
The Role of Long-Range Non-Specific Electrostatic Interactions in Inhibiting the Pre-Fusion Proteolytic Processing of the SARS-CoV-2 S Glycoprotein by Heparin

Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Biologicals
2.2. Mass Spectrometry (MS)
2.3. Molecular Modeling
3. Results
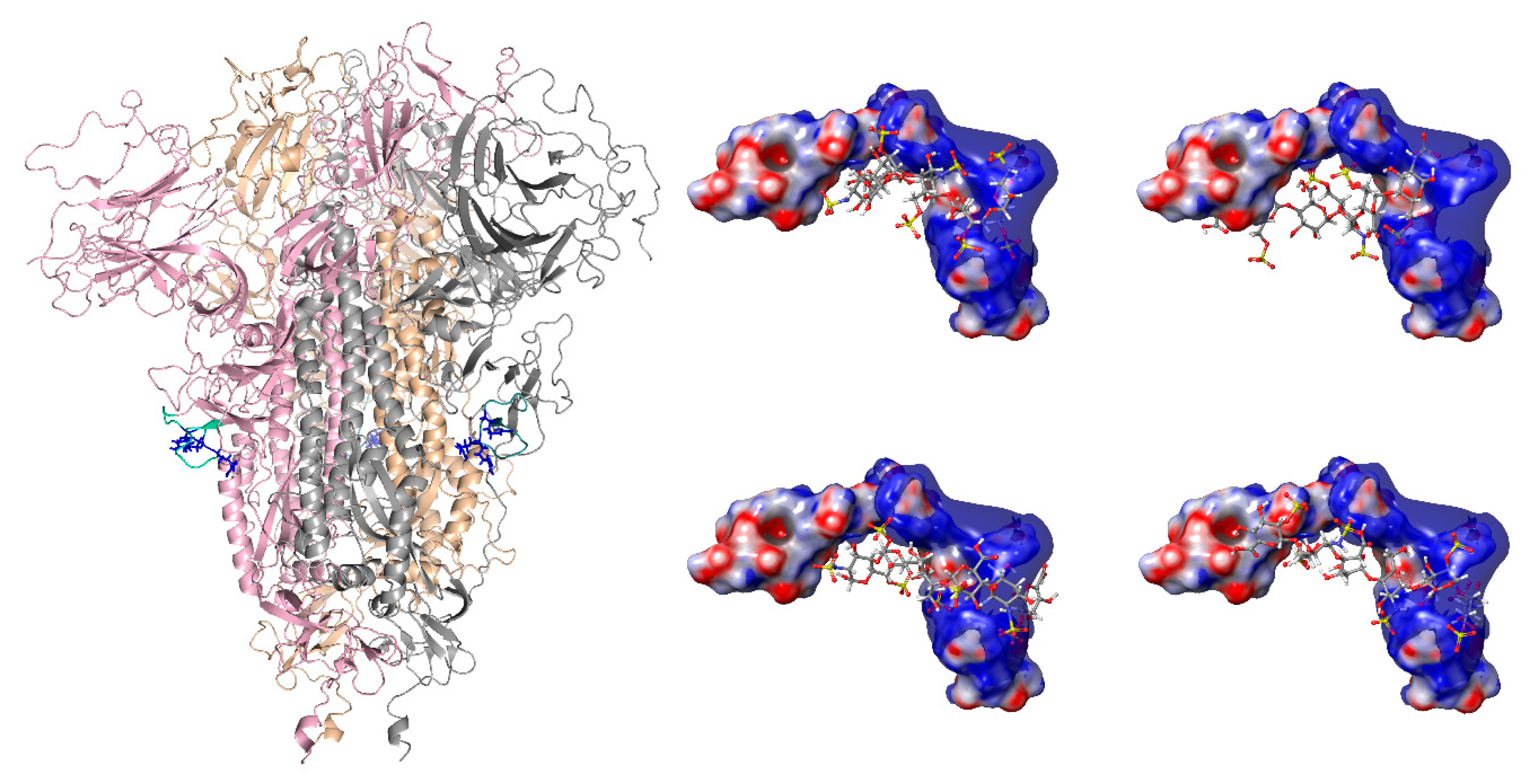
3.1. Molecular Modeling of the Interaction Between Heparin Fragments and the Furin Cleavage Site of the SARS-CoV-2 S Glycoprotein
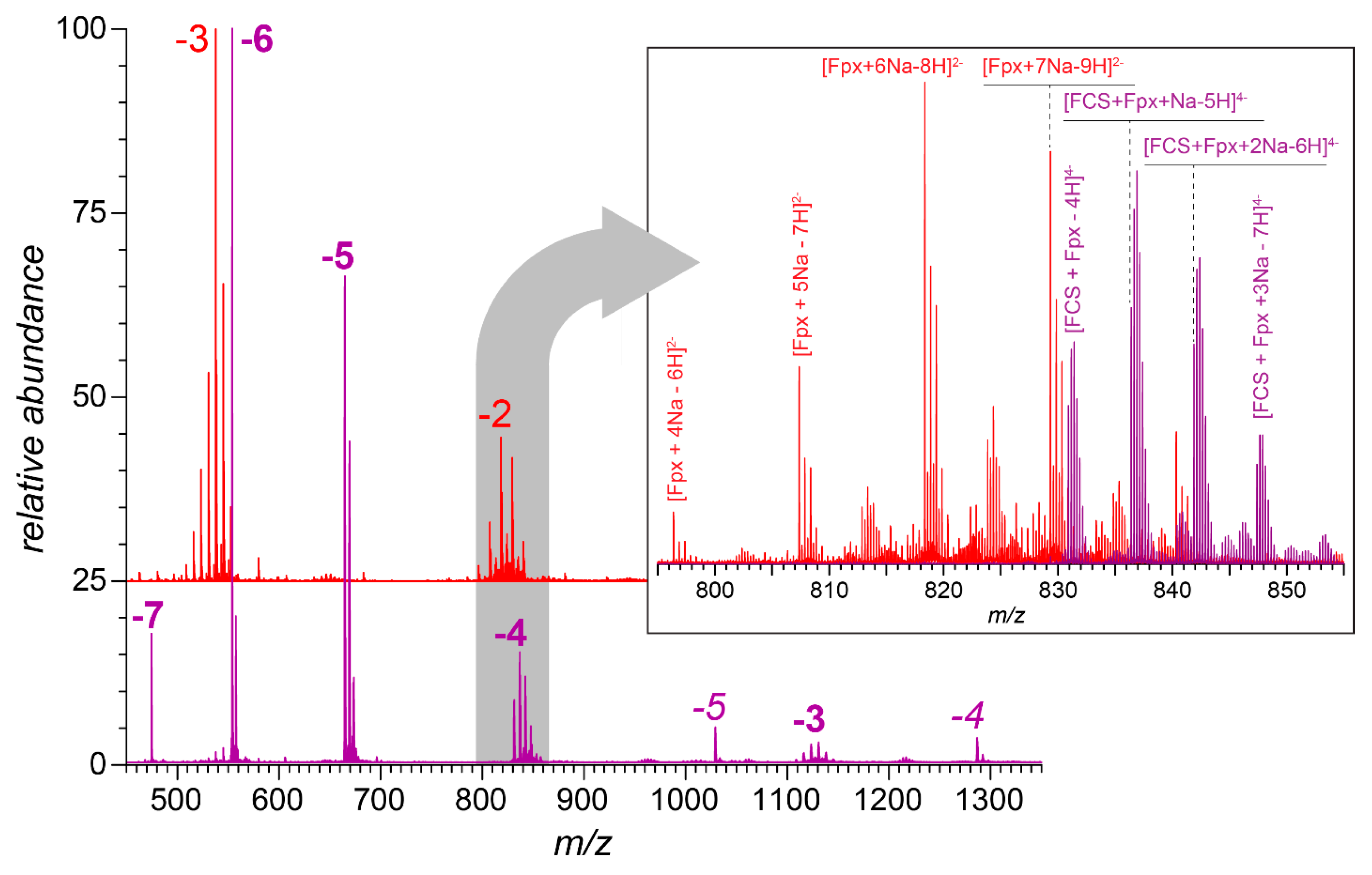
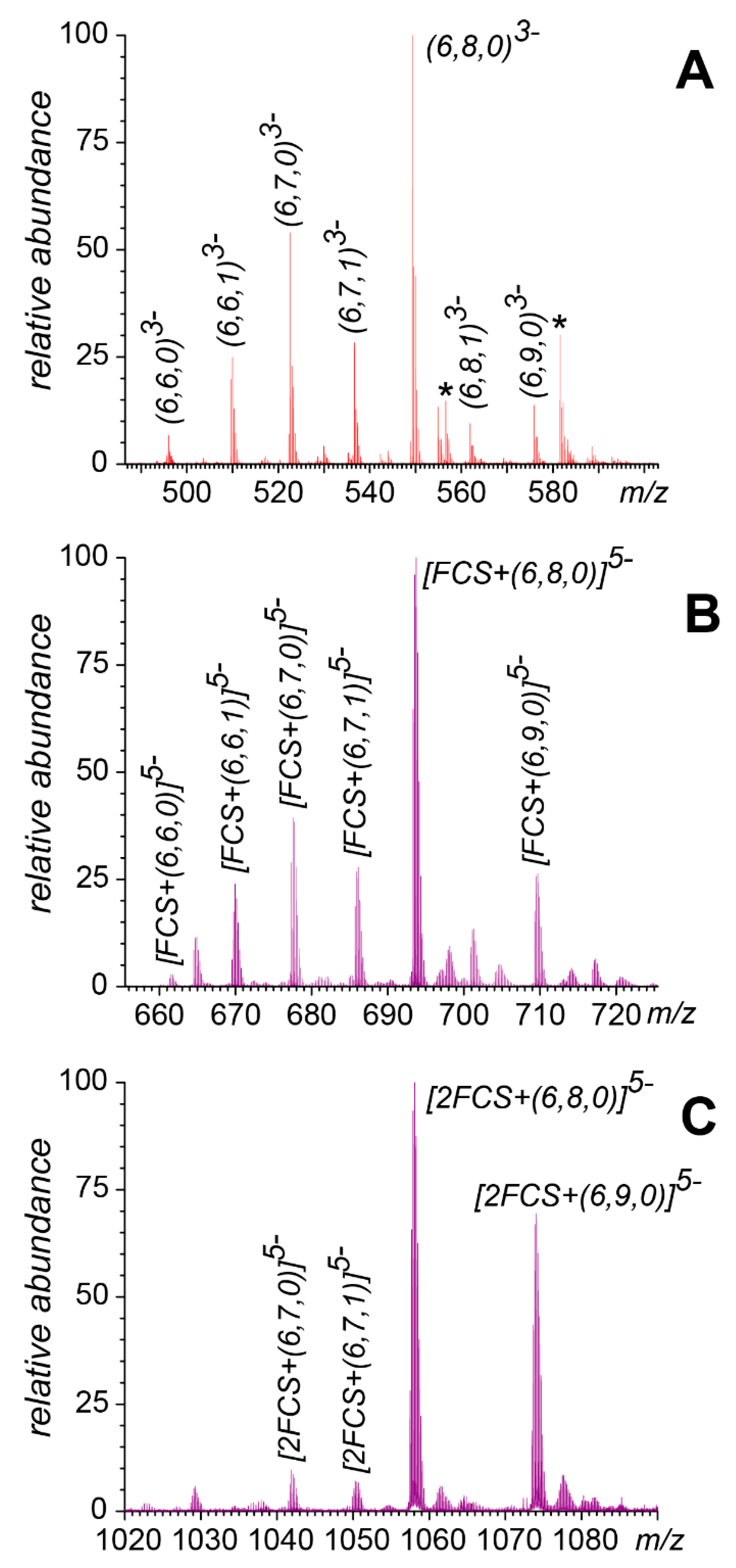
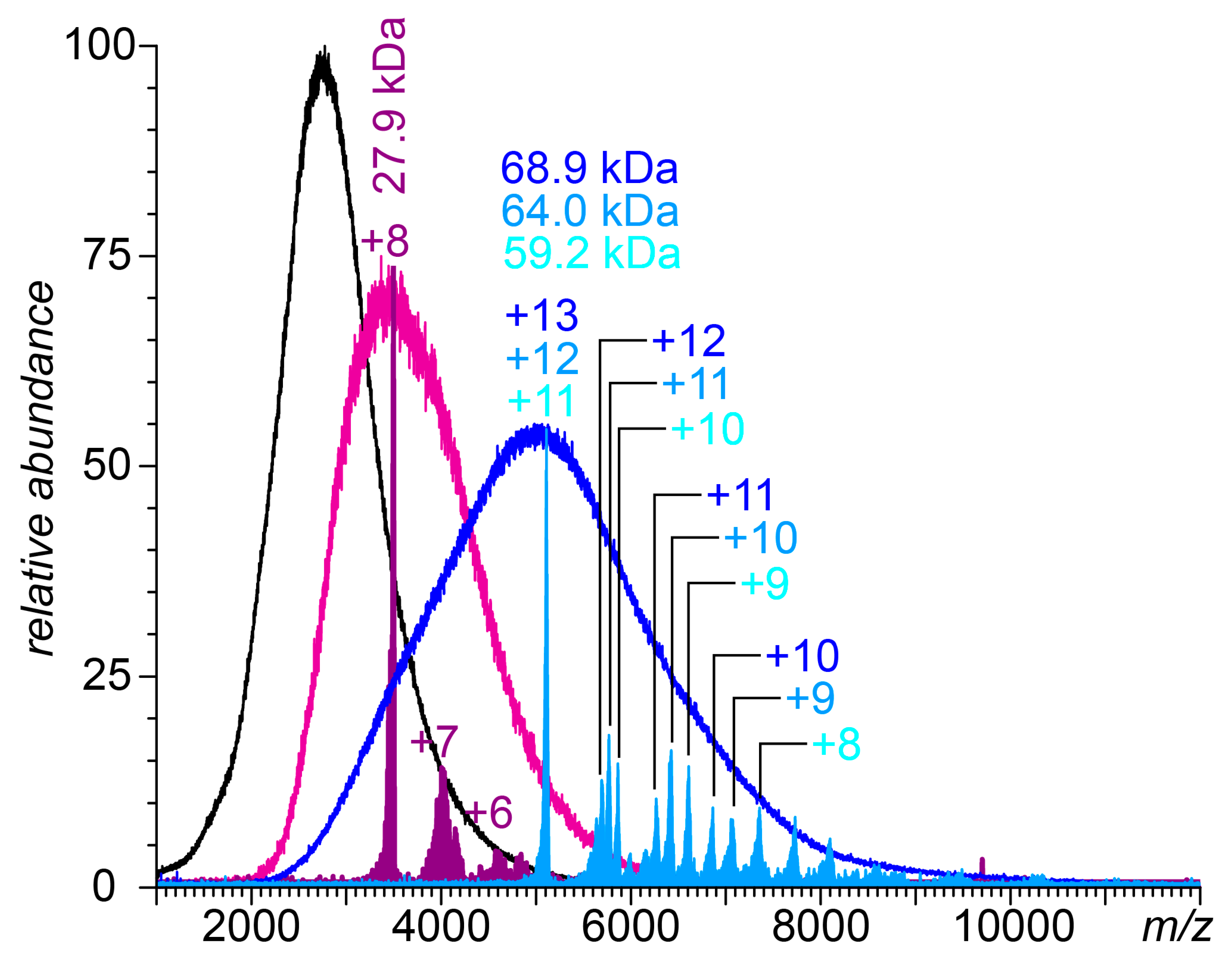
3.2. Native MS of FCS Interactions with Short-Chain GAGs
3.3. Native MS of FCS Interactions with UFH
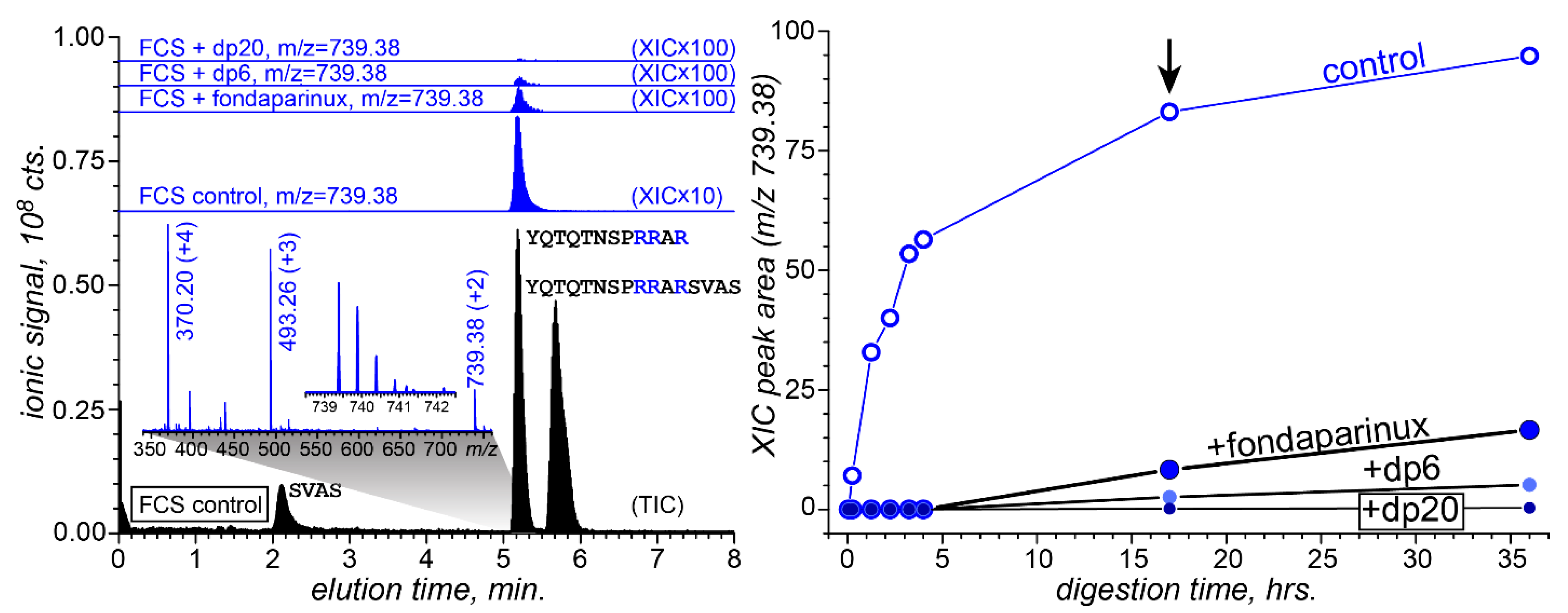
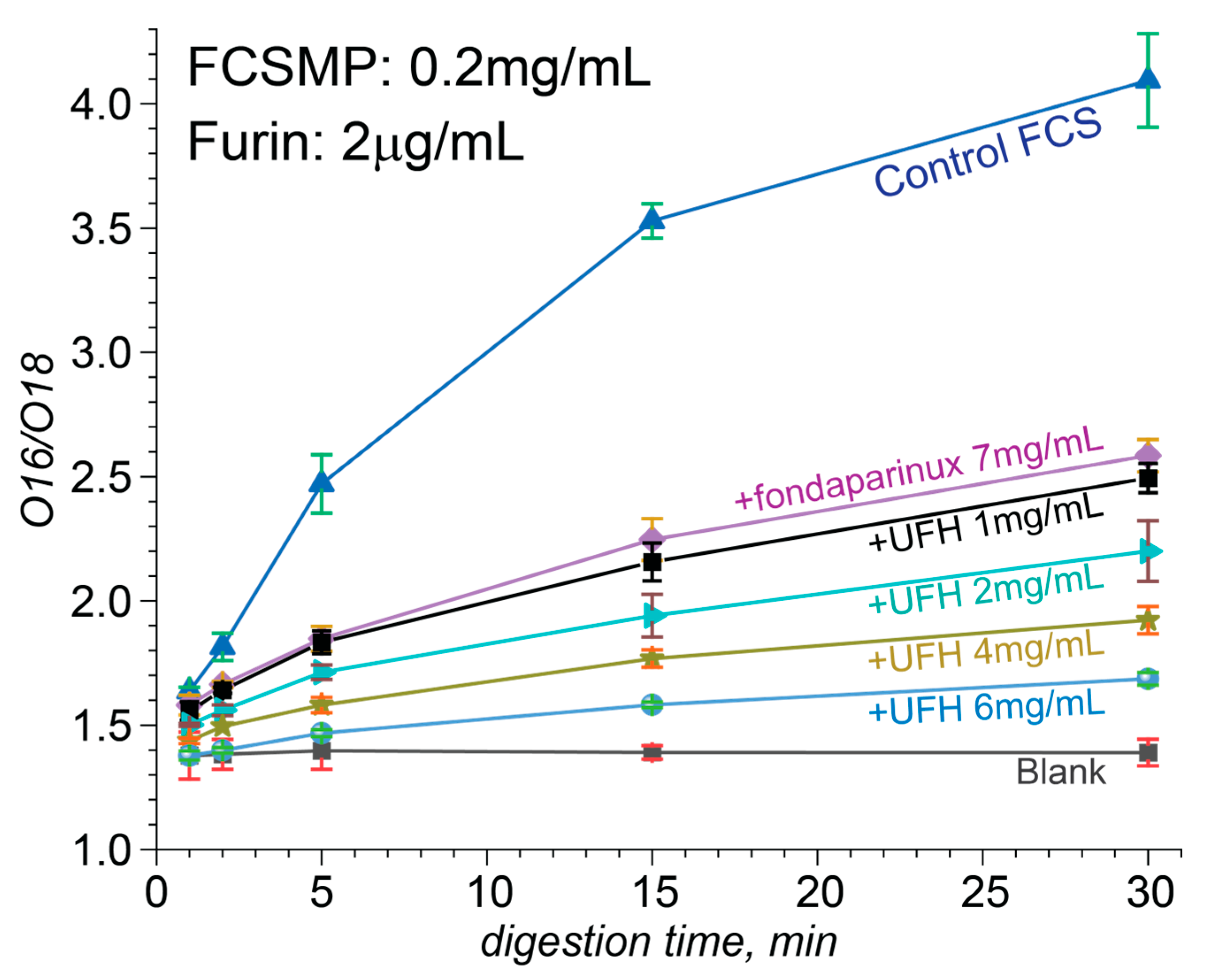
3.4. Attenuation of FCS Proteolysis with Furin by Fondaparinux, Fixed-Length Heparin Fragments, and UFH
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carvajal, J.J.; García-Castillo, V.; Cuellar, S.V.; Campillay-Véliz, C.P.; Salazar-Ardiles, C.; Avellaneda, A.M.; Muñoz, C.A.; Retamal-Díaz, A.; Bueno, S.M.; González, P.A.; et al. New insights into the pathogenesis of SARS-CoV-2 during and after the COVID-19 pandemic. Front. Immunol. 2024, 15, 1363572. [Google Scholar] [CrossRef] [PubMed]
- Maison, D.P.; Tasissa, H.; Deitchman, A.; Peluso, M.J.; Deng, Y.; Miller, F.D.; Henrich, T.J.; Gerschenson, M. COVID-19 clinical presentation, management, and epidemiology: A concise compendium. Front. Public Health 2025, 13, 1498445. [Google Scholar] [CrossRef] [PubMed]
- Vicenzi, E.; Canducci, F.; Pinna, D.; Mancini, N.; Carletti, S.; Lazzarin, A.; Bordignon, C.; Poli, G.; Clementi, M. Coronaviridae and SARS-associated coronavirus strain HSR1. Emerg. Infect. Dis. 2004, 10, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, J.; Arnold, K.; Pawlinski, R.; Key, N.S. Using heparin molecules to manage COVID-2019. Res. Pract. Thromb. Haemost. 2020, 4, 518–523. [Google Scholar] [CrossRef]
- Hippensteel, J.A.; LaRiviere, W.B.; Colbert, J.F.; Langouët-Astrié, C.J.; Schmidt, E.P. Heparin as a Therapy for COVID-19: Current Evidence and Future Possibilities. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2020, 319, L211–L217. [Google Scholar] [CrossRef]
- Miesbach, W.; Makris, M. COVID-19: Coagulopathy, Risk of Thrombosis, and the Rationale for Anticoagulation. Clin. Appl. Thromb. Hemost. 2020, 26, 1076029620938149. [Google Scholar] [CrossRef]
- Lindahl, U.; Li, J.P. Heparin—An old drug with multiple potential targets in COVID-19 therapy. J. Thromb. Haemost. 2020, 18, 2422–2424. [Google Scholar] [CrossRef]
- Lawler, P.R.; Goligher, E.C.; Berger, J.S.; Neal, M.D.; McVerry, B.J.; Nicolau, J.C.; Gong, M.N.; Carrier, M.; Rosenson, R.S.; Reynolds, H.R.; et al. Therapeutic Anticoagulation with Heparin in Noncritically Ill Patients with COVID-19. N. Engl. J. Med. 2021, 385, 790–802. [Google Scholar]
- Mycroft-West, C.J.; Su, D.; Pagani, I.; Rudd, T.R.; Elli, S.; Gandhi, N.S.; Guimond, S.E.; Miller, G.J.; Meneghetti, M.C.Z.; Nader, H.B.; et al. Heparin Inhibits Cellular Invasion by SARS-CoV-2: Structural Dependence of the Interaction of the Spike S1 Receptor-Binding Domain with Heparin. Thromb. Haemost. 2020, 120, 1700–1715. [Google Scholar] [CrossRef]
- Yang, Y.; Du, Y.; Kaltashov, I.A. The utility of native MS for understanding the mechanism of action of repurposed therapeutics in COVID-19: Heparin as a disruptor of the SARS-CoV-2 interaction with its host cell receptor. Anal. Chem. 2020, 92, 10930–10934. [Google Scholar] [CrossRef]
- Yang, Y.; Ivanov, D.G.; Kaltashov, I.A. The challenge of structural heterogeneity in the native mass spectrometry studies of the SARS-CoV-2 spike protein interactions with its host cell-surface receptor. Anal. Bioanal. Chem. 2021, 413, 7205–7214. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Jin, W.; Sood, A.; Montgomery, D.W.; Grant, O.C.; Fuster, M.M.; Fu, L.; Dordick, J.S.; Woods, R.J.; Zhang, F.; et al. Characterization of heparin and severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) spike glycoprotein binding interactions. Antivir. Res. 2020, 181, 104873. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784. [Google Scholar] [CrossRef]
- Cardin, A.D.; Weintraub, H.J. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 1989, 9, 21–32. [Google Scholar] [CrossRef]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; Robertson, D.L. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef]
- Barile, E.; Baggio, C.; Gambini, L.; Shiryaev, S.A.; Strongin, A.Y.; Pellecchia, M. Potential Therapeutic Targeting of Coronavirus Spike Glycoprotein Priming. Molecules 2020, 25, 2424. [Google Scholar] [CrossRef]
- Ivachtchenko, A.V.; Khvat, A.V.; Shkil, D.O. Development and Prospects of Furin Inhibitors for Therapeutic Applications. Int. J. Mol. Sci. 2024, 25, 9199. [Google Scholar] [CrossRef]
- Seidah, N.G.; Prat, A. The biology and therapeutic targeting of the proprotein convertases. Nat. Rev. Drug Discov. 2012, 11, 367–383. [Google Scholar] [CrossRef]
- Johnson, B.A.; Xie, X.; Bailey, A.L.; Kalveram, B.; Lokugamage, K.G.; Muruato, A.; Zou, J.; Zhang, X.; Juelich, T.; Smith, J.K.; et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature 2021, 591, 293–299. [Google Scholar] [CrossRef]
- Sasaki, M.; Toba, S.; Itakura, Y.; Chambaro, H.M.; Kishimoto, M.; Tabata, K.; Intaruck, K.; Uemura, K.; Sanaki, T.; Sato, A.; et al. SARS-CoV-2 Bearing a Mutation at the S1/S2 Cleavage Site Exhibits Attenuated Virulence and Confers Protective Immunity. mBio 2021, 12, e0141521. [Google Scholar] [CrossRef]
- Willett, B.J.; Grove, J.; MacLean, O.A.; Wilkie, C.; De Lorenzo, G.; Furnon, W.; Cantoni, D.; Scott, S.; Logan, N.; Ashraf, S.; et al. SARS-CoV-2 Omicron is an immune escape variant with an altered cell entry pathway. Nat. Microbiol. 2022, 7, 1161–1179. [Google Scholar] [CrossRef] [PubMed]
- Molloy, S.S.; Bresnahan, P.A.; Leppla, S.H.; Klimpel, K.R.; Thomas, G. Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Arg-X-X-Arg and efficiently cleaves anthrax toxin protective antigen. J. Biol. Chem. 1992, 267, 16396–16402. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory. Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]
- Obradovic, Z.; Peng, K.; Vucetic, S.; Radivojac, P.; Brown, C.J.; Dunker, A.K. Predicting intrinsic disorder from amino acid sequence. Proteins 2003, 53, 566–572. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- Petitou, M.; van Boeckel, C.A.A. A synthetic antithrombin III binding pentasaccharide is now a drug! What comes next? Angew. Chem. Int. Ed. Engl. 2004, 43, 3118–3133. [Google Scholar] [CrossRef]
- Kayitmazer, A.B.; Quinn, B.; Kimura, K.; Ryan, G.L.; Tate, A.J.; Pink, D.A.; Dubin, P.L. Protein Specificity of Charged Sequences in Polyanions and Heparins. Biomacromolecules 2010, 11, 3325–3331. [Google Scholar] [CrossRef]
- Niu, C.; Zhao, Y.; Bobst, C.E.; Savinov, S.N.; Kaltashov, I.A. Identification of Protein Recognition Elements within Heparin Chains Using Enzymatic Foot-Printing in Solution and Online SEC/MS. Anal. Chem. 2020, 92, 7565–7573. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, J.; Ringborg, L.H.; Roepstorrf, P. On-line size-exclusion chromatography/mass spectrometry of low molecular mass heparin. J. Mass Spectrom. 2004, 39, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Abzalimov, R.R.; Kaltashov, I.A. Electrospray ionization mass spectrometry of highly heterogeneous protein systems: Protein ion charge state assignment via incomplete charge reduction. Anal. Chem. 2010, 82, 7523–7526. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Abzalimov, R.R.; Kaltashov, I.A. Interactions of Intact Unfractionated Heparin with Its Client Proteins Can Be Probed Directly Using Native Electrospray Ionization Mass Spectrometry. Anal. Chem. 2016, 88, 1711–1718. [Google Scholar] [CrossRef]
- Yang, Y.; Du, Y.; Ivanov, D.; Niu, C.; Clare, R.; Smith, J.W.; Nazy, I.; Kaltashov, I.A. Molecular architecture and platelet-activating properties of small immune complexes assembled on heparin and platelet factor 4. Commun. Biol. 2024, 7, 308. [Google Scholar] [CrossRef]
- Fenselau, C.; Yao, X. 18O2-Labeling in quantitative proteomic strategies: A status report. J. Proteome Res. 2009, 8, 2140–2143. [Google Scholar] [CrossRef]
- Hogwood, J.; Mulloy, B.; Lever, R.; Gray, E.; Page, C.P. Pharmacology of Heparin and Related Drugs: An Update. Pharmacol. Rev. 2023, 75, 328–379. [Google Scholar] [CrossRef]
- Tritschler, T.; Le Gal, G.; Brosnahan, S.; Carrier, M. POINT: Should Therapeutic Heparin Be Administered to Acutely Ill Hospitalized Patients with COVID-19? Yes. Chest 2022, 161, 1446–1448. [Google Scholar] [CrossRef]
- Sugraliyev, A.B. Heparin-Induced Thrombocytopenia. Kardiologiia 2024, 64, 18–25. [Google Scholar] [CrossRef]
- Favaloro, E.J.; Henry, B.M.; Lippi, G. The complicated relationships of heparin-induced thrombocytopenia and platelet factor 4 antibodies with COVID-19. Int. J. Lab. Hematol. 2021, 43, 547–558. [Google Scholar] [CrossRef]
- Greinacher, A. CLINICAL PRACTICE. Heparin-Induced Thrombocytopenia. N. Engl. J. Med. 2015, 373, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Toida, T.; Yoshida, H.; Toyoda, H.; Koshiishi, I.; Imanari, T.; Hileman, R.E.; Fromm, J.R.; Linhardt, R.J. Structural differences and the presence of unsubstituted amino groups in heparan sulphates from different tissues and species. Biochem. J. 1997, 322, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Paiardi, G.; Richter, S.; Oreste, P.; Urbinati, C.; Rusnati, M.; Wade, R.C. The binding of heparin to spike glycoprotein inhibits SARS-CoV-2 infection by three mechanisms. J. Biol. Chem. 2022, 298, 101507. [Google Scholar] [CrossRef]
- Pavlov, G.; Finet, S.; Tatarenko, K.; Korneeva, E.; Ebel, C. Conformation of heparin studied with macromolecular hydrodynamic methods and X-ray scattering. Eur. Biophys. J. 2003, 32, 437–449. [Google Scholar] [CrossRef]
- Guo, C.; Tsai, S.J.; Ai, Y.; Li, M.; Anaya, E.; Pekosz, A.; Cox, A.; Gould, S.J. The D614G mutation redirects SARS-CoV-2 spike to lysosomes and suppresses deleterious traits of the furin cleavage site insertion mutation. Sci. Adv. 2022, 8, eade5085. [Google Scholar] [CrossRef]
- Higgins, W.J.; Fox, D.M.; Kowalski, P.S.; Nielsen, J.E.; Worrall, D.M. Heparin enhances serpin inhibition of the cysteine protease cathepsin L. J. Biol. Chem. 2010, 285, 3722–3729. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Mercadante, M.; Nilsson-Payant, B.; Johnson, J.L.; Jaimes, J.A.; Muecksch, F.; Weisblum, Y.; Bram, Y.; Chandar, V.; Whittaker, G.R.; et al. Coagulation factors directly cleave SARS-CoV-2 spike and enhance viral entry. eLife 2022, 11, e77444. [Google Scholar] [CrossRef]
- Spyropoulos, A.C.; Goldin, M.; Giannis, D.; Diab, W.; Wang, J.; Khanijo, S.; Mignatti, A.; Gianos, E.; Cohen, M.; Sharifova, G.; et al. Efficacy and Safety of Therapeutic-Dose Heparin vs Standard Prophylactic or Intermediate-Dose Heparins for Thromboprophylaxis in High-risk Hospitalized Patients with COVID-19: The HEP-COVID Randomized Clinical Trial. JAMA Intern. Med. 2021, 181, 1612–1620. [Google Scholar] [CrossRef]
- Sachs, J.D.; Karim, S.S.A.; Aknin, L.; Allen, J.; Brosbøl, K.; Colombo, F.; Barron, G.C.; Espinosa, M.F.; Gaspar, V.; Gaviria, A.; et al. The Lancet Commission on lessons for the future from the COVID-19 pandemic. Lancet 2022, 400, 1224–1280. [Google Scholar] [CrossRef]
- Tubiana, S.; Rontani, M.; Herlemont, P.; Dray-Spira, R.; Zureik, M.; Weill, A.; Duval, X.; Burdet, C. Long-term health outcomes following hospitalisation for COVID-19: A 30- month cohort analysis. Infect. Dis. 2025, 57, 433–443. [Google Scholar] [CrossRef]
- Lee, L.Y.Y.; Suryadinata, R.; McCafferty, C.; Ignjatovic, V.; Purcell, D.F.J.; Robinson, P.; Morton, C.J.; Parker, M.W.; Anderson, G.P.; Monagle, P.; et al. Heparin Inhibits SARS-CoV-2 Replication in Human Nasal Epithelial Cells. Viruses 2022, 14, 2620. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chain Length (Number of Saccharide Residues) | Number of Negative Charges at pH 7/Number of Sulfate Groups | Max n (FCS Molecules Bound to a Single Chain | ||
|---|---|---|---|---|
| fondaparinux | 5 | 10/8 | 2 | |
| dp6 | (6,6,0) | 6 | 9/6 | 1 |
| (6,7,0) | 6 | 10/7 | 2 (low abundance) | |
| (6,7,1) | 6 | 10/7 | 2 | |
| (6,8,0) | 6 | 11/8 | 2 | |
| (6,8,1) | 6 | 11/8 | 2 | |
| (6,9,0) | 6 | 12/9 | 2 | |
| dp20 | (20,17,0) | 20 | 27/17 | 8 |
| (20,24,n) | 20 | 34/24 | 8 | |
| (20,27,0) | 20 | 37/27 | 8 | |
| (20,28,1) | 20 | 38/28 | 8 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, Y.; Yang, Y.; Nguyen, S.N.; Kaltashov, I.A. The Role of Long-Range Non-Specific Electrostatic Interactions in Inhibiting the Pre-Fusion Proteolytic Processing of the SARS-CoV-2 S Glycoprotein by Heparin. Biomolecules 2025, 15, 778. https://doi.org/10.3390/biom15060778
Du Y, Yang Y, Nguyen SN, Kaltashov IA. The Role of Long-Range Non-Specific Electrostatic Interactions in Inhibiting the Pre-Fusion Proteolytic Processing of the SARS-CoV-2 S Glycoprotein by Heparin. Biomolecules. 2025; 15(6):778. https://doi.org/10.3390/biom15060778
Chicago/Turabian StyleDu, Yi, Yang Yang, Son N. Nguyen, and Igor A. Kaltashov. 2025. "The Role of Long-Range Non-Specific Electrostatic Interactions in Inhibiting the Pre-Fusion Proteolytic Processing of the SARS-CoV-2 S Glycoprotein by Heparin" Biomolecules 15, no. 6: 778. https://doi.org/10.3390/biom15060778
APA StyleDu, Y., Yang, Y., Nguyen, S. N., & Kaltashov, I. A. (2025). The Role of Long-Range Non-Specific Electrostatic Interactions in Inhibiting the Pre-Fusion Proteolytic Processing of the SARS-CoV-2 S Glycoprotein by Heparin. Biomolecules, 15(6), 778. https://doi.org/10.3390/biom15060778