
Identification of Splicing Regulatory Activity of ATXN1 and Its Associated Domains


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Cell Culture
2.3. Cellular Splicing Assay
2.4. SDS-PAGE and Western Blotting
2.5. Intracellular Localization of ATXN1
2.6. RNA Immunoprecipitation (RIP)
2.7. RNA Pull-Down Assay
2.8. Statistical Analysis
3. Results
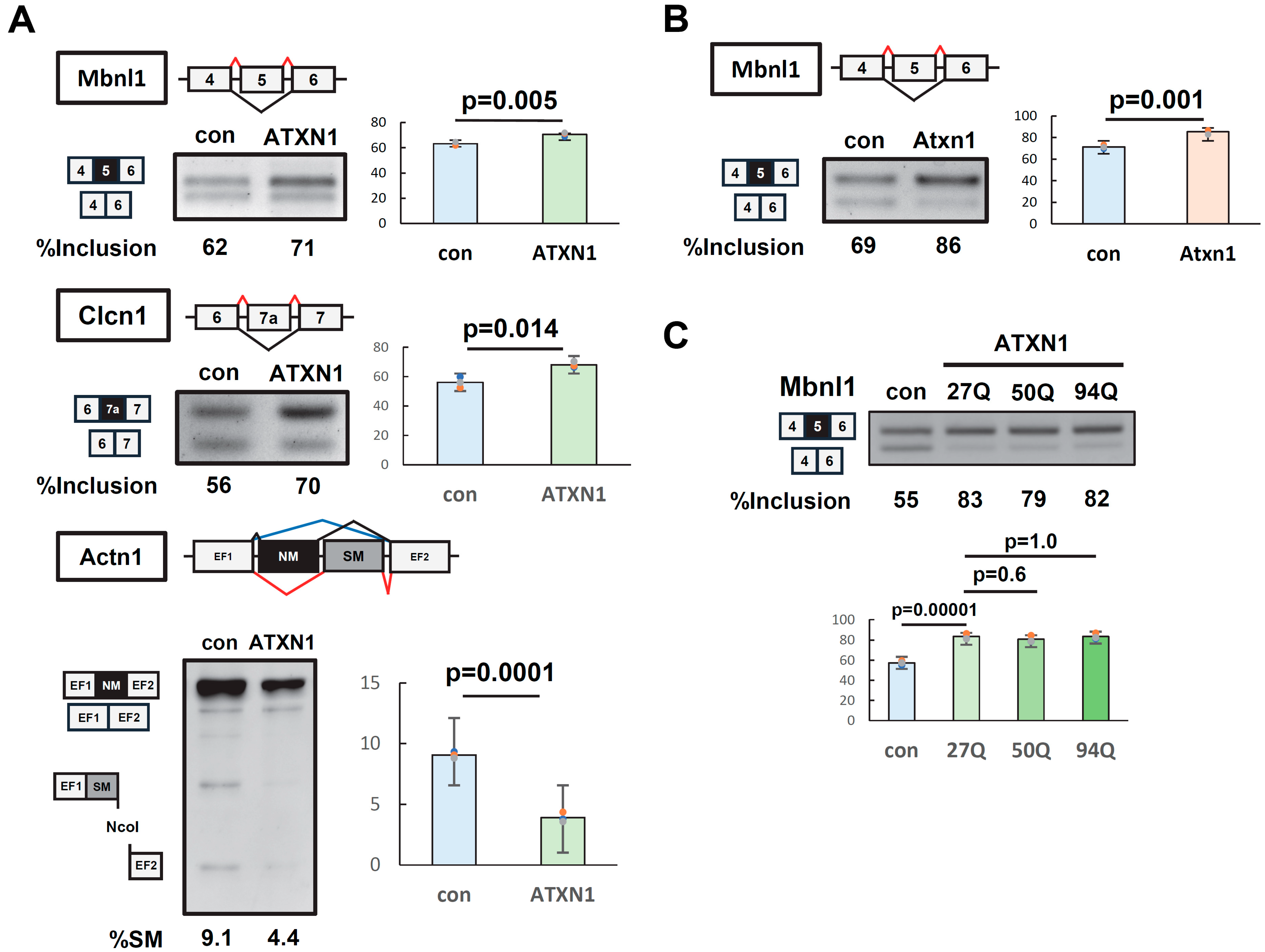
3.1. ATXN1 Regulates Alternative Splicing
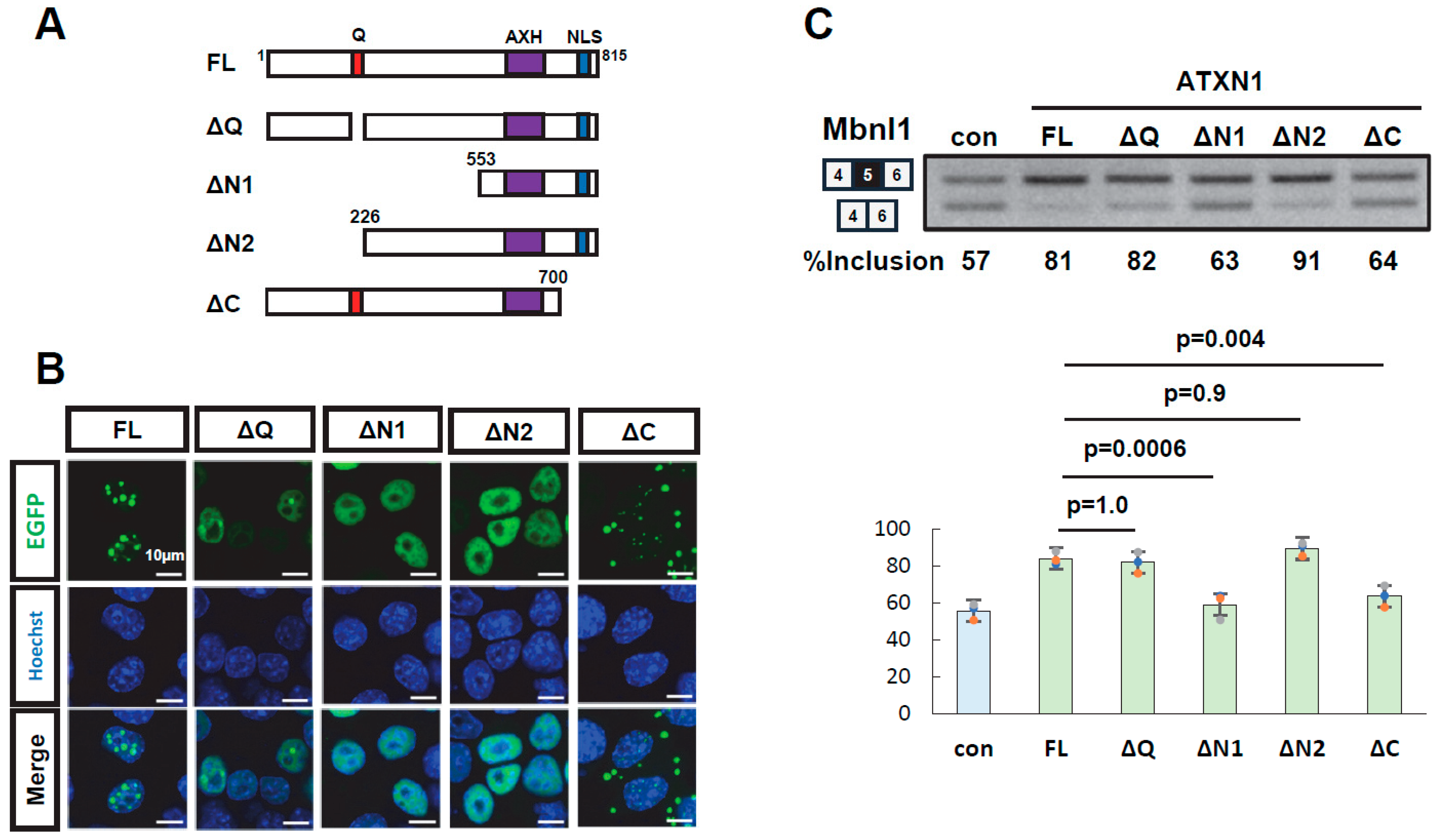
3.2. Multiple Regions of ATXN1 Are Involved in Its Splicing Regulatory Activity
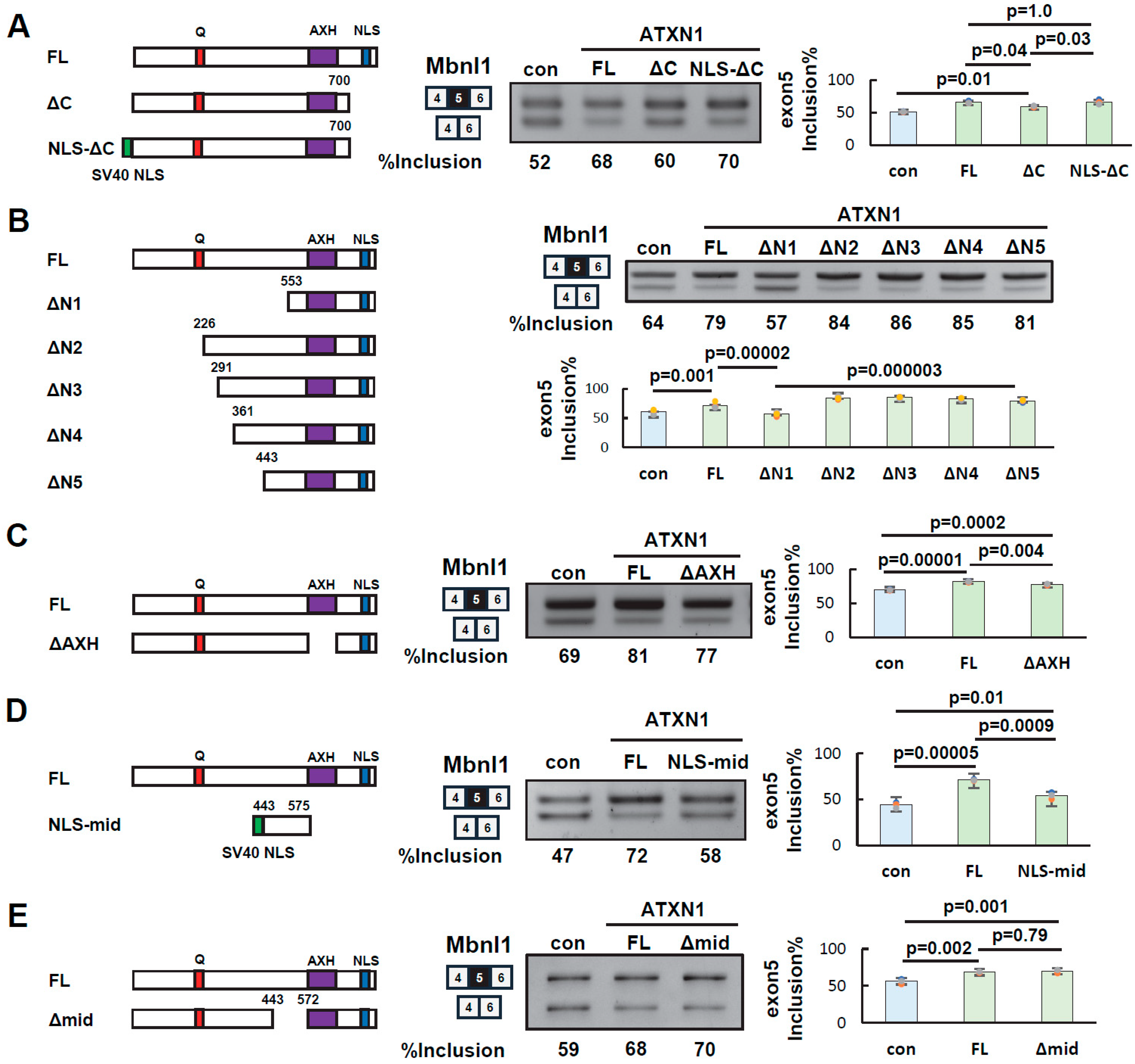
3.3. Identification of Splicing Regulatory Regions of ATXN1
3.4. Binding of ATXN1 Protein to RNA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AXH | ATXN1-HBP1 |
| EGFP | enhanced green fluorescence protein |
| NLS | nuclear localization signal |
| polyQ | polyglutamine |
| RIP | RNA immunoprecipitation |
| SCA1 | spinocerebellar ataxia type 1 |
References
- Chung, M.Y.; Ranum, L.P.; Duvick, L.A.; Servadio, A.; Zoghbi, H.Y.; Orr, H.T. Evidence for a mechanism predisposing to intergenerational CAG repeat instability in spinocerebellar ataxia type I. Nat. Genet. 1993, 5, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Nethisinghe, S.; Pigazzini, M.L.; Pemble, S.; Sweeney, M.G.; Labrum, R.; Manso, K.; Moore, D.; Warner, J.; Davis, M.B.; Giunti, P. PolyQ Tract Toxicity in SCA1 is Length Dependent in the Absence of CAG Repeat Interruption. Front. Cell. Neurosci. 2018, 12, 200. [Google Scholar] [CrossRef]
- Watase, K.; Weeber, E.J.; Xu, B.; Antalffy, B.; Yuva-Paylor, L.; Hashimoto, K.; Kano, M.; Atkinson, R.; Sun, Y.; Armstrong, D.L.; et al. A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron 2002, 34, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Burright, E.N.; Clark, H.B.; Servadio, A.; Matilla, T.; Feddersen, R.M.; Yunis, W.S.; Duvick, L.A.; Zoghbi, H.Y.; Orr, H.T. SCA1 transgenic mice: A model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell 1995, 82, 937–948. [Google Scholar] [CrossRef]
- Matilla, A.; Roberson, E.D.; Banfi, S.; Morales, J.; Armstrong, D.L.; Burright, E.N.; Orr, H.T.; Sweatt, J.D.; Zoghbi, H.Y.; Matzuk, M.M. Mice lacking ataxin-1 display learning deficits and decreased hippocampal paired-pulse facilitation. J. Neurosci. 1998, 18, 5508–5516. [Google Scholar] [CrossRef]
- Crespo-Barreto, J.; Fryer, J.D.; Shaw, C.A.; Orr, H.T.; Zoghbi, H.Y. Partial loss of ataxin-1 function contributes to transcriptional dysregulation in spinocerebellar ataxia type 1 pathogenesis. PLoS Genet. 2010, 6, e1001021. [Google Scholar] [CrossRef]
- Suh, J.; Romano, D.M.; Nitschke, L.; Herrick, S.P.; DiMarzio, B.A.; Dzhala, V.; Bae, J.S.; Oram, M.K.; Zheng, Y.; Hooli, B.; et al. Loss of Ataxin-1 Potentiates Alzheimer’s Pathogenesis by Elevating Cerebral BACE1 Transcription. Cell 2019, 178, 1159–1175.e1117. [Google Scholar] [CrossRef]
- Didonna, A.; Canto Puig, E.; Ma, Q.; Matsunaga, A.; Ho, B.; Caillier, S.J.; Shams, H.; Lee, N.; Hauser, S.L.; Tan, Q.; et al. Ataxin-1 regulates B cell function and the severity of autoimmune experimental encephalomyelitis. Proc. Natl. Acad. Sci. USA 2020, 117, 23742–23750. [Google Scholar] [CrossRef] [PubMed]
- Kang, A.R.; An, H.T.; Ko, J.; Choi, E.J.; Kang, S. Ataxin-1 is involved in tumorigenesis of cervical cancer cells via the EGFR-RAS-MAPK signaling pathway. Oncotarget 2017, 8, 94606–94618. [Google Scholar] [CrossRef]
- Xu, F.; Viaene, A.N.; Ruiz, J.; Schubert, J.; Wu, J.; Chen, J.; Cao, K.; Fu, W.; Bagatell, R.; Fan, Z.; et al. Novel ATXN1/ATXN1L::NUTM2A fusions identified in aggressive infant sarcomas with gene expression and methylation patterns similar to CIC-rearranged sarcoma. Acta Neuropathol. Commun. 2022, 10, 102. [Google Scholar] [CrossRef]
- Lam, Y.C.; Bowman, A.B.; Jafar-Nejad, P.; Lim, J.; Richman, R.; Fryer, J.D.; Hyun, E.D.; Duvick, L.A.; Orr, H.T.; Botas, J.; et al. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell 2006, 127, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Rousseaux, M.W.C.; Tschumperlin, T.; Lu, H.C.; Lackey, E.P.; Bondar, V.V.; Wan, Y.W.; Tan, Q.; Adamski, C.J.; Friedrich, J.; Twaroski, K.; et al. ATXN1-CIC Complex Is the Primary Driver of Cerebellar Pathology in Spinocerebellar Ataxia Type 1 through a Gain-of-Function Mechanism. Neuron 2018, 97, 1235–1243.e1235. [Google Scholar] [CrossRef]
- Klement, I.A.; Skinner, P.J.; Kaytor, M.D.; Yi, H.; Hersch, S.M.; Clark, H.B.; Zoghbi, H.Y.; Orr, H.T. Ataxin-1 nuclear localization and aggregation: Role in polyglutamine-induced disease in SCA1 transgenic mice. Cell 1998, 95, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Handler, H.P.; Duvick, L.; Mitchell, J.S.; Cvetanovic, M.; Reighard, M.; Soles, A.; Mather, K.B.; Rainwater, O.; Serres, S.; Nichols-Meade, T.; et al. Decreasing mutant ATXN1 nuclear localization improves a spectrum of SCA1-like phenotypes and brain region transcriptomic profiles. Neuron 2023, 111, 493–507.e496. [Google Scholar] [CrossRef]
- Duvick, L.; Barnes, J.; Ebner, B.; Agrawal, S.; Andresen, M.; Lim, J.; Giesler, G.J.; Zoghbi, H.Y.; Orr, H.T. SCA1-like disease in mice expressing wild-type ataxin-1 with a serine to aspartic acid replacement at residue 776. Neuron 2010, 67, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Crespo-Barreto, J.; Jafar-Nejad, P.; Bowman, A.B.; Richman, R.; Hill, D.E.; Orr, H.T.; Zoghbi, H.Y. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature 2008, 452, 713–718. [Google Scholar] [CrossRef]
- Yue, S.; Serra, H.G.; Zoghbi, H.Y.; Orr, H.T. The spinocerebellar ataxia type 1 protein, ataxin-1, has RNA-binding activity that is inversely affected by the length of its polyglutamine tract. Hum. Mol. Genet. 2001, 10, 25–30. [Google Scholar] [CrossRef]
- de Chiara, C.; Giannini, C.; Adinolfi, S.; de Boer, J.; Guida, S.; Ramos, A.; Jodice, C.; Kioussis, D.; Pastore, A. The AXH module: An independently folded domain common to ataxin-1 and HBP1. FEBS Lett. 2003, 551, 107–112. [Google Scholar] [CrossRef]
- Shorrock, H.K.; Lennon, C.D.; Aliyeva, A.; Davey, E.E.; DeMeo, C.C.; Pritchard, C.E.; Planco, L.; Velez, J.M.; Mascorro-Huamancaja, A.; Shin, D.S.; et al. Widespread alternative splicing dysregulation occurs presymptomatically in CAG expansion spinocerebellar ataxias. Brain 2024, 147, 486–504. [Google Scholar] [CrossRef]
- Aliyeva, A.; Lennon, C.D.; Cleary, J.D.; Shorrock, H.K.; Berglund, J.A. Dysregulation of alternative splicing is a transcriptomic feature of patient-derived fibroblasts from CAG repeat expansion spinocerebellar ataxias. Hum. Mol. Genet. 2025, 34, 239–250. [Google Scholar] [CrossRef]
- Kino, Y.; Washizu, C.; Oma, Y.; Onishi, H.; Nezu, Y.; Sasagawa, N.; Nukina, N.; Ishiura, S. MBNL and CELF proteins regulate alternative splicing of the skeletal muscle chloride channel CLCN1. Nucleic Acids Res. 2009, 37, 6477–6490. [Google Scholar] [CrossRef] [PubMed]
- Kino, Y.; Washizu, C.; Kurosawa, M.; Yamada, M.; Miyazaki, H.; Akagi, T.; Hashikawa, T.; Doi, H.; Takumi, T.; Hicks, G.G.; et al. FUS/TLS deficiency causes behavioral and pathological abnormalities distinct from amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2015, 3, 24. [Google Scholar] [CrossRef] [PubMed]
- Kino, Y.; Washizu, C.; Aquilanti, E.; Okuno, M.; Kurosawa, M.; Yamada, M.; Doi, H.; Nukina, N. Intracellular localization and splicing regulation of FUS/TLS are variably affected by amyotrophic lateral sclerosis-linked mutations. Nucleic Acids Res. 2011, 39, 2781–2798. [Google Scholar] [CrossRef]
- Yanaizu, M.; Washizu, C.; Nukina, N.; Satoh, J.I.; Kino, Y. CELF2 regulates the species-specific alternative splicing of TREM2. Sci. Rep. 2020, 10, 17995. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Yanaizu, M.; Ohki, A.; Nishimiya, K.; Kino, Y. CUG repeat RNA-dependent proteasomal degradation of MBNL1 in a cellular model of myotonic dystrophy type 1. Biochem. Biophys. Res. Commun. 2024, 733, 150729. [Google Scholar] [CrossRef]
- Komuro, R.; Honda, Y.; Yanaizu, M.; Nagahama, M.; Kino, Y. Alzheimer’s Disease-Associated Alternative Splicing of CD33 Is Regulated by the HNRNPA Family Proteins. Cells 2023, 12, 602. [Google Scholar] [CrossRef]
- Chen, J.M.; Chen, S.K.; Jin, P.P.; Sun, S.C. Identification of the ataxin-1 interaction network and its impact on spinocerebellar ataxia type 1. Hum. Genom. 2022, 16, 29. [Google Scholar] [CrossRef]
- Tsuda, H.; Jafar-Nejad, H.; Patel, A.J.; Sun, Y.; Chen, H.K.; Rose, M.F.; Venken, K.J.; Botas, J.; Orr, H.T.; Bellen, H.J.; et al. The AXH domain of Ataxin-1 mediates neurodegeneration through its interaction with Gfi-1/Senseless proteins. Cell 2005, 122, 633–644. [Google Scholar] [CrossRef]
- Kim, E.; Lee, Y.; Choi, S.; Song, J.J. Structural basis of the phosphorylation dependent complex formation of neurodegenerative disease protein Ataxin-1 and RBM17. Biochem. Biophys. Res. Commun. 2014, 449, 399–404. [Google Scholar] [CrossRef]
- Olmos, V.; Thompson, E.N.; Gogia, N.; Luttik, K.; Veeranki, V.; Ni, L.; Sim, S.; Chen, K.; Krause, D.S.; Lim, J. Dysregulation of alternative splicing in spinocerebellar ataxia type 1. Hum. Mol. Genet. 2024, 33, 138–149. [Google Scholar] [CrossRef]
- Lai, S.; O’Callaghan, B.; Zoghbi, H.Y.; Orr, H.T. 14-3-3 Binding to ataxin-1(ATXN1) regulates its dephosphorylation at Ser-776 and transport to the nucleus. J. Biol. Chem. 2011, 286, 34606–34616. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Hao, T.; Shaw, C.; Patel, A.J.; Szabó, G.; Rual, J.F.; Fisk, C.J.; Li, N.; Smolyar, A.; Hill, D.E.; et al. A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell 2006, 125, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Hinde, E.; Parkyn Schneider, M.; Jans, D.A.; Bogoyevitch, M.A. Nuclear bodies formed by polyQ-ataxin-1 protein are liquid RNA/protein droplets with tunable dynamics. Sci. Rep. 2020, 10, 1557. [Google Scholar] [CrossRef]
- Gkekas, I.; Vagiona, A.C.; Pechlivanis, N.; Kastrinaki, G.; Pliatsika, K.; Iben, S.; Xanthopoulos, K.; Psomopoulos, F.E.; Andrade-Navarro, M.A.; Petrakis, S. Intranuclear inclusions of polyQ-expanded ATXN1 sequester RNA molecules. Front. Mol. Neurosci. 2023, 16, 1280546. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohki, A.; Kato, M.; Aoki, Y.; Kubokawa, A.; Yanaizu, M.; Kino, Y. Identification of Splicing Regulatory Activity of ATXN1 and Its Associated Domains. Biomolecules 2025, 15, 782. https://doi.org/10.3390/biom15060782
Ohki A, Kato M, Aoki Y, Kubokawa A, Yanaizu M, Kino Y. Identification of Splicing Regulatory Activity of ATXN1 and Its Associated Domains. Biomolecules. 2025; 15(6):782. https://doi.org/10.3390/biom15060782
Chicago/Turabian StyleOhki, Ai, Masahide Kato, Yoshitaka Aoki, Arisa Kubokawa, Motoaki Yanaizu, and Yoshihiro Kino. 2025. "Identification of Splicing Regulatory Activity of ATXN1 and Its Associated Domains" Biomolecules 15, no. 6: 782. https://doi.org/10.3390/biom15060782
APA StyleOhki, A., Kato, M., Aoki, Y., Kubokawa, A., Yanaizu, M., & Kino, Y. (2025). Identification of Splicing Regulatory Activity of ATXN1 and Its Associated Domains. Biomolecules, 15(6), 782. https://doi.org/10.3390/biom15060782