXL2Δ13. J. Biol. Chem. 2024, 300, 107414. [Google Scholar] [CrossRef]
- Emilius, L.; Bremm, F.; Binder, A.K.; Schaft, N.; Dörrie, J. Tumor Antigens beyond the Human Exome. Int. J. Mol. Sci. 2024, 25, 4673. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Miao, K.; Sun, H.; Deng, C.X. Tumor heterogeneity reshapes the tumor microenvironment to influence drug resistance. Int. J. Biol. Sci. 2022, 18, 3019–3033. [Google Scholar] [CrossRef] [PubMed]
- Manabile, M.A.; Hull, R.; Khanyile, R.; Molefi, T.; Damane, B.P.; Mongan, N.P.; Bates, D.O.; Dlamini, Z. Alternative Splicing Events and Their Clinical Significance in Colorectal Cancer: Targeted Therapeutic Opportunities. Cancers 2023, 15, 3999. [Google Scholar] [CrossRef]
- Bhuiyan, S.A.; Ly, S.; Phan, M.; Huntington, B.; Hogan, E.; Liu, C.C.; Liu, J.; Pavlidis, P. Systematic evaluation of isoform function in literature reports of alternative splicing. BMC Genomics 2018, 19, 637. [Google Scholar] [CrossRef]
- Gueroussov, S.; Gonatopoulos-Pournatzis, T.; Irimia, M.; Raj, B.; Lin, Z.Y.; Gingras, A.C.; Blencowe, B.J. An alternative splicing event amplifies evolutionary differences between vertebrates. Science 2015, 349, 868–873. [Google Scholar] [CrossRef]
- Fair, B.; Buen Abad Najar, C.F.; Zhao, J.; Lozano, S.; Reilly, A.; Mossian, G.; Staley, J.P.; Wang, J.; Li, Y.I. Global impact of unproductive splicing on human gene expression. Nat. Genet. 2024, 56, 1851–1861. [Google Scholar] [CrossRef]
- Li, S.; Guo, W.; Dewey, C.N.; Greaser, M.L. Rbm20 regulates titin alternative splicing as a splicing repressor. Nucleic Acids Res. 2013, 41, 2659–2672. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.C.; Yan, Y.T.; Hsieh, W.K.; Peng, P.J.; Su, C.H.; Tarn, W.Y. RBM4 promotes pancreas cell differentiation and insulin expression. Mol. Cell Biol. 2013, 33, 319–327. [Google Scholar] [CrossRef]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Yano, M.; Hayakawa-Yano, Y.; Mele, A.; Darnell, R.B. Nova2 regulates neuronal migration through an RNA switch in disabled-1 signaling. Neuron 2010, 66, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Nam, J.; Mukouyama, Y.S.; Kawamoto, S. Rbfox3-regulated alternative splicing of Numb promotes neuronal differentiation during development. J. Cell Biol. 2013, 200, 443–458. [Google Scholar] [CrossRef]
- Sen, S.; Jumaa, H.; Webster, N.J. Splicing factor SRSF3 is crucial for hepatocyte differentiation and metabolic function. Nat. Commun. 2013, 4, 1336. [Google Scholar] [CrossRef]
- Rothrock, C.R.; House, A.E.; Lynch, K.W. HnRNP L represses exon splicing via a regulated exonic splicing silencer. Embo J. 2005, 24, 2792–2802. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Galarza-Muñoz, G.; Garcia-Blanco, M.A. Role of RNA Alternative Splicing in T Cell Function and Disease. Genes 2023, 14, 1896. [Google Scholar] [CrossRef]
- Hirano, M.; Galarza-Muñoz, G.; Nagasawa, C.; Schott, G.; Wang, L.; Antonia, A.L.; Jain, V.; Yu, X.; Widen, S.G.; Briggs, F.B.S.; et al. The RNA helicase DDX39B activates FOXP3 RNA splicing to control T regulatory cell fate. eLife 2023, 12, e76927. [Google Scholar] [CrossRef]
- Yamamoto, M.L.; Clark, T.A.; Gee, S.L.; Kang, J.A.; Schweitzer, A.C.; Wickrema, A.; Conboy, J.G. Alternative pre-mRNA splicing switches modulate gene expression in late erythropoiesis. Blood 2009, 113, 3363–3370. [Google Scholar] [CrossRef]
- Shi, W.; Tang, J.; Xiang, J. Therapeutic strategies for aberrant splicing in cancer and genetic disorders. Clin. Genet. 2024, 105, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef]
- Deng, K.; Yao, J.; Huang, J.; Ding, Y.; Zuo, J. Abnormal alternative splicing promotes tumor resistance in targeted therapy and immunotherapy. Transl. Oncol. 2021, 14, 101077. [Google Scholar] [CrossRef] [PubMed]
- Kahles, A.; Lehmann, K.V.; Toussaint, N.C.; Hüser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Rätsch, G. Comprehensive Analysis of Alternative Splicing Across Tumors from 8705 Patients. Cancer Cell 2018, 34, 211–224.e6. [Google Scholar] [CrossRef]
- Zammarchi, F.; de Stanchina, E.; Bournazou, E.; Supakorndej, T.; Martires, K.; Riedel, E.; Corben, A.D.; Bromberg, J.F.; Cartegni, L. Antitumorigenic potential of STAT3 alternative splicing modulation. Proc. Natl. Acad. Sci. USA 2011, 108, 17779–17784. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yin, N.; Shi, W.; Xie, Y.; Yi, J.; Tang, Z.; Tang, J.; Xiang, J. Splicing inhibition mediated by reduced splicing factors and helicases is associated with the cellular response of lung cancer cells to cisplatin. Comput. Struct. Biotechnol. J. 2024, 23, 648–658. [Google Scholar] [CrossRef]
- Xie, N.; Shen, G.; Gao, W.; Huang, Z.; Huang, C.; Fu, L. Neoantigens: Promising targets for cancer therapy. Signal Transduct. Target. Ther. 2023, 8, 9. [Google Scholar] [CrossRef]
- Park, E.; Pan, Z.; Zhang, Z.; Lin, L.; Xing, Y. The Expanding Landscape of Alternative Splicing Variation in Human Populations. Am. J. Hum. Genet. 2018, 102, 11–26. [Google Scholar] [CrossRef]
- Cortés-López, M.; Schulz, L.; Enculescu, M.; Paret, C.; Spiekermann, B.; Quesnel-Vallières, M.; Torres-Diz, M.; Unic, S.; Busch, A.; Orekhova, A.; et al. High-throughput mutagenesis identifies mutations and RNA-binding proteins controlling CD19 splicing and CART-19 therapy resistance. Nat. Commun. 2022, 13, 5570. [Google Scholar] [CrossRef]
- Shlien, A.; Raine, K.; Fuligni, F.; Arnold, R.; Nik-Zainal, S.; Dronov, S.; Mamanova, L.; Rosic, A.; Ju, Y.S.; Cooke, S.L.; et al. Direct Transcriptional Consequences of Somatic Mutation in Breast Cancer. Cell Rep. 2016, 16, 2032–2046. [Google Scholar] [CrossRef]
- Luo, J.; Chen, C.; Liu, Z.; Wang, X. The mutation in splicing factor genes correlates with unfavorable prognosis, genomic instability, anti-tumor immunosuppression and increased immunotherapy response in pan-cancer. Front. Cell Dev. Biol. 2022, 10, 1045130. [Google Scholar] [CrossRef] [PubMed]
- Van Der Steen, N.; Giovannetti, E.; Pauwels, P.; Peters, G.J.; Hong, D.S.; Cappuzzo, F.; Hirsch, F.R.; Rolfo, C. cMET Exon 14 Skipping: From the Structure to the Clinic. J. Thorac. Oncol. 2016, 11, 1423–1432. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Kataoka, K.; Chiba, K.; Okada, A.; Kogure, Y.; Tanaka, H.; Ogawa, S.; Miyano, S. A comprehensive characterization of cis-acting splicing-associated variants in human cancer. Genome Res. 2018, 28, 1111–1125. [Google Scholar] [CrossRef] [PubMed]
- Ullah, F.; Jabeen, S.; Salton, M.; Reddy, A.S.N.; Ben-Hur, A. Evidence for the role of transcription factors in the co-transcriptional regulation of intron retention. Genome Biol. 2023, 24, 53. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.L.; Luo, G.; Wise, J.A.; Lou, H. Regulation of alternative splicing by local histone modifications: Potential roles for RNA-guided mechanisms. Nucleic Acids Res. 2014, 42, 701–713. [Google Scholar] [CrossRef]
- Bar-Ziv, R.; Voichek, Y.; Barkai, N. Chromatin dynamics during DNA replication. Genome Res. 2016, 26, 1245–1256. [Google Scholar] [CrossRef]
- Do, H.T.T.; Shanak, S.; Barghash, A.; Helms, V. Differential exon usage of developmental genes is associated with deregulated epigenetic marks. Sci. Rep. 2023, 13, 12256. [Google Scholar] [CrossRef]
- Yang, H.; Wang, Y.; Zhang, Y. Characterization of H3K9me3 and DNA methylation co-marked CpG-rich regions during mouse development. BMC Genomics 2023, 24, 663. [Google Scholar] [CrossRef]
- Batsché, E.; Yi, J.; Mauger, O.; Kornobis, E.; Hopkins, B.; Hanmer-Lloyd, C.; Muchardt, C. CD44 alternative splicing senses intragenic DNA methylation in tumors via direct and indirect mechanisms. Nucleic Acids Res. 2021, 49, 6213–6237. [Google Scholar] [CrossRef]
- Buratti, E.; Baralle, F.E. Influence of RNA secondary structure on the pre-mRNA splicing process. Mol. Cell Biol. 2004, 24, 10505–10514. [Google Scholar] [CrossRef]
- Zhu, Z.M.; Huo, F.C.; Zhang, J.; Shan, H.J.; Pei, D.S. Crosstalk between m6A modification and alternative splicing during cancer progression. Clin. Transl. Med. 2023, 13, e1460. [Google Scholar] [CrossRef]
- Zhao, B.; Xiang, Z.; Wu, B.; Zhang, X.; Feng, N.; Wei, Y.; Zhang, W. Use of Novel m6A Regulator-mediated Methylation Modification Patterns in Distinct Tumor Microenvironment Profiles to Identify and Predict Glioma Prognosis and Progression, T-cell Dysfunction, and Clinical Response to ICI Immunotherapy. Curr. Pharm. Des. 2023, 29, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Yan, Y.; Ren, Y.; Bao, Y.; Wang, Y. RNA splicing alterations in lung cancer pathogenesis and therapy. Cancer Pathog. Ther. 2023, 1, 272–283. [Google Scholar] [CrossRef]
- Alors-Perez, E.; Blázquez-Encinas, R.; Alcalá, S.; Viyuela-García, C.; Pedraza-Arevalo, S.; Herrero-Aguayo, V.; Jiménez-Vacas, J.M.; Mafficini, A.; Sánchez-Frías, M.E.; Cano, M.T.; et al. Dysregulated splicing factor SF3B1 unveils a dual therapeutic vulnerability to target pancreatic cancer cells and cancer stem cells with an anti-splicing drug. J. Exp. Clin. Cancer Res. 2021, 40, 382. [Google Scholar] [CrossRef]
- Jyotsana, N.; Heuser, M. Exploiting differential RNA splicing patterns: A potential new group of therapeutic targets in cancer. Expert Opin. Ther. Targets 2018, 22, 107–121. [Google Scholar] [CrossRef]
- Wen, W.X.; Mead, A.J.; Thongjuea, S. MARVEL: An integrated alternative splicing analysis platform for single-cell RNA sequencing data. Nucleic Acids Res. 2023, 51, e29. [Google Scholar] [CrossRef] [PubMed]
- Alsafadi, S.; Dayot, S.; Tarin, M.; Houy, A.; Bellanger, D.; Cornella, M.; Wassef, M.; Waterfall, J.J.; Lehnert, E.; Roman-Roman, S.; et al. Genetic alterations of SUGP1 mimic mutant-SF3B1 splice pattern in lung adenocarcinoma and other cancers. Oncogene 2021, 40, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi, Y.; Sugihara, Y.; Uneda, A.; Nakashima, T.; Suzuki, H. Recent advances in the molecular understanding of medulloblastoma. Cancer Sci. 2023, 114, 741–749. [Google Scholar] [CrossRef]
- Zhang, Z.; Huang, R.; Lai, Y. Expression signature of ten small nuclear RNAs serves as novel biomarker for prognosis prediction of acute myeloid leukemia. Sci. Rep. 2023, 13, 18489. [Google Scholar] [CrossRef]
- George, J.; Chen, Y.; Abdelfattah, N.; Yamamoto, K.; Gallup, T.D.; Adamson, S.I.; Rybinski, B.; Srivastava, A.; Kumar, P.; Lee, M.G.; et al. Cancer stem cells, not bulk tumor cells, determine mechanisms of resistance to SMO inhibitors. Cancer Res. Commun. 2022, 2, 402–416. [Google Scholar] [CrossRef] [PubMed]
- Eisemann, T.; Wechsler-Reya, R.J. Coming in from the cold: Overcoming the hostile immune microenvironment of medulloblastoma. Genes Dev. 2022, 36, 514–532. [Google Scholar] [CrossRef] [PubMed]
- Richard, S.A. The pivotal role of irradiation-induced apoptosis in the pathogenesis and therapy of medulloblastoma. Cancer Rep. 2024, 7, e2048. [Google Scholar] [CrossRef]
- Padariya, M.; Vojtesek, B.; Hupp, T.; Kalathiya, U. In Vitro Cross-Linking MS Reveals SMG1-UPF2-SMG7 Assembly as Molecular Partners within the NMD Surveillance. Int. J. Mol. Sci. 2024, 25, 3182. [Google Scholar] [CrossRef]
- Ge, Y.; Porse, B.T. The functional consequences of intron retention: Alternative splicing coupled to NMD as a regulator of gene expression. Bioessays 2014, 36, 236–243. [Google Scholar] [CrossRef]
- Seo, J.; Singh, N.N.; Ottesen, E.W.; Lee, B.M.; Singh, R.N. A novel human-specific splice isoform alters the critical C-terminus of Survival Motor Neuron protein. Sci. Rep. 2016, 6, 30778. [Google Scholar] [CrossRef] [PubMed]
- Usuki, F.; Fujimura, M.; Yamashita, A. Endoplasmic reticulum stress preconditioning modifies intracellular mercury content by upregulating membrane transporters. Sci. Rep. 2017, 7, 12390. [Google Scholar] [CrossRef]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.B.; Murphy, M.E.; et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef]
- Inoue, K.; Fry, E.A. Aberrant Splicing of Estrogen Receptor, HER2, and CD44 Genes in Breast Cancer. Genet. Epigenet. 2015, 7, 19–32. [Google Scholar] [CrossRef]
- Yae, T.; Tsuchihashi, K.; Ishimoto, T.; Motohara, T.; Yoshikawa, M.; Yoshida, G.J.; Wada, T.; Masuko, T.; Mogushi, K.; Tanaka, H.; et al. Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat. Commun. 2012, 3, 883. [Google Scholar] [CrossRef]
- Wallach-Dayan, S.B.; Rubinstein, A.M.; Hand, C.; Breuer, R.; Naor, D. DNA vaccination with CD44 variant isoform reduces mammary tumor local growth and lung metastasis. Mol. Cancer Ther. 2008, 7, 1615–1623. [Google Scholar] [CrossRef] [PubMed]
- Sandström, K.; Haylock, A.K.; Spiegelberg, D.; Qvarnström, F.; Wester, K.; Nestor, M. A novel CD44v6 targeting antibody fragment with improved tumor-to-blood ratio. Int. J. Oncol. 2012, 40, 1525–1532. [Google Scholar] [CrossRef] [PubMed]
- Chopra, A. Humanized anti-CD44v6 monoclonal antibody labeled with IRDye800CW. In Molecular Imaging and Contrast Agent Database (MICAD); National Center for Biotechnology Information: Bethesda, MD, USA, 2004. [Google Scholar]
- Lv, X.; Lu, X.; Cao, J.; Luo, Q.; Ding, Y.; Peng, F.; Pataer, A.; Lu, D.; Han, D.; Malmberg, E.; et al. Modulation of the proteostasis network promotes tumor resistance to oncogenic KRAS inhibitors. Science 2023, 381, eabn4180. [Google Scholar] [CrossRef]
- Whitley, M.J.; Tran, T.H.; Rigby, M.; Yi, M.; Dharmaiah, S.; Waybright, T.J.; Ramakrishnan, N.; Perkins, S.; Taylor, T.; Messing, S.; et al. Comparative analysis of KRAS4a and KRAS4b splice variants reveals distinctive structural and functional properties. Sci. Adv. 2024, 10, eadj4137. [Google Scholar] [CrossRef]
- Chen, W.C.; To, M.D.; Westcott, P.M.K.; Delrosario, R.; Kim, I.J.; Philips, M.; Tran, Q.; Bollam, S.R.; Goodarzi, H.; Bayani, N.; et al. Targeting KRAS4A splicing through the RBM39/DCAF15 pathway inhibits cancer stem cells. Nat. Commun. 2021, 12, 4288. [Google Scholar] [CrossRef]
- Yochum, Z.A.; Villaruz, L.C. Alternative splicing of HER2: A novel mediator of EGFR TKI resistance. Transl. Lung Cancer Res. 2020, 9, 1606–1612. [Google Scholar] [CrossRef] [PubMed]
- Weinholdt, C.; Wichmann, H.; Kotrba, J.; Ardell, D.H.; Kappler, M.; Eckert, A.W.; Vordermark, D.; Grosse, I. Prediction of regulatory targets of alternative isoforms of the epidermal growth factor receptor in a glioblastoma cell line. BMC Bioinform. 2019, 20, 434. [Google Scholar] [CrossRef]
- Takezawa, K.; Pirazzoli, V.; Arcila, M.E.; Nebhan, C.A.; Song, X.; de Stanchina, E.; Ohashi, K.; Janjigian, Y.Y.; Spitzler, P.J.; Melnick, M.A.; et al. HER2 amplification: A potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012, 2, 922–933. [Google Scholar] [CrossRef]
- Ray Das, S.; Delahunt, B.; Lasham, A.; Li, K.; Wright, D.; Print, C.; Slatter, T.; Braithwaite, A.; Mehta, S. Combining TP53 mutation and isoform has the potential to improve clinical practice. Pathology 2024, 56, 473–483. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, D.; Qin, X.; Owzar, K.; McCann, J.J.; Kastan, M.B. DNA-Damage-Induced Alternative Splicing of p53. Cancers 2021, 13, 251. [Google Scholar] [CrossRef]
- Antonio-Vejar, V.; Ortiz-Sanchez, E.; Rosendo-Chalma, P.; Patino-Morales, C.C.; Guido-Jimenez, M.C.; Alvarado-Ortiz, E.; Hernandez, G.; Garcia-Carranca, A. New insights into the interactions of HPV-16 E6*I and E6*II with p53 isoforms and induction of apoptosis in cancer-derived cell lines. Pathol. Res. Pract. 2022, 234, 153890. [Google Scholar] [CrossRef] [PubMed]
- Legscha, K.J.; Antunes, E.; Guezguez, B.; Theobald, M.; Echchannaoui, H. p53 Isoform D133p53a: A Novel Transcriptional Enhancer of T-Cell Effector Function to Improve T-Cell Based Cancer Immunotherapy. Blood Adv. 2018, 132 (Suppl. S1), 3489. [Google Scholar] [CrossRef]
- Ho, J.N.; Kang, G.Y.; Lee, S.S.; Kim, J.; Bae, I.H.; Hwang, S.G.; Um, H.D. Bcl-XL and STAT3 mediate malignant actions of gamma-irradiation in lung cancer cells. Cancer Sci. 2010, 101, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.K.; Alex, D.; Bosdet, I.; Hughesman, C.; Karsan, A.; Yip, S.; Ho, C. MET exon 14 skipping mutation positive non-small cell lung cancer: Response to systemic therapy. Lung Cancer 2021, 154, 142–145. [Google Scholar] [CrossRef]
- Warren, C.F.A.; Wong-Brown, M.W.; Bowden, N.A. BCL-2 family isoforms in apoptosis and cancer. Cell Death Dis. 2019, 10, 177. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Chen, X.; Shen, Y.; Lei, Y.; Mu, L.; Wang, Z.; Xiang, R.; Gao, W.; Wang, L.; Wang, L.; et al. A combinatorial therapeutic approach to enhance FLT3-ITD AML treatment. Cell Rep. Med. 2023, 4, 101286. [Google Scholar] [CrossRef]
- Zheng, L.; Luthra, R.; Alvarez, H.A.; San Lucas, F.A.; Duose, D.Y.; Wistuba, I.I.; Fuller, G.N.; Ballester, L.Y.; Roy-Chowdhuri, S.; Sweeney, K.J.; et al. Intragenic EGFR::EGFR.E1E8 Fusion (EGFRvIII) in 4331 Solid Tumors. Cancers 2023, 16, 6. [Google Scholar] [CrossRef]
- Babic, I.; Anderson, E.S.; Tanaka, K.; Guo, D.; Masui, K.; Li, B.; Zhu, S.; Gu, Y.; Villa, G.R.; Akhavan, D.; et al. EGFR mutation-induced alternative splicing of Max contributes to growth of glycolytic tumors in brain cancer. Cell Metab. 2013, 17, 1000–1008. [Google Scholar] [CrossRef]
- Chaudhri, R.A.; Schwartz, N.; Elbaradie, K.; Schwartz, Z.; Boyan, B.D. Role of ERα36 in membrane-associated signaling by estrogen. Steroids 2014, 81, 74–80. [Google Scholar] [CrossRef]
- Chantalat, E.; Boudou, F.; Laurell, H.; Palierne, G.; Houtman, R.; Melchers, D.; Rochaix, P.; Filleron, T.; Stella, A.; Burlet-Schiltz, O.; et al. The AF-1-deficient estrogen receptor ERα46 isoform is frequently expressed in human breast tumors. Breast Cancer Res. 2016, 18, 123. [Google Scholar] [CrossRef]
- Anamthathmakula, P.; Kyathanahalli, C.; Ingles, J.; Hassan, S.S.; Condon, J.C.; Jeyasuria, P. Estrogen receptor alpha isoform ERdelta7 in myometrium modulates uterine quiescence during pregnancy. eBioMedicine 2019, 39, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Ke, H.; Zhang, H.; Zou, L.; Yang, Q.; Lu, X.; Zhao, L.; Jiao, B. TDP43 promotes stemness of breast cancer stem cells through CD44 variant splicing isoforms. Cell Death Dis. 2022, 13, 428. [Google Scholar] [CrossRef] [PubMed]
- Eilertsen, I.A.; Sveen, A.; Strømme, J.M.; Skotheim, R.I.; Nesbakken, A.; Lothe, R.A. Alternative splicing expands the prognostic impact of KRAS in microsatellite stable primary colorectal cancer. Int. J. Cancer 2019, 144, 841–847. [Google Scholar] [CrossRef]
- Zhang, B.; Guo, D.D.; Zheng, J.Y.; Wu, Y.A. Expression of KLF6-SV2 in colorectal cancer and its impact on proliferation and apoptosis. Eur. J. Cancer Prev. 2018, 27, 20–26. [Google Scholar] [CrossRef]
- Giménez-Capitán, A.; Sánchez-Herrero, E.; Robado de Lope, L.; Aguilar-Hernández, A.; Sullivan, I.; Calvo, V.; Moya-Horno, I.; Viteri, S.; Cabrera, C.; Aguado, C.; et al. Detecting ALK, ROS1, and RET fusions and the METΔex14 splicing variant in liquid biopsies of non-small-cell lung cancer patients using RNA-based techniques. Mol. Oncol. 2023, 17, 1884–1897. [Google Scholar] [CrossRef]
- Hsu, C.C.; Liao, B.C.; Liao, W.Y.; Markovets, A.; Stetson, D.; Thress, K.; Yang, J.C. Exon 16-Skipping HER2 as a Novel Mechanism of Osimertinib Resistance in EGFR L858R/T790M-Positive Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2020, 15, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Gudikote, J.P.; Cascone, T.; Poteete, A.; Sitthideatphaiboon, P.; Wu, Q.; Morikawa, N.; Zhang, F.; Peng, S.; Tong, P.; Li, L.; et al. Inhibition of nonsense-mediated decay rescues p53β/γ isoform expression and activates the p53 pathway in MDM2-overexpressing and select p53-mutant cancers. J. Biol. Chem. 2021, 297, 101163. [Google Scholar] [CrossRef]
- Senturk, S.; Yao, Z.; Camiolo, M.; Stiles, B.; Rathod, T.; Walsh, A.M.; Nemajerova, A.; Lazzara, M.J.; Altorki, N.K.; Krainer, A.; et al. p53Ψ is a transcriptionally inactive p53 isoform able to reprogram cells toward a metastatic-like state. Proc. Natl. Acad. Sci. USA 2014, 111, E3287–E3296. [Google Scholar] [CrossRef]
- Joruiz, S.M.; Bourdon, J.C. p53 Isoforms: Key Regulators of the Cell Fate Decision. Cold Spring Harb. Perspect. Med. 2016, 6, a026039. [Google Scholar] [CrossRef]
- Le Sénéchal, R.; Keruzoré, M.; Quillévéré, A.; Loaëc, N.; Dinh, V.T.; Reznichenko, O.; Guixens-Gallardo, P.; Corcos, L.; Teulade-Fichou, M.P.; Granzhan, A.; et al. Alternative splicing of BCL-x is controlled by RBM25 binding to a G-quadruplex in BCL-x pre-mRNA. Nucleic Acids Res. 2023, 51, 11239–11257. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, S.; Qin, J.; Zhang, X.; Lu, G.; Liu, H.; Guo, H.; Wu, L.; Shender, V.O.; Shao, C.; et al. Splicing factor BUD31 promotes ovarian cancer progression through sustaining the expression of anti-apoptotic BCL2L12. Nat. Commun. 2022, 13, 6246. [Google Scholar] [CrossRef]
- Liang, W.C.; Wang, Y.; Xiao, L.J.; Wang, Y.B.; Fu, W.M.; Wang, W.M.; Jiang, H.Q.; Qi, W.; Wan, D.C.; Zhang, J.F.; et al. Identification of miRNAs that specifically target tumor suppressive KLF6-FL rather than oncogenic KLF6-SV1 isoform. RNA Biol. 2014, 11, 845–854. [Google Scholar] [CrossRef]
- Liu, X.; Zheng, Y.; Xiao, M.; Chen, X.; Zhu, Y.; Xu, C.; Wang, F.; Liu, Z.; Cao, K. SRSF10 stabilizes CDC25A by triggering exon 6 skipping to promote hepatocarcinogenesis. J. Exp. Clin. Cancer Res. 2022, 41, 353. [Google Scholar] [CrossRef]
- Sun, Y.; Xu, M.; Lee Wan, H.; Ding, X.; Wong, A.M.; Pu, D.; Ng, K.K.; Wong, N. Spliced exon9 ADRM1 promotes liver oncogenicity via selective degradation of tumor suppressor FBXW7. J. Hepatol. 2025. [Google Scholar] [CrossRef]
- Kwok, D.W.; Stevers, N.O.; Etxeberria, I.; Nejo, T.; Colton Cove, M.; Chen, L.H.; Jung, J.; Okada, K.; Lakshmanachetty, S.; Gallus, M.; et al. Tumour-wide RNA splicing aberrations generate actionable public neoantigens. Nature 2025, 639, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Dighe, A.; Maziarz, J.; Neumann, E.; Erkenbrack, E.; Hei, Y.Y.; Liu, Y.; Suhail, Y.; Pak, I.; Levchenko, A.; et al. Evolution of higher mesenchymal CD44 expression in the human lineage: A gene linked to cancer malignancy. Evol. Med. Public Health 2022, 10, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Elliott, K.; Nilsson, J.; Van den Eynden, J. Pharmacologic RNA splicing modulation: A novel mechanism to enhance neoantigen-directed anti-tumor immunity and immunotherapy response. Signal Transduct. Target. Ther. 2021, 6, 373. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.X.; De Neef, E.; Thomas, J.D.; Sabio, E.; Rousseau, B.; Gigoux, M.; Knorr, D.A.; Greenbaum, B.; Elhanati, Y.; Hogg, S.J.; et al. Pharmacologic modulation of RNA splicing enhances anti-tumor immunity. Cell 2021, 184, 4032–4047.e31. [Google Scholar] [CrossRef]
- Lo, A.; McSharry, M.; Berger, A.H. Oncogenic KRAS alters splicing factor phosphorylation and alternative splicing in lung cancer. BMC Cancer 2022, 22, 1315. [Google Scholar] [CrossRef]
- Lang, F.; Schrörs, B.; Löwer, M.; Türeci, Ö.; Sahin, U. Identification of neoantigens for individualized therapeutic cancer vaccines. Nat. Rev. Drug Discov. 2022, 21, 261–282. [Google Scholar] [CrossRef]
- Fuentes-Rodriguez, A.; Mitchell, A.; Guérin, S.L.; Landreville, S. Recent Advances in Molecular and Genetic Research on Uveal Melanoma. Cells 2024, 13, 1023. [Google Scholar] [CrossRef]
- Cai, Y.; Chen, R.; Gao, S.; Li, W.; Liu, Y.; Su, G.; Song, M.; Jiang, M.; Jiang, C.; Zhang, X. Artificial intelligence applied in neoantigen identification facilitates personalized cancer immunotherapy. Front. Oncol. 2022, 12, 1054231. [Google Scholar] [CrossRef] [PubMed]
- Peltier, D.C.; Roberts, A.; Reddy, P. LNCing RNA to immunity. Trends Immunol. 2022, 43, 478–495. [Google Scholar] [CrossRef]
- Pilger, D.; Seymour, L.W.; Jackson, S.P. Interfaces between cellular responses to DNA damage and cancer immunotherapy. Genes Dev. 2021, 35, 602–618. [Google Scholar] [CrossRef] [PubMed]
- Pelizzaro, F.; Farinati, F.; Trevisani, F. Immune Checkpoint Inhibitors in Hepatocellular Carcinoma: Current Strategies and Biomarkers Predicting Response and/or Resistance. Biomedicines 2023, 11, 1020. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Chi, H.; Gou, S.; Guo, X.; Li, L.; Peng, G.; Zhang, J.; Xu, J.; Nian, S.; Yuan, Q. An Aggrephagy-Related LncRNA Signature for the Prognosis of Pancreatic Adenocarcinoma. Genes 2023, 14, 124. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef]
- Liu, L.; Sun, J.; Zhong, C.; Zhang, A.; Wang, G.; Chen, S.; Zhang, S.; Wang, M.; Li, L. Identification of a fatty acid metabolism-related gene signature to predict prognosis in stomach adenocarcinoma. Aging 2024, 16, 8552–8571. [Google Scholar] [CrossRef]
- Zhang, Y.; Qian, J.; Gu, C.; Yang, Y. Alternative splicing and cancer: A systematic review. Signal Transduct. Target. Ther. 2021, 6, 78. [Google Scholar] [CrossRef]
- Advani, R.; Luzzi, S.; Scott, E.; Dalgliesh, C.; Weischenfeldt, J.; Munkley, J.; Elliott, D.J. Epithelial specific splicing regulator proteins as emerging oncogenes in aggressive prostate cancer. Oncogene 2023, 42, 3161–3168. [Google Scholar] [CrossRef]
- Pan, J.; Zhou, T.; Na, K.; Xu, K.; Yan, C.; Song, H.; Han, Y. Identification of hub modules and therapeutic targets associated with CD8(+)T-cells in HF and their pan-cancer analysis. Sci. Rep. 2024, 14, 18823. [Google Scholar] [CrossRef]
- Jiang, H.; Awuti, G.; Guo, X. Construction of an Immunophenoscore-Related Signature for Evaluating Prognosis and Immunotherapy Sensitivity in Ovarian Cancer. ACS Omega 2023, 8, 33017–33031. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Wang, P.; Li, Y.; Xu, J.; Yin, T.; Teng, F. Using MRI radiomics to predict the efficacy of immunotherapy for brain metastasis in patients with small cell lung cancer. Thorac. Cancer 2024, 15, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Wang, L.; Ma, P.; Ju, D.; Zhao, M.; Shi, Y. Enhancing anti-tumor immune responses through combination therapies: Epigenetic drugs and immune checkpoint inhibitors. Front. Immunol. 2023, 14, 1308264. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, L.; Sasso, E.; Feola, S.; Coluccino, L.; Vitale, M.; Leoni, G.; Szomolay, B.; Pastore, L.; Cerullo, V. Systems Biology Approaches for the Improvement of Oncolytic Virus-Based Immunotherapies. Cancers 2023, 15, 1297. [Google Scholar] [CrossRef]
- Boussaad, I.; Obermaier, C.D.; Hanss, Z.; Bobbili, D.R.; Bolognin, S.; Glaab, E.; Wołyńska, K.; Weisschuh, N.; De Conti, L.; May, C.; et al. A patient-based model of RNA mis-splicing uncovers treatment targets in Parkinson’s disease. Sci. Transl. Med. 2020, 12, eaau3960. [Google Scholar] [CrossRef]
- Matsushima, S.; Ajiro, M.; Iida, K.; Chamoto, K.; Honjo, T.; Hagiwara, M. Chemical induction of splice-neoantigens attenuates tumor growth in a preclinical model of colorectal cancer. Sci. Transl. Med. 2022, 14, eabn6056. [Google Scholar] [CrossRef]
- Gordon, E.D.; Simpson, L.J.; Rios, C.L.; Ringel, L.; Lachowicz-Scroggins, M.E.; Peters, M.C.; Wesolowska-Andersen, A.; Gonzalez, J.R.; MacLeod, H.J.; Christian, L.S.; et al. Alternative splicing of interleukin-33 and type 2 inflammation in asthma. Proc. Natl. Acad. Sci. USA 2016, 113, 8765–8770. [Google Scholar] [CrossRef]
- Ren, P.; Lu, L.; Cai, S.; Chen, J.; Lin, W.; Han, F. Alternative Splicing: A New Cause and Potential Therapeutic Target in Autoimmune Disease. Front. Immunol. 2021, 12, 713540. [Google Scholar] [CrossRef]
- Reiter, S.; Schroeder, C.; Broche, J.; Sinnberg, T.; Bonzheim, I.; Süsskind, D.; Flatz, L.; Forschner, A. Successful treatment of metastatic uveal melanoma with ipilimumab and nivolumab after severe progression under tebentafusp: A case report. Front. Oncol. 2023, 13, 1167791. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, H.; Xiang, T.; Wang, G. Clinical Application of Adaptive Immune Therapy in MSS Colorectal Cancer Patients. Front. Immunol. 2021, 12, 762341. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Joung, J.G.; Choi, Y.L.; Lee, S.H.; Park, B.J.; Choi, Y.S.; Ryu, D.; Nam, J.Y.; Lee, M.S.; Park, W.Y.; et al. Earlier-Phased Cancer Immunity Cycle Strongly Influences Cancer Immunity in Operable Never-Smoker Lung Adenocarcinoma. iScience 2020, 23, 101386. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020, 581, 100–105. [Google Scholar] [CrossRef]
- Müller, L.; Kschischo, M.; Vokuhl, C.; Stahl, D.; Gütgemann, I. Stemness Correlates Inversely with MHC Class I Expression in Pediatric Small Round Blue Cell Tumors. Cancers 2022, 14, 3584. [Google Scholar] [CrossRef]
- Fu, X.; Liu, S.; Cao, D.; Li, C.; Ji, H.; Wang, G. Med23 deficiency reprograms the tumor microenvironment to promote lung tumorigenesis. Br. J. Cancer 2024, 130, 716–727. [Google Scholar] [CrossRef]
- Niu, M.; Liu, Y.; Yi, M.; Jiao, D.; Wu, K. Biological Characteristics and Clinical Significance of Soluble PD-1/PD-L1 and Exosomal PD-L1 in Cancer. Front. Immunol. 2022, 13, 827921. [Google Scholar] [CrossRef] [PubMed]
- Maebele, L.T.; Mulaudzi, T.V.; Yasasve, M.; Dlamini, Z.; Damane, B.P. Immunomodulatory Gene-Splicing Dysregulation in Tumorigenesis: Unmasking the Complexity. Molecules 2023, 28, 5984. [Google Scholar] [CrossRef]
- Raza, A.; Mohsen, R.; Kanbour, A.; Zar Gul, A.R.; Philip, A.; Vijayakumar, S.; Hydrose, S.; Prabhu, K.S.; Al-Suwaidi, A.K.; Inchakalody, V.P.; et al. Serum immune mediators as novel predictors of response to anti-PD-1/PD-L1 therapy in non-small cell lung cancer patients with high tissue-PD-L1 expression. Front. Immunol. 2023, 14, 1157100. [Google Scholar] [CrossRef]
- Hassounah, N.B.; Malladi, V.S.; Huang, Y.; Freeman, S.S.; Beauchamp, E.M.; Koyama, S.; Souders, N.; Martin, S.; Dranoff, G.; Wong, K.K.; et al. Identification and characterization of an alternative cancer-derived PD-L1 splice variant. Cancer Immunol. Immunother. 2019, 68, 407–420. [Google Scholar] [CrossRef]
- Gong, B.; Kiyotani, K.; Sakata, S.; Nagano, S.; Kumehara, S.; Baba, S.; Besse, B.; Yanagitani, N.; Friboulet, L.; Nishio, M.; et al. Secreted PD-L1 variants mediate resistance to PD-L1 blockade therapy in non-small cell lung cancer. J. Exp. Med. 2019, 216, 982–1000. [Google Scholar] [CrossRef]
- Incorvaia, L.; Fanale, D.; Badalamenti, G.; Porta, C.; Olive, D.; De Luca, I.; Brando, C.; Rizzo, M.; Messina, C.; Rediti, M.; et al. Baseline plasma levels of soluble PD-1, PD-L1, and BTN3A1 predict response to nivolumab treatment in patients with metastatic renal cell carcinoma: A step toward a biomarker for therapeutic decisions. Oncoimmunology 2020, 9, 1832348. [Google Scholar] [CrossRef]
- Bernard, A.; Boidot, R.; Végran, F. Alternative Splicing in Cancer and Immune Cells. Cancers 2022, 14, 1726. [Google Scholar] [CrossRef]
- Dursun, H.G.; Yılmaz, H.O.; Dursun, R.; Kulaksızoğlu, S. Association of Cytotoxic T Lymphocyte Antigen-4 Gene Polymorphisms with Psoriasis Vulgaris: A Case-Control Study in Turkish Population. J. Immunol. Res. 2018, 2018, 1643906. [Google Scholar] [CrossRef]
- Zhao, C.; Zhao, J.W.; Zhang, Y.H.; Zhu, Y.D.; Yang, Z.Y.; Liu, S.L.; Tang, Q.Y.; Yang, Y.; Wang, H.K.; Shu, Y.J.; et al. PTBP3 Mediates IL-18 Exon Skipping to Promote Immune Escape in Gallbladder Cancer. Adv. Sci. 2024, 11, e2406633. [Google Scholar] [CrossRef]
- Angarola, B.L.; Anczuków, O. Splicing alterations in healthy aging and disease. Wiley Interdiscip. Rev. RNA 2021, 12, e1643. [Google Scholar] [CrossRef]
- Martinez, N.M.; Lynch, K.W. Control of alternative splicing in immune responses: Many regulators, many predictions, much still to learn. Immunol. Rev. 2013, 253, 216–236. [Google Scholar] [CrossRef]
- Leowattana, W.; Leowattana, P.; Leowattana, T. Systemic treatments for resectable carcinoma of the esophagus. World J. Gastroenterol. 2023, 29, 4628–4641. [Google Scholar] [CrossRef]
- Furmanski, A.L.; Barbarulo, A.; Solanki, A.; Lau, C.I.; Sahni, H.; Saldana, J.I.; D’Acquisto, F.; Crompton, T. The transcriptional activator Gli2 modulates T-cell receptor signalling through attenuation of AP-1 and NFκB activity. J. Cell Sci. 2015, 128, 2085–2095. [Google Scholar] [CrossRef]
- Conserva, M.R.; Redavid, I.; Anelli, L.; Zagaria, A.; Tarantini, F.; Cumbo, C.; Tota, G.; Parciante, E.; Coccaro, N.; Minervini, C.F.; et al. IKAROS in Acute Leukemia: A Positive Influencer or a Mean Hater? Int. J. Mol. Sci. 2023, 24, 3282. [Google Scholar] [CrossRef]
- McNamee, E.N.; Korns Johnson, D.; Homann, D.; Clambey, E.T. Hypoxia and hypoxia-inducible factors as regulators of T cell development, differentiation, and function. Immunol. Res. 2013, 55, 58–70. [Google Scholar] [CrossRef]
- Du, J.; Wang, Q.; Yang, S.; Chen, S.; Fu, Y.; Spath, S.; Domeier, P.; Hagin, D.; Anover-Sombke, S.; Haouili, M.; et al. FOXP3 exon 2 controls T(reg) stability and autoimmunity. Sci. Immunol. 2022, 7, eabo5407. [Google Scholar] [CrossRef]
- Legrand, J.M.D.; Chan, A.L.; La, H.M.; Rossello, F.J.; Änkö, M.L.; Fuller-Pace, F.V.; Hobbs, R.M. DDX5 plays essential transcriptional and post-transcriptional roles in the maintenance and function of spermatogonia. Nat. Commun. 2019, 10, 2278. [Google Scholar] [CrossRef] [PubMed]
- Roganowicz, M.; Bär, D.; Bersaglieri, C.; Aprigliano, R.; Santoro, R. BAZ2A-RNA mediated association with TOP2A and KDM1A represses genes implicated in prostate cancer. Life Sci. Alliance 2023, 6, e202301950. [Google Scholar] [CrossRef]
- Mailer, R.K.; Joly, A.L.; Liu, S.; Elias, S.; Tegner, J.; Andersson, J. IL-1beta promotes Th17 differentiation by inducing alternative splicing of FOXP3. Sci. Rep. 2015, 5, 14674. [Google Scholar] [CrossRef] [PubMed]
- Ding, R.; Yu, X.; Hu, Z.; Dong, Y.; Huang, H.; Zhang, Y.; Han, Q.; Ni, Z.Y.; Zhao, R.; Ye, Y.; et al. Lactate modulates RNA splicing to promote CTLA-4 expression in tumor-infiltrating regulatory T cells. Immunity 2024, 57, 528–540.e6. [Google Scholar] [CrossRef] [PubMed]
- Marima, R.; Basera, A.; Miya, T.; Damane, B.P.; Kandhavelu, J.; Mirza, S.; Penny, C.; Dlamini, Z. Exosomal long non-coding RNAs in cancer: Interplay, modulation, and therapeutic avenues. Noncoding RNA Res. 2024, 9, 887–900. [Google Scholar] [CrossRef]
- Lin, H.; Li, C.; Zhang, W.; Wu, B.; Wang, Y.; Wang, S.; Wang, D.; Li, X.; Huang, H. Synthetic Cells and Molecules in Cellular Immunotherapy. Int. J. Biol. Sci. 2024, 20, 2833–2859. [Google Scholar] [CrossRef]
- Hwang, A.; Mehra, V.; Chhetri, J.; Ali, S.; Tran, M.; Roddie, C. Current Treatment Options for Renal Cell Carcinoma: Focus on Cell-Based Immunotherapy. Cancers 2024, 16, 1209. [Google Scholar] [CrossRef]
- Yu, G.; Wang, W.; He, X.; Xu, J.; Xu, R.; Wan, T.; Wu, Y. Synergistic Therapeutic Effects of Low Dose Decitabine and NY-ESO-1 Specific TCR-T Cells for the Colorectal Cancer with Microsatellite Stability. Front. Oncol. 2022, 12, 895103. [Google Scholar] [CrossRef]
- Lu, F.; Ma, X.J.; Jin, W.L.; Luo, Y.; Li, X. Neoantigen Specific T Cells Derived from T Cell-Derived Induced Pluripotent Stem Cells for the Treatment of Hepatocellular Carcinoma: Potential and Challenges. Front. Immunol. 2021, 12, 690565. [Google Scholar] [CrossRef]
- Filin, I.Y.; Mayasin, Y.P.; Kharisova, C.B.; Gorodilova, A.V.; Kitaeva, K.V.; Chulpanova, D.S.; Solovyeva, V.V.; Rizvanov, A.A. Cell Immunotherapy against Melanoma: Clinical Trials Review. Int. J. Mol. Sci. 2023, 24, 2413. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, C.; Liu, C.; Shao, B.; Wang, D.; Wu, P. Exploring the Complexity and Promise of Tumor Immunotherapy in Drug Development. Int. J. Mol. Sci. 2024, 25, 6444. [Google Scholar] [CrossRef]
- Nejo, T.; Wang, L.; Leung, K.K.; Wang, A.; Lakshmanachetty, S.; Gallus, M.; Kwok, D.W.; Hong, C.; Chen, L.H.; Carrera, D.A.; et al. Challenges in the discovery of tumor-specific alternative splicing-derived cell-surface antigens in glioma. Sci. Rep. 2024, 14, 6362. [Google Scholar] [CrossRef] [PubMed]
- Choi, N.; Jang, H.N.; Oh, J.; Ha, J.; Park, H.; Zheng, X.; Lee, S.; Shen, H. SRSF6 Regulates the Alternative Splicing of the Apoptotic Fas Gene by Targeting a Novel RNA Sequence. Cancers 2022, 14, 1990. [Google Scholar] [CrossRef]
- Izquierdo, J.M.; Majós, N.; Bonnal, S.; Martínez, C.; Castelo, R.; Guigó, R.; Bilbao, D.; Valcárcel, J. Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol. Cell 2005, 19, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Takeiwa, T.; Ikeda, K.; Horie, K.; Inoue, S. Role of RNA binding proteins of the Drosophila behavior and human splicing (DBHS) family in health and cancer. RNA Biol. 2024, 21, 1–17. [Google Scholar] [CrossRef]
- Ji, C.; Bao, L.; Yuan, S.; Qi, Z.; Wang, F.; You, M.; Yu, G.; Liu, J.; Cui, X.; Wang, Z.; et al. SRSF1 Deficiency Impairs the Late Thymocyte Maturation and the CD8 Single-Positive Lineage Fate Decision. Front. Immunol. 2022, 13, 838719. [Google Scholar] [CrossRef]
- Lee, K.; Hyung, D.; Cho, S.Y.; Yu, N.; Hong, S.; Kim, J.; Kim, S.; Han, J.Y.; Park, C. Splicing signature database development to delineate cancer pathways using literature mining and transcriptome machine learning. Comput. Struct. Biotechnol. J. 2023, 21, 1978–1988. [Google Scholar] [CrossRef]
- Mickleburgh, I.; Kafasla, P.; Cherny, D.; Llorian, M.; Curry, S.; Jackson, R.J.; Smith, C.W. The organization of RNA contacts by PTB for regulation of FAS splicing. Nucleic Acids Res. 2014, 42, 8605–8620. [Google Scholar] [CrossRef]
- Chen, T.; Dai, X.; Dai, J.; Ding, C.; Zhang, Z.; Lin, Z.; Hu, J.; Lu, M.; Wang, Z.; Qi, Y.; et al. AFP promotes HCC progression by suppressing the HuR-mediated Fas/FADD apoptotic pathway. Cell Death Dis. 2020, 11, 822. [Google Scholar] [CrossRef]
- Gu, Z.; Yang, Y.; Ma, Q.; Wang, H.; Zhao, S.; Qi, Y.; Li, Y. HNRNPC, a predictor of prognosis and immunotherapy response based on bioinformatics analysis, is related to proliferation and invasion of NSCLC cells. Respir. Res. 2022, 23, 362. [Google Scholar] [CrossRef] [PubMed]
- Soni, K.; Martínez-Lumbreras, S.; Sattler, M. Conformational Dynamics from Ambiguous Zinc Coordination in the RanBP2-Type Zinc Finger of RBM5. J. Mol. Biol. 2020, 432, 4127–4138. [Google Scholar] [CrossRef]
- Cheng, Y.; Li, L.; Wei, X.; Xu, F.; Huang, X.; Qi, F.; Zhang, Y.; Li, X. HNRNPC suppresses tumor immune microenvironment by activating Treg cells promoting the progression of prostate cancer. Cancer Sci. 2023, 114, 1830–1845. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, C.K.; Bailey, A.O.; Russell, W.K.; Garcia-Blanco, M.A. Inefficient recruitment of DDX39B impedes pre-spliceosome assembly on FOXP3 introns. RNA 2024, 30, 824–838. [Google Scholar] [CrossRef]
- Subramani, P.G.; Fraszczak, J.; Helness, A.; Estall, J.L.; Möröy, T.; Di Noia, J.M. Conserved role of hnRNPL in alternative splicing of epigenetic modifiers enables B cell activation. EMBO Rep. 2024, 25, 2662–2697. [Google Scholar] [CrossRef] [PubMed]
- Szelest, M.; Giannopoulos, K. Biological relevance of alternative splicing in hematologic malignancies. Mol. Med. 2024, 30, 62. [Google Scholar] [CrossRef]
- Liu, Z.; Yoshimi, A.; Wang, J.; Cho, H.; Chun-Wei Lee, S.; Ki, M.; Bitner, L.; Chu, T.; Shah, H.; Liu, B.; et al. Mutations in the RNA Splicing Factor SF3B1 Promote Tumorigenesis through MYC Stabilization. Cancer Discov. 2020, 10, 806–821. [Google Scholar] [CrossRef]
- Tangye, S.G.; Nguyen, T.; Deenick, E.K.; Bryant, V.L.; Ma, C.S. Inborn errors of human B cell development, differentiation, and function. J. Exp. Med. 2023, 220, e20221105. [Google Scholar] [CrossRef]
- Escherich, C.; Chen, W.; Miyamoto, S.; Namikawa, Y.; Yang, W.; Teachey, D.T.; Li, Z.; Raetz, E.A.; Larsen, E.; Devidas, M.; et al. Identification of TCF3 germline variants in pediatric B-cell acute lymphoblastic leukemia. Blood Adv. 2023, 7, 2177–2180. [Google Scholar] [CrossRef]
- Bruijnesteijn, J.; van der Wiel, M.K.H.; de Groot, N.; Otting, N.; de Vos-Rouweler, A.J.M.; Lardy, N.M.; de Groot, N.G.; Bontrop, R.E. Extensive Alternative Splicing of KIR Transcripts. Front. Immunol. 2018, 9, 2846. [Google Scholar] [CrossRef]
- Geng, G.; Xu, C.; Peng, N.; Li, Y.; Liu, J.; Wu, J.; Liang, J.; Zhu, Y.; Shi, L. PTBP1 is necessary for dendritic cells to regulate T-cell homeostasis and antitumour immunity. Immunology 2021, 163, 74–85. [Google Scholar] [CrossRef]
- Chen, W.; Geng, D.; Chen, J.; Han, X.; Xie, Q.; Guo, G.; Chen, X.; Zhang, W.; Tang, S.; Zhong, X. Roles and mechanisms of aberrant alternative splicing in melanoma—Implications for targeted therapy and immunotherapy resistance. Cancer Cell Int. 2024, 24, 101. [Google Scholar] [CrossRef] [PubMed]
- Ando, T.; Okamoto, K.; Ueda, Y.; Kataoka, N.; Shintani, T.; Yanamoto, S.; Miyauchi, M.; Kajiya, M. YAP/TAZ interacts with RBM39 to confer resistance against indisulam. Oncogenesis 2024, 13, 25. [Google Scholar] [CrossRef] [PubMed]
- Donisi, C.; Pretta, A.; Pusceddu, V.; Ziranu, P.; Lai, E.; Puzzoni, M.; Mariani, S.; Massa, E.; Madeddu, C.; Scartozzi, M. Immunotherapy and Cancer: The Multi-Omics Perspective. Int. J. Mol. Sci. 2024, 25, 3563. [Google Scholar] [CrossRef]
- Wang, B.; Han, Y.; Zhang, Y.; Zhao, Q.; Wang, H.; Wei, J.; Meng, L.; Xin, Y.; Jiang, X. Overcoming acquired resistance to cancer immune checkpoint therapy: Potential strategies based on molecular mechanisms. Cell Biosci. 2023, 13, 120. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Yang, X.; Chen, S.; Wang, J.; Fan, X.; Fu, S.; Yang, L. The predictive efficacy of tumor mutation burden in immunotherapy across multiple cancer types: A meta-analysis and bioinformatics analysis. Transl. Oncol. 2022, 20, 101375. [Google Scholar] [CrossRef]
- Bauman, J.A.; Li, S.D.; Yang, A.; Huang, L.; Kole, R. Anti-tumor activity of splice-switching oligonucleotides. Nucleic Acids Res. 2010, 38, 8348–8356. [Google Scholar] [CrossRef]
- Sen, S.; Talukdar, I.; Webster, N.J. SRp20 and CUG-BP1 modulate insulin receptor exon 11 alternative splicing. Mol. Cell Biol. 2009, 29, 871–880. [Google Scholar] [CrossRef]
- Khurshid, S.; Venkataramany, A.S.; Montes, M.; Kipp, J.F.; Roberts, R.D.; Wein, N.; Rigo, F.; Wang, P.Y.; Cripe, T.P.; Chandler, D.S. Employing splice-switching oligonucleotides and AAVrh74.U7 snRNA to target insulin receptor splicing and cancer hallmarks in osteosarcoma. Mol. Ther. Oncol. 2024, 32, 200908. [Google Scholar] [CrossRef]
- Xu, Y.; Spear, S.; Ma, Y.; Lorentzen, M.P.; Gruet, M.; McKinney, F.; Xu, Y.; Wickremesinghe, C.; Shepherd, M.R.; McNeish, I.; et al. Pharmacological depletion of RNA splicing factor RBM39 by indisulam synergizes with PARP inhibitors in high-grade serous ovarian carcinoma. Cell Rep. 2023, 42, 113307. [Google Scholar] [CrossRef]
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; Mazieres, J.; Viteri, S.; Senellart, H.; Van Meerbeeck, J.; et al. Tepotinib in Non-Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. N. Engl. J. Med. 2020, 383, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Calderon-Aparicio, A.; Wang, B.D. Prostate cancer: Alternatively spliced mRNA transcripts in tumor progression and their uses as therapeutic targets. Int. J. Biochem. Cell Biol. 2021, 141, 106096. [Google Scholar] [CrossRef] [PubMed]
- Mossmann, D.; Müller, C.; Park, S.; Ryback, B.; Colombi, M.; Ritter, N.; Weißenberger, D.; Dazert, E.; Coto-Llerena, M.; Nuciforo, S.; et al. Arginine reprograms metabolism in liver cancer via RBM39. Cell 2023, 186, 5068–5083.e23. [Google Scholar] [CrossRef]
- Huang, L.; Zeng, X.; Ma, H.; Yang, Y.; Akimoto, Y.; Wei, G.; Ni, T. Pan-Cancer Profiling of Intron Retention and Its Clinical Significance in Diagnosis and Prognosis. Cancers 2023, 15, 5689. [Google Scholar] [CrossRef]
- Li, G.; Iyer, B.; Prasath, V.B.S.; Ni, Y.; Salomonis, N. DeepImmuno: Deep learning-empowered prediction and generation of immunogenic peptides for T-cell immunity. Brief. Bioinform. 2021, 22, bbab160. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Park, J.W.; Lu, Z.X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef]
- Sterne-Weiler, T.; Weatheritt, R.J.; Best, A.J.; Ha, K.C.H.; Blencowe, B.J. Efficient and Accurate Quantitative Profiling of Alternative Splicing Patterns of Any Complexity on a Laptop. Mol. Cell 2018, 72, 187–200.e6. [Google Scholar] [CrossRef]
- Saraiva-Agostinho, N.; Barbosa-Morais, N.L. psichomics: Graphical application for alternative splicing quantification and analysis. Nucleic Acids Res. 2019, 47, e7. [Google Scholar] [CrossRef]
- Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2023. Nucleic Acids Res. 2023, 51, D18–D28. [CrossRef]
- Li, G.; Mahajan, S.; Ma, S.; Jeffery, E.D.; Zhang, X.; Bhattacharjee, A.; Venkatasubramanian, M.; Weirauch, M.T.; Miraldi, E.R.; Grimes, H.L.; et al. Splicing neoantigen discovery with SNAF reveals shared targets for cancer immunotherapy. Sci. Transl. Med. 2024, 16, eade2886. [Google Scholar] [CrossRef]
- Pan, Y.; Phillips, J.W.; Zhang, B.D.; Noguchi, M.; Kutschera, E.; McLaughlin, J.; Nesterenko, P.A.; Mao, Z.; Bangayan, N.J.; Wang, R.; et al. IRIS: Discovery of cancer immunotherapy targets arising from pre-mRNA alternative splicing. Proc. Natl. Acad. Sci. USA 2023, 120, e2221116120. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shamardani, K.; Babikir, H.; Catalan, F.; Nejo, T.; Chang, S.; Phillips, J.J.; Okada, H.; Diaz, A.A. The evolution of alternative splicing in glioblastoma under therapy. Genome Biol. 2021, 22, 48. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Huber, F.; Arnaud, M.; Kraemer, A.I.; Altimiras, E.R.; Michaux, J.; Taillandier-Coindard, M.; Chiffelle, J.; Murgues, B.; Gehret, T.; et al. Machine learning methods and harmonized datasets improve immunogenic neoantigen prediction. Immunity 2023, 56, 2650–2663.e6. [Google Scholar] [CrossRef]
- Yang, R.L.; Qian, G.L.; Wu, D.W.; Miao, J.K.; Yang, X.; Wu, B.Q.; Yan, Y.Q.; Li, H.B.; Mao, X.M.; He, J.; et al. A multicenter prospective study of next-generation sequencing-based newborn screening for monogenic genetic diseases in China. World J. Pediatr. 2023, 19, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Ngiam, K.Y. How data science and AI-based technologies impact genomics. Singap. Med. J. 2023, 64, 59–66. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, C.; Shao, M.W.; Chen, Y.L.; Liu, P.; Chen, G.Q. The roadmap of bioeconomy in China. Eng. Biol. 2022, 6, 71–81. [Google Scholar] [CrossRef]
- Hashemi, S.; Hashemi, S.E.; Lien, K.M.; Lamb, J.J. Molecular Microbial Community Analysis as an Analysis Tool for Optimal Biogas Production. Microorganisms 2021, 9, 1162. [Google Scholar] [CrossRef]
- Chokr, N.; Pine, A.B.; Bewersdorf, J.P.; Shallis, R.M.; Stahl, M.; Zeidan, A.M. Getting personal with myelodysplastic syndromes: Is now the right time? Expert Rev. Hematol. 2019, 12, 215–224. [Google Scholar] [CrossRef]
- Zhao, Y.C.; Ding, Y.Z.; Zhao, X.; Fu, G.W.; Huang, M.J.; Li, X.X.; Sun, Q.Q.; Kan, Y.B.; Li, J.; Wang, S.L.; et al. Role and Clinical Application of Metagenomic Next-Generation Sequencing in Immunocompromised Patients with Acute Respiratory Failure During Veno-Venous Extracorporeal Membrane Oxygenation. Front. Cell Infect. Microbiol. 2022, 12, 877205. [Google Scholar] [CrossRef]
- Peng, W.; Cui, S.; Song, C. One-time-pad cipher algorithm based on confusion mapping and DNA storage technology. PLoS ONE 2021, 16, e0245506. [Google Scholar] [CrossRef]
- Ma, J.; Xiang, Y.; Xiong, Y.; Lin, Z.; Xue, Y.; Mao, M.; Sun, L.; Zhou, Y.; Li, X.; Huang, Z. SMRT sequencing analysis reveals the full-length transcripts and alternative splicing patterns in Ananas comosus var. bracteatus. PeerJ 2019, 7, e7062. [Google Scholar] [CrossRef]
- Zhang, F.; Chen, J.Y. A method for identifying discriminative isoform-specific peptides for clinical proteomics application. BMC Genomics 2016, 17 (Suppl. 7), 522. [Google Scholar] [CrossRef]
- Liu, H.; Liu, H.; Wang, L.; Song, L.; Jiang, G.; Lu, Q.; Yang, T.; Peng, H.; Cai, R.; Zhao, X.; et al. Cochlear transcript diversity and its role in auditory functions implied by an otoferlin short isoform. Nat. Commun. 2023, 14, 3085. [Google Scholar] [CrossRef]
- Chang, J.; Zhang, Y.; Zhou, T.; Qiao, Q.; Shan, J.; Chen, Y.; Jiang, W.; Wang, Y.; Liu, S.; Wang, Y.; et al. RBM10 C761Y mutation induced oncogenic ASPM isoforms and regulated β-catenin signaling in cholangiocarcinoma. J. Exp. Clin. Cancer Res. 2024, 43, 104. [Google Scholar] [CrossRef] [PubMed]
- Su, T.; Hollas, M.A.R.; Fellers, R.T.; Kelleher, N.L. Identification of Splice Variants and Isoforms in Transcriptomics and Proteomics. Annu. Rev. Biomed. Data Sci. 2023, 6, 357–376. [Google Scholar] [CrossRef] [PubMed]
- Trincado, J.L.; Entizne, J.C.; Hysenaj, G.; Singh, B.; Skalic, M.; Elliott, D.J.; Eyras, E. SUPPA2: Fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 2018, 19, 40. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.I.; Knowles, D.A.; Humphrey, J.; Barbeira, A.N.; Dickinson, S.P.; Im, H.K.; Pritchard, J.K. Annotation-free quantification of RNA splicing using LeafCutter. Nat. Genet. 2018, 50, 151–158. [Google Scholar] [CrossRef]
- Hundal, J.; Kiwala, S.; McMichael, J.; Miller, C.A.; Xia, H.; Wollam, A.T.; Liu, C.J.; Zhao, S.; Feng, Y.Y.; Graubert, A.P.; et al. pVACtools: A Computational Toolkit to Identify and Visualize Cancer Neoantigens. Cancer Immunol. Res. 2020, 8, 409–420. [Google Scholar] [CrossRef]
- O’Donnell, T.J.; Rubinsteyn, A.; Laserson, U. MHCflurry 2.0: Improved Pan-Allele Prediction of MHC Class I-Presented Peptides by Incorporating Antigen Processing. Cell Syst. 2020, 11, 42–48.e7. [Google Scholar] [CrossRef]

{kind=link}
| Cancer | Target Gene | AS Isoform | Exon/Intron | AS Type | Function | Reference |
|---|---|---|---|---|---|---|
| Breast Cancer | ER | ERα36 | exon 7, 8 | Exon skipping, Inclusion | Resistance to endocrine therapies. | [90,91,92] |
| ERα46 | exon 1, exon 9 | |||||
| ERΔ7 | exon 7 | |||||
| HER2 | Δ16HER2 | exon 20 | Exon skipping | Increased transforming ability. | [69] | |
| CD44 | CD44v2-v10 | exon v2-v10 | Exon Inclusion | BC growth and metastasis. | [72,93] | |
| CD44v3-v10 | exon v3-v10 | |||||
| CD44v8-v10 | exon v8-v10 | |||||
| CD44v6 | exon v6 | |||||
| Colorectal Cancer | KRAS | KRAS4A | exon 4A | Exon Inclusion | Cancer stemness. | [94] |
| KRAS4B | exon 4A | Exon skipping | Response to endoplasmic reticulum stress. | |||
| KLF6 | KLF6-SV2 | exon 2 | Exon skipping | CRC cell proliferation and apoptosis. | [95] | |
| Lung Cancer | RBM4 | RBM4-S | exon3 | Exon skipping | Activation of the SRSF1-mTORC1 pathway promotes NSCLC cell growth. | [2] |
| MET | METΔex14 | exon 14 | Exon skipping | Oncogenic activity. | [96] | |
| HER2 | HER2D16 | exon 16 | Exon skipping | A mediator of osimertinib resistance in patients with metastatic EGFR-mutant NSCLC. | [97] | |
| TP53 | P53β | exon 9β | Exon Inclusion | Cellular senescence. | [98,99,100] | |
| p53γ | exon 9γ | Cell differentiation/Antioxidant response. | ||||
| p53Ψ | intron 6 | Alternative 3′ splicing | Epigenetic regulation. | |||
| Δ40p53 | intron 2 | Intron Retention | Apoptosis. | |||
| Δ133p53 | intron 4 | A novel transcriptional enhancer of T-cell effector function. | ||||
| Δ160p53 | intron 4 | Tumor cell migration and invasion. | ||||
| Bcl—X | Bcl-Xs | exon 2 | Alternative 5′ splicing | Resistance against chemotherapeutic agents. | [101] | |
| Hematological Malignancies | BCL—2 | BCL-2β | exon 3 | Exon skipping | Antiapoptotic. | [86] |
| Ovarian Cancer | BCL—2 | BCL2L12-L | exon 3 | Exon Inclusion | Apoptosis. | [102] |
| BCL2L12-S | exon 3 | Exon skipping | ||||
| Hepatocellular Carcinoma | KLF6 | KLF6-SV1 | exon 2 | Alternative 5′ splicing | Cancer metastasis, progression. | [103] |
| CDC25A | CDC25A ΔE6 | exon 6 | Exon skipping | HCC growth. | [104] | |
| ADRM1 | ADRM1-ΔEx9 | exon 9 | Exon skipping | Ubiquitin proteasome specificity. | [105] |
| Splicing Factors | Types of Immune Cells | Affected Genes | References |
|---|---|---|---|
| SRSF1, SFPQ, CELF2 | T cell | Irf7, Il27ra, CD45 | [167,168,169] |
| PTB, HuR, hnRNPC, RBM5 | T cell | Fas, PD-L1, CTLA-4 | [170,171,172,173,174] |
| DDX39B | T cell | FOXP3 | [175] |
| hnRNPL | B cell | MYC, E2F | [176] |
| SF3B1 | B cell | BCL2, MYC | [177,178] |
| TCF3 | B cell | E12, E47 | [179,180] |
| KIR | NK cell | ___ | [181] |
| PTBP1 | DCs | MHC II | [182,183] |
| Technology/Database | Core Functions/ Technical Features | Advantages | Limitation | Complementary Technologies | Clinical Translational Value | References |
|---|---|---|---|---|---|---|
| rMATS | Quantifies 5 classical splicing events using ΔPSI | High sensitivity, standardized pipeline | Requires replicate samples, cannot resolve complex isoforms | Whippet | Initial screening of splicing events linked to high-frequency mutations | [197] |
| Whippet | Lightweight algorithm for high-entropy AS detection | Rapid single-sample analysis, covers 40% human genes | Low sensitivity for weakly expressed genes, lacks clinical data integration | PacBio SMRT | Large-scale screening of potential therapeutic targets | [198] |
| Psichomics | TCGA integration with Cox survival modeling | Direct patient prognosis association, target prioritization | Public database dependency, low flexibility | AS Cancer Atlas | Prognostic biomarker discovery | [199] |
| AS Cancer Atlas | Pan-cancer AS event database integrating TCGA/GTEx (33 cancer types) with survival-mutation links | Interactive visualization of splicing-clinical correlations | Infrequent updates, sparse data for rare cancers | Psichomics | Identification of pan-cancer splicing targets and therapy-response biomarkers | [200] |
| SNAF | DeepImmuno-CNN + BayesTS framework for splicing-derived neoantigens | High specificity (>85%), high shared antigen ratio (>90%) | RNA-seq coverage dependency, lacks MS validation | IRIS | Development of universal TCR therapies | [201] |
| IRIS | Differential expression + HLA-I affinity filtering | 30% reduced false positives, high verifiability | Requires matched normal samples, HLA typing constraints | ML | Personalized TCR therapy development | [202,203] |
| ML | XGBoost + immunogenicity classifier | 30% AUC improvement, strong generalizability | Data-hungry, computationally intensive | Orbitrap Astral MS | Enhanced personalized vaccine design | [204] |
| NGS | Short-read detection of high-frequency mutations/Differential genes | Cost-effective, standardized workflow | Misses splicing drivers | rMATS/Whippet | Foundational mutation profiling | [205,206,207,208,209,210,211] |
| PacBio SMRT | Long-read (10–15 kb) resolution of complex SVs | Error-free assembly, full-length isoform detection | High cost, low throughput | PacBio HiFi | Guidance for fusion protein targeting | [212] |
| PacBio HiFi | High-fidelity long-reads (≥99.9%) for rare isoform validation | 0.1 attomolar sensitivity | Large storage requirements | Orbitrap Astral MS | Enhanced neoantigen authenticity validation | [212] |
| Orbitrap Astral MS | DIA-MS detection of frameshift peptides | Antibody-free direct validation | Database dependency, low-abundance peptide challenges | Spectronaut | Confirmation of neoantigen presentation | [213,214,215,216] |
| SUPPA2 | AS analysis via transcript abundance (PSI calculation) | Single-sample compatibility, no replicates required | Limited by transcript reconstruction accuracy, low sensitivity for rare isoforms | Salmon | Rapid identification of prognosis-associated splicing events | [217] |
| LeafCutter | Reference-free splicing analysis using intron excision sites | Novel isoform discovery without exon annotation | High sequencing depth requirement, limited complex SV resolution | STAR | Detection of non-canonical splicing drivers | [218] |
| pVACtools | NetMHCpan + expression filtering + immunogenicity scoring | Open-source multi-threading support | Manual parameter tuning, lacks long-read integration | NeoPredPipe | Personalized vaccine candidate prioritization | [219] |
| MHCflurry2.0 | DL-based HLA-I/II affinity prediction | Covers >10,000 HLA alleles, cross-validation support | Reduced accuracy for rare HLA types | PrimeRank | Improved T-cell response prediction | [220] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Tang, J.; Xiang, J. Alternative Splicing in Tumorigenesis and Cancer Therapy. Biomolecules 2025, 15, 789. https://doi.org/10.3390/biom15060789
Chen H, Tang J, Xiang J. Alternative Splicing in Tumorigenesis and Cancer Therapy. Biomolecules. 2025; 15(6):789. https://doi.org/10.3390/biom15060789
Chicago/Turabian StyleChen, Huiping, Jingqun Tang, and Juanjuan Xiang. 2025. "Alternative Splicing in Tumorigenesis and Cancer Therapy" Biomolecules 15, no. 6: 789. https://doi.org/10.3390/biom15060789
APA StyleChen, H., Tang, J., & Xiang, J. (2025). Alternative Splicing in Tumorigenesis and Cancer Therapy. Biomolecules, 15(6), 789. https://doi.org/10.3390/biom15060789